
Whole Exome Sequencing Identifies APCDD1 and HDAC5 Genes as Potentially Cancer Predisposing in Familial Colorectal Cancer

 , , ,
, , , 
 and
and 
Abstract
:1. Introduction
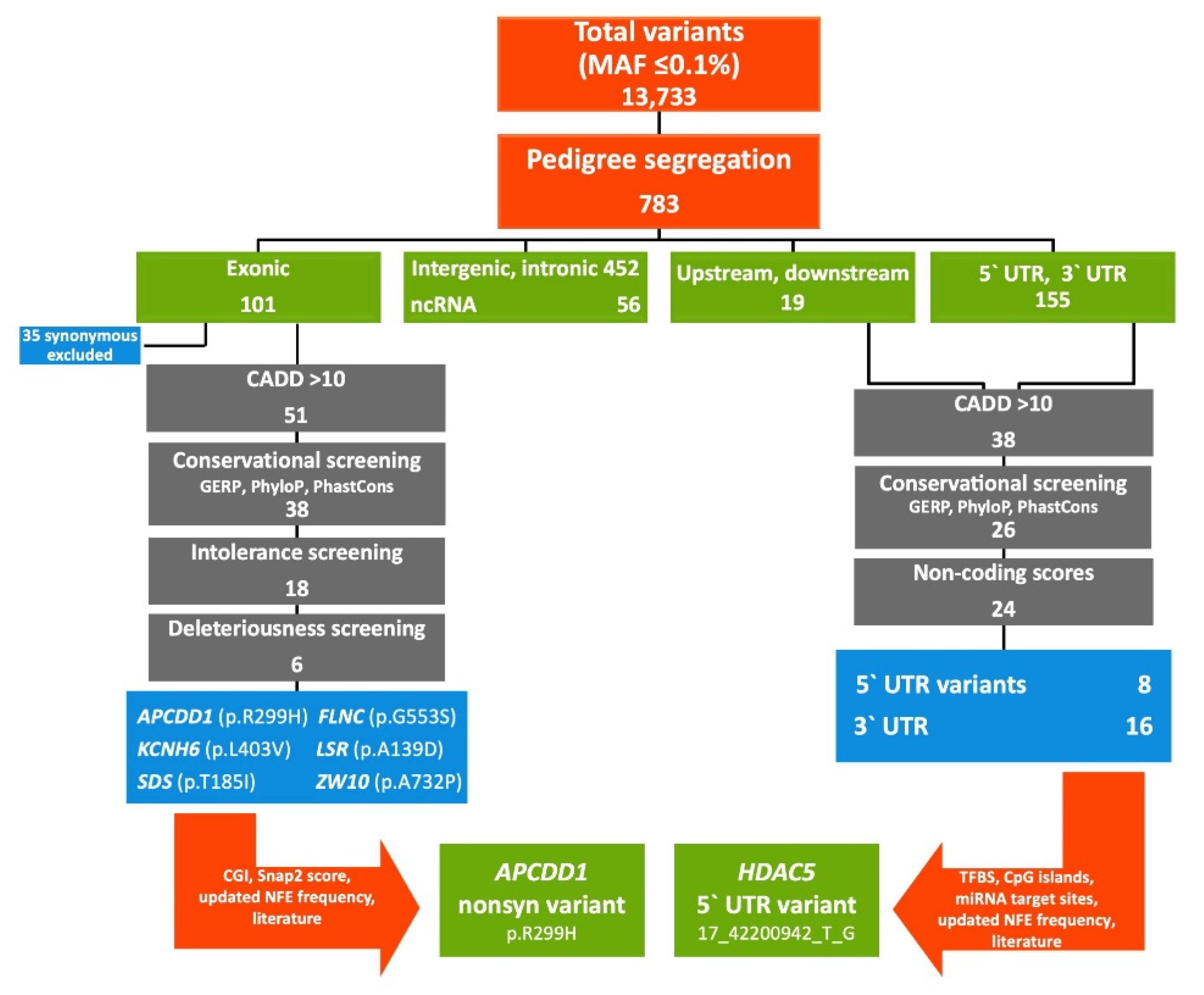
2. Results
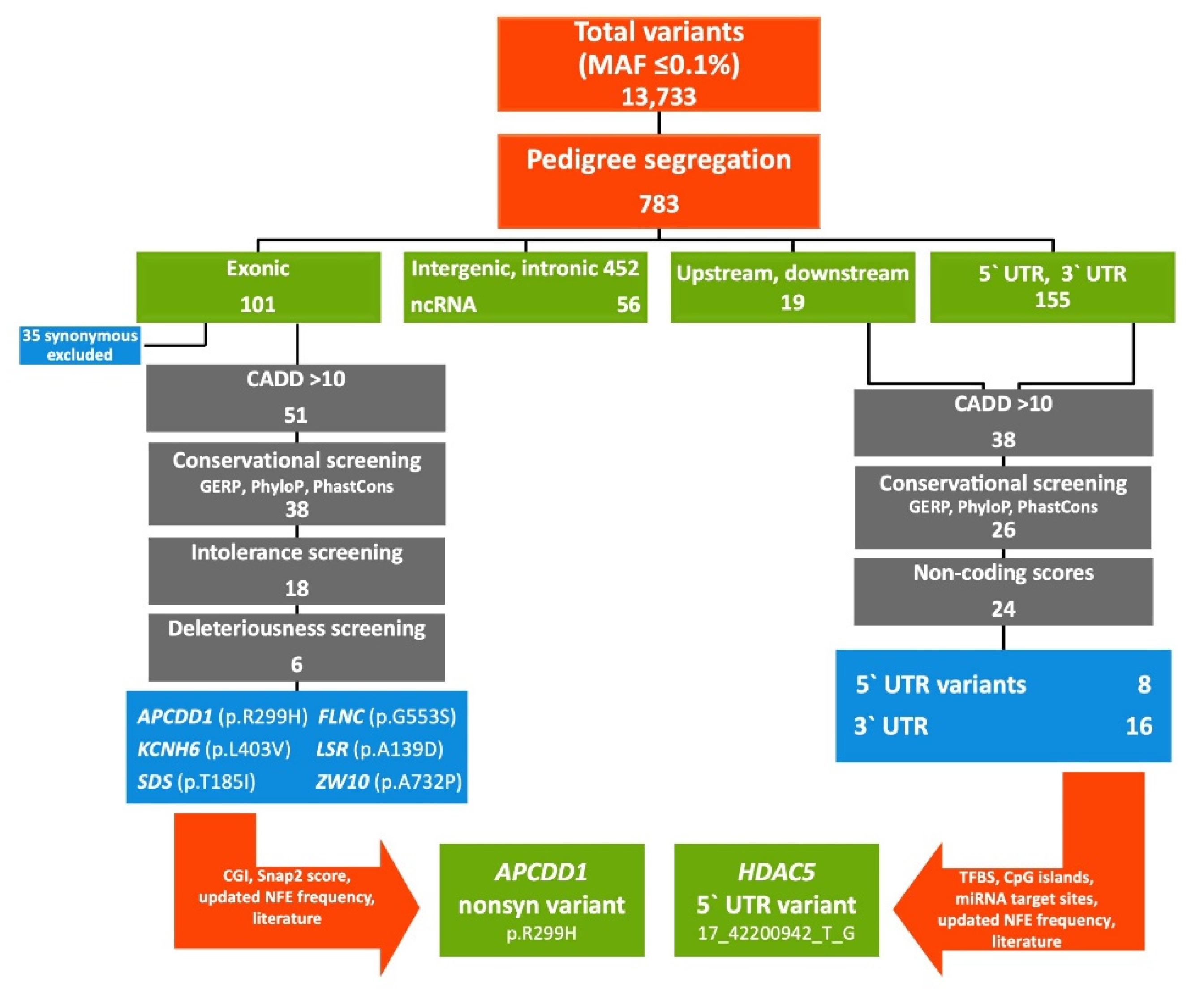
2.1. FCVPPv2 Analysis of Coding Variants Prioritized a Missense Variant in APCDD1 Gene
2.2. FCVPPv2 Analysis of Non-Coding Variants Prioritized a 5′UTR Variant in HDAC5 Gene
2.3. Allele Frequency in a Large Familial CRC Cohort
2.4. APCDD1 Variant Did Not Show a Significant Effect on Proliferation of HEK293T and HT-29 Cells
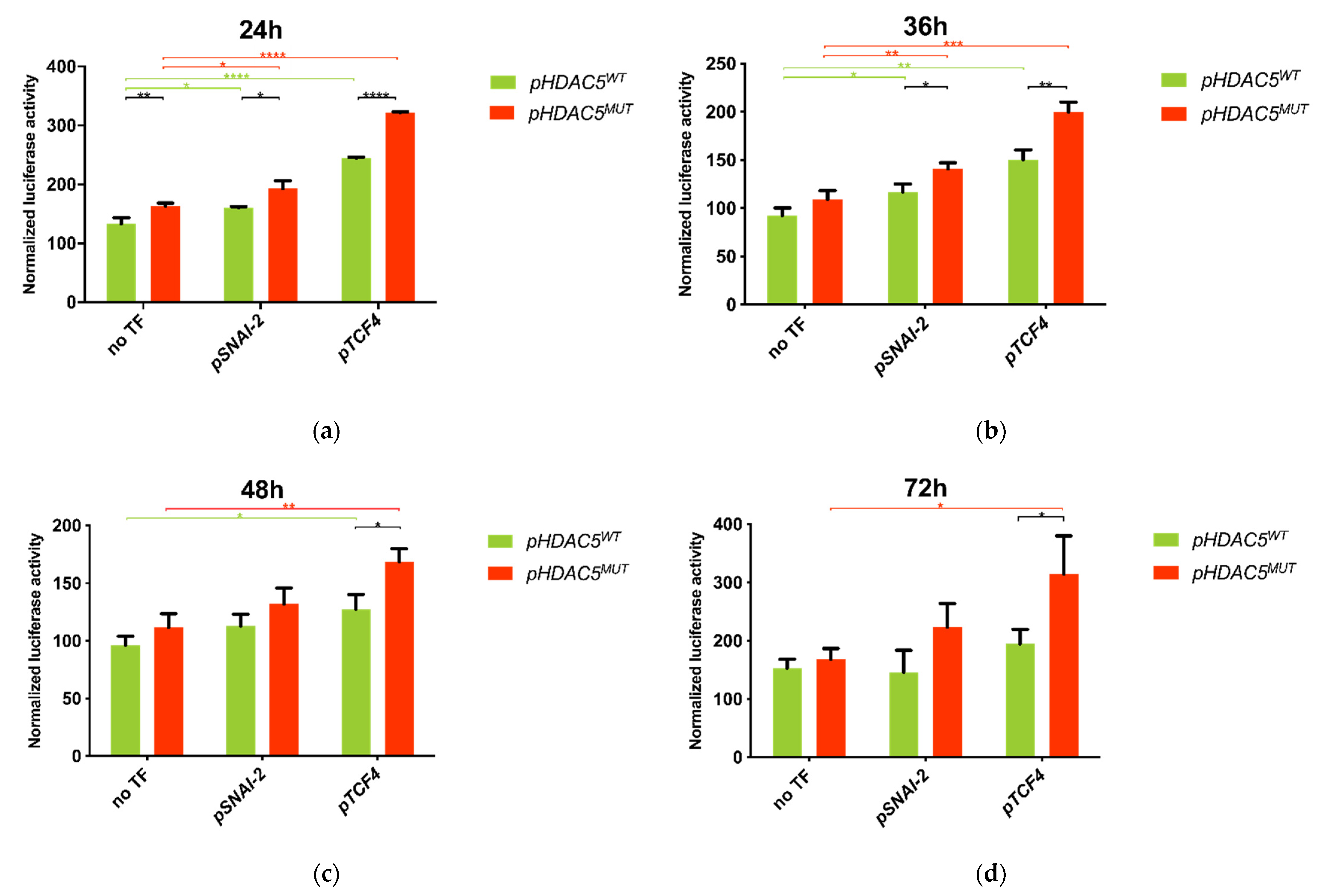
2.5. 5′ UTR Variant of HDAC5 Gene Enhances Promoter Activity
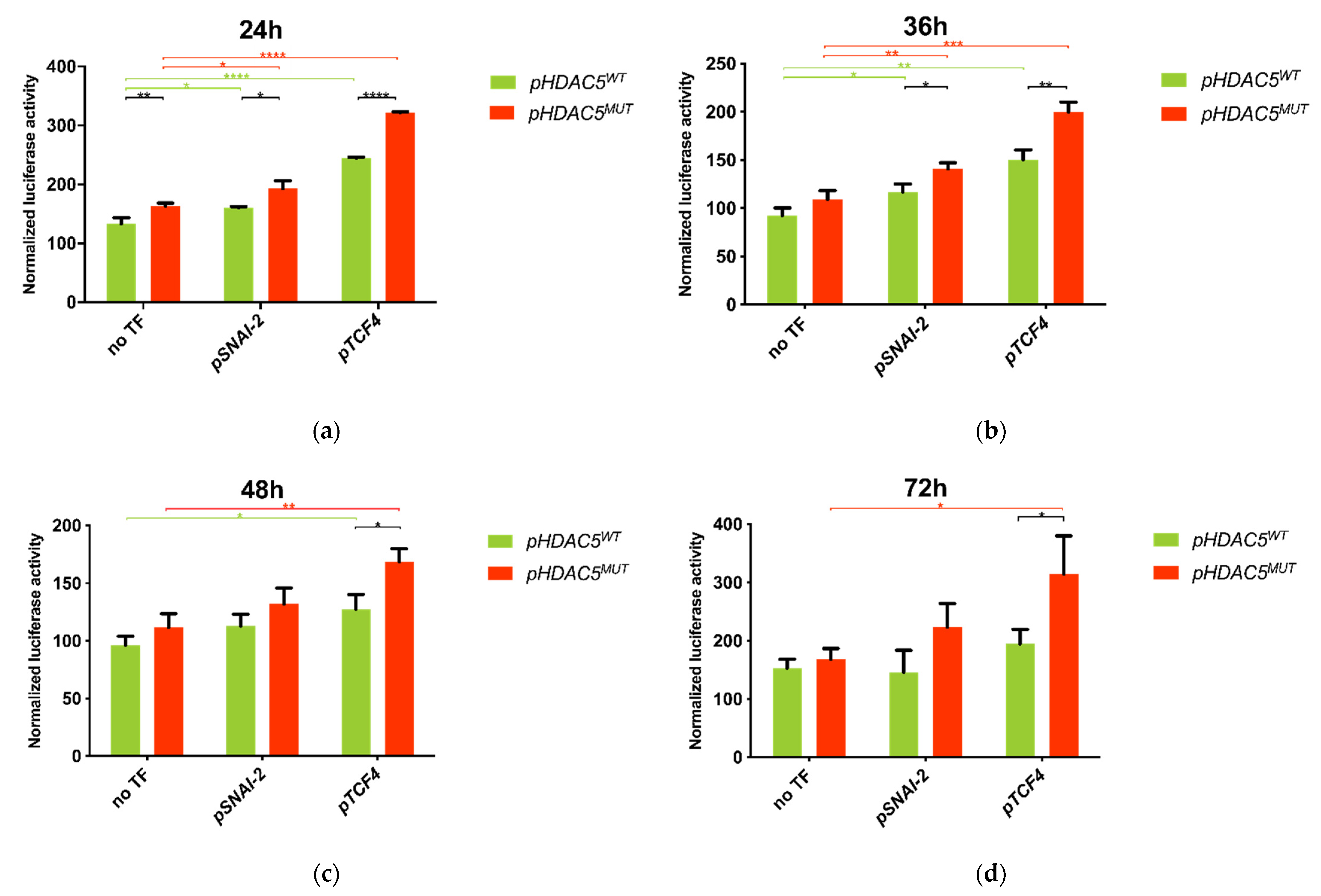
2.6. 5′ UTR Variant of HDAC5 Disrupts SNAI-2 and TCF4 Transcription Factor Binding Sites
2.7. Co-Transfection of HDAC5 and TCF4 Increases Promoter Activity Due to 5′UTR Variant of HDAC5 Gene
3. Discussion
4. Materials and Methods
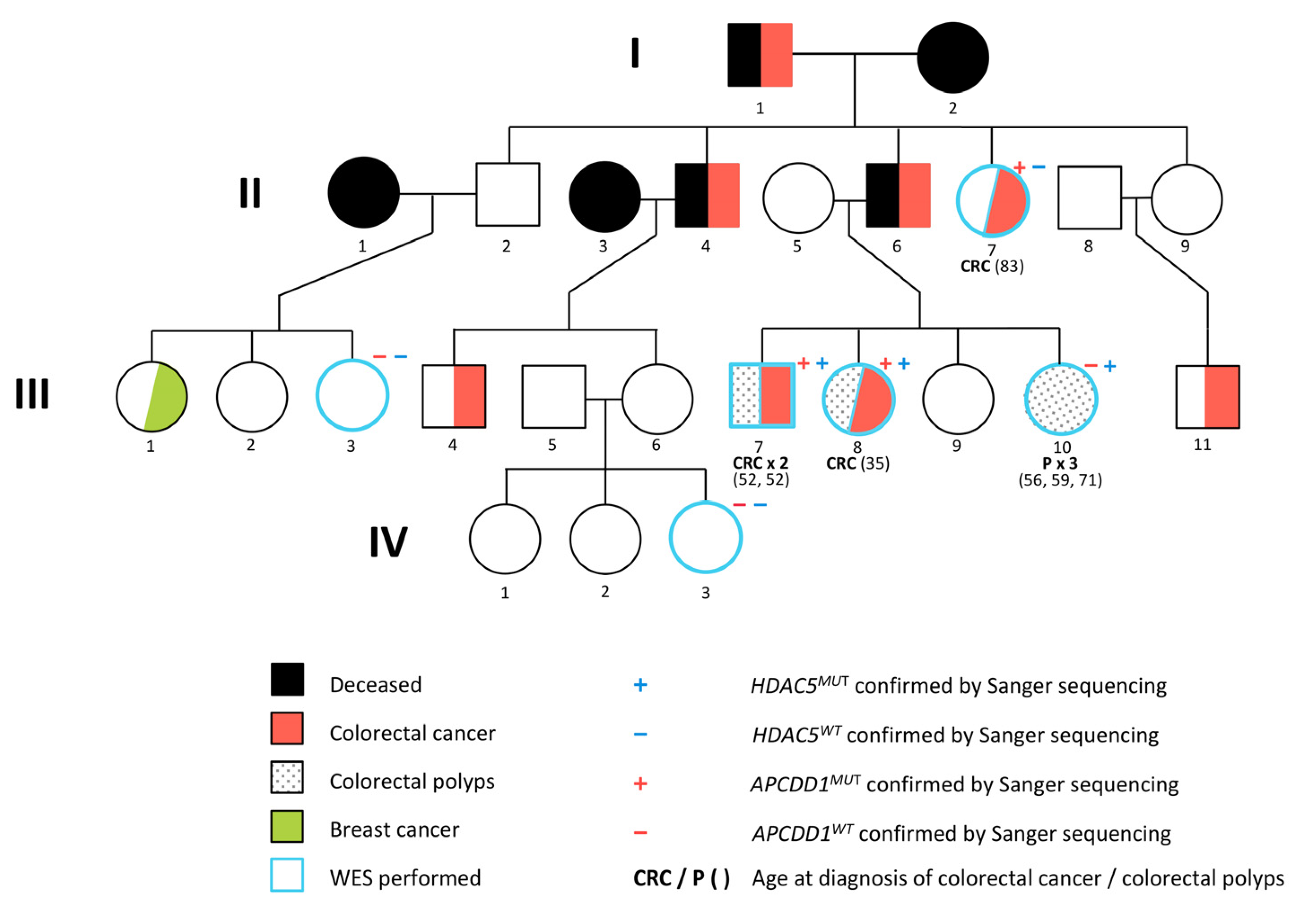
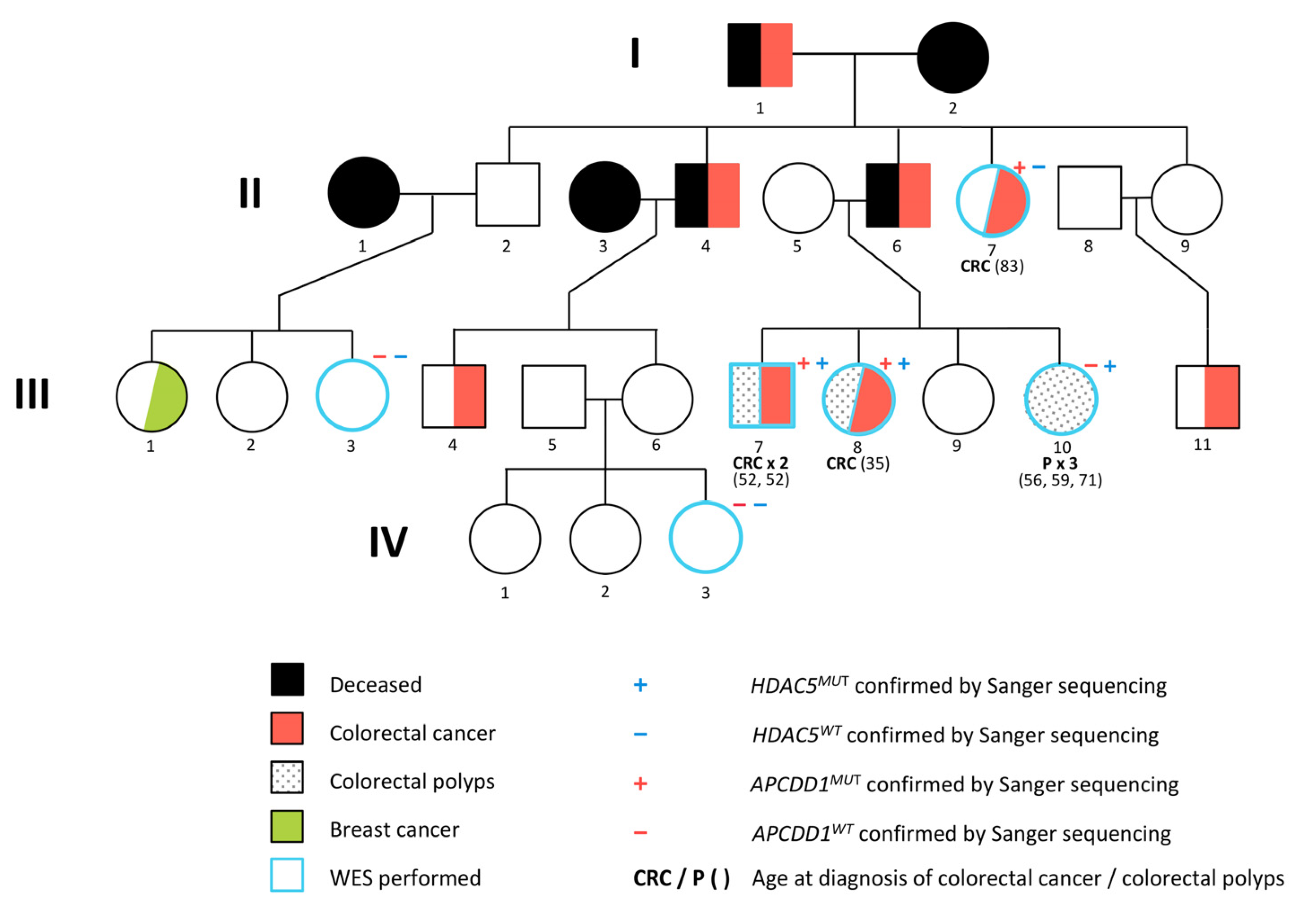
4.1. Patient Samples
4.2. Whole Exome Sequencing and Variant Evaluation
4.3. Variant Calling, Annotation and Filtering
4.4. Variant Filtering According to FCVPPv2
4.4.1. Familial Segregation of the Cancer Predisposing Variants
4.4.2. Analysis of Coding Variants
4.4.3. Analysis of Non-Coding Variants
4.5. Analysis of Transcription Factor Binding Sites
4.6. Variant Validation with IGV
4.7. Confirmation of Familial Segregation by Sanger Sequencing
4.8. Screening of Large Case and Control Cohorts
4.9. PCR-Based Cloning of Gene Reporter Constructs
4.10. Cloning of SNAI-2, TCF4 and APCDD1
4.11. Plasmid Amplification and Extraction
4.12. Cell Line and Culture Conditions
4.13. Cell Proliferation Assay—APCDD1
4.14. Luciferase Reporter Assay—HDAC5
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and Heritable Factors in the Causation of Cancer—Analyses of Cohorts of Twins from Sweden, Denmark, and Finland. N. Engl. J. Med. 2000, 343, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Jasperson, K.W.; Tuohy, T.M.; Neklason, D.W.; Burt, R.W. Hereditary and Familial Colon Cancer. Gastroenterology 2010, 138, 2044–2058. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, F.C.; Wernhoff, P.; Dominguez-Barrera, C.; Dominguez-Valentin, M. Update on Hereditary Colorectal Cancer. Anticancer. Res. 2016, 36, 4399–4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Peng, B.; Han, Y.; Chen, W.V.; Rother, J.; Tomlinson, G.E.; Boland, C.R.; Chaussabel, M.; Frazier, M.L.; Amos, C.I. Mutations of HNRNPA0 and WIF1 predispose members of a large family to multiple cancers. Fam. Cancer 2015, 14, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, R.P.; Hoogerbrugge, N. NTHL1 defines novel cancer syndrome. Oncotarget 2015, 6, 34069–34070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weren, R.D.; Ligtenberg, M.J.; Kets, C.M.; De Voer, R.M.; Verwiel, E.T.; Spruijt, L.; van Zelst-Stams, W.A.; Jongmans, M.C.; Gilissen, C.; Hehir-Kwa, J.Y.; et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 2015, 47, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Briggs, S.; Tomlinson, I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J. Pathol. 2013, 230, 148–153. [Google Scholar] [CrossRef] [Green Version]
- Palles, C.; Cazier, J.-B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorans, M.; Dow, E.; Macrae, F.A.; Winship, I.M.; Buchanan, D.D. Update on Hereditary Colorectal Cancer: Improving the Clinical Utility of Multigene Panel Testing. Clin. Color. Cancer 2018, 17, e293–e305. [Google Scholar] [CrossRef] [Green Version]
- Gloss, B.S.; Dinger, M.E. Realizing the significance of noncoding functionality in clinical genomics. Exp. Mol. Med. 2018, 50, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mucaki, E.J.; Caminsky, N.G.; Perri, A.M.; Lu, R.; Laederach, A.; Halvorsen, M.; Knoll, J.H.M.; Rogan, P.K. A unified analytic framework for prioritization of non-coding variants of uncertain significance in heritable breast and ovarian cancer. BMC Med. Genom. 2016, 9, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alanazi, I.O.; Al Shehri, Z.S.; Ebrahimie, E.; Giahi, H.; Mohammadi-Dehcheshmeh, M. Non-coding and coding genomic variants distinguish prostate cancer, castration-resistant prostate cancer, familial prostate cancer, and metastatic castration-resistant prostate cancer from each other. Mol. Carcinog. 2019, 58, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, S.A.; Spiridonov, N.A. The mammalian transcriptome and the function of non-coding DNA sequences. Genome Biol. 2004, 5, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Bandapalli, O.R.; Paramasivam, N.; Giangiobbe, S.; Diquigiovanni, C.; Bonora, E.; Eils, R.; Schlesner, M.; Hemminki, K.; Försti, A. Familial Cancer Variant Prioritization Pipeline version 2 (FCVPPv2) applied to a papillary thyroid cancer family. Sci. Rep. 2018, 8, 11635. [Google Scholar] [CrossRef] [Green Version]
- Bandapalli, O.R.; Paramasivam, N.; Giangiobbe, S.; Kumar, A.; Benisch, W.; Engert, A.; Witzens-Harig, M.; Schlesner, M.; Hemminki, K.; Försti, A. Whole genome sequencing reveals DICER1 as a candidate predisposing gene in familial Hodgkin lymphoma. Int. J. Cancer 2018, 143, 2076–2078. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Kumar, A.; Giangiobbe, S.; Bonora, E.; Hemminki, K.; Försti, A.; Bandapalli, O.R. Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways. Biomolecules 2019, 9, 605. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Giangiobbe, S.; Kumar, A.; Paramasivam, N.; Dymerska, D.; Behnisch, W.; Witzens-Harig, M.; Lubinski, J.; Hemminki, K.; Försti, A.; et al. Identification of Familial Hodgkin Lymphoma Predisposing Genes Using Whole Genome Sequencing. Front. Bioeng. Biotechnol. 2020, 8, 179. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Dayem Ullah, A.Z.; Oscanoa, J.; Wang, J.; Nagano, A.; Lemoine, N.R.; Chelala, C. SNPnexus: Assessing the functional relevance of genetic variation to facilitate the promise of precision medicine. Nucleic Acids Res. 2018, 46, W109–W113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef]
- Wang, Z.; Cummins, J.M.; Shen, D.; Cahill, D.P.; Jallepalli, P.V.; Wang, T.-L.; Parsons, D.W.; Traverso, G.; Awad, M.; Silliman, N.; et al. Three Classes of Genes Mutated In Colorectal Cancers with Chromosomal Instability. Cancer Res. 2004, 64, 2998–3001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridge, A.; Brown, S.D.; Chang, H.-Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 2018, 46, D493–D496. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Fujita, M.; Furukawa, Y.; Hamamoto, R.; Shimokawa, T.; Miwa, N.; Ogawa, M.; Nakamura, Y. Isolation of a novel human gene, APCDD1, as a direct target of the beta-Catenin/T-cell factor 4 complex with probable involvement in colorectal carcinogenesis. Cancer Res. 2002, 62, 5651–5656. [Google Scholar]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar]
- Hoffman, M.M.; Buske, O.J.; Wang, J.; Weng, Z.; Bilmes, J.A.; Noble, W.S. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods 2012, 9, 473–476. [Google Scholar] [CrossRef] [Green Version]
- Fre, S.; Pallavi, S.K.; Huyghe, M.; Laé, M.; Janssen, K.-P.; Robine, S.; Artavanis-Tsakonas, S.; Louvard, D. Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proc. Natl. Acad. Sci. USA 2009, 106, 6309–6314. [Google Scholar] [CrossRef] [Green Version]
- Badenes, M.; Trindade, A.; Pissarra, H.; Lopes-da-Costa, L.; Duarte, A. Delta-like 4/Notch signaling promotes Apc (Min/+) tumor initiation through angiogenic and non-angiogenic related mechanisms. BMC Cancer 2017, 17, 50. [Google Scholar]
- He, P.; Liang, J.; Shao, T.; Guo, Y.; Hou, Y.; Li, Y. HDAC5 promotes colorectal cancer cell proliferation by up-regulating DLL4 expression. Int. J. Clin. Exp. Med. 2015, 8, 6510–6516. [Google Scholar]
- Stypula-Cyrus, Y.; Damania, D.; Kunte, D.P.; Cruz, M.D.; Subramanian, H.; Roy, H.K.; Backman, V. HDAC Up-Regulation in Early Colon Field Carcinogenesis Is Involved in Cell Tumorigenicity through Regulation of Chromatin Structure. PLoS ONE 2013, 8, e64600. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Rennoll, S.A.; Raup-Konsavage, W.M.; Yochum, G.S. A dynamic exchange of TCF3 and TCF4 transcription factors controls MYC expression in colorectal cancer cells. Cell Cycle 2015, 14, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Findlay, V.J.; Wang, C.; Nogueira, L.M.; Hurst, K.; Quirk, D.; Ethier, S.P.; O’Carroll, K.F.S.; Watson, D.K.; Camp, E.R. SNAI2 Modulates Colorectal Cancer 5-Fluorouracil Sensitivity through miR145 Repression. Mol. Cancer Ther. 2014, 13, 2713–2726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Sousa, E.M.F.; Colak, S.; Buikhuisen, J.; Koster, J.; Cameron, K.; de Jong, J.H.; Tuynman, J.B.; Prasetyanti, P.R.; Fessler, E.; van den Bergh, S.P.; et al. Methylation of cancer-stem-cell-associated Wnt target genes predicts poor prognosis in colorectal cancer patients. Cell Stem Cell 2011, 9, 476–485. [Google Scholar]
- Shimomura, Y.; Agalliu, D.; Vonica, A.; Luria, V.; Wajid, M.; Baumer, A.; Belli, S.; Petukhova, L.; Schinzel, A.; Brivanlou, A.H.; et al. APCDD1 is a novel Wnt inhibitor mutated in hereditary hypotrichosis simplex. Nature 2010, 464, 1043–1047. [Google Scholar] [CrossRef] [Green Version]
- Ordóñez-Morán, P.; Dafflon, C.; Imajo, M.; Nishida, E.; Huelsken, J. HOXA5 Counteracts Stem Cell Traits by Inhibiting Wnt Signaling in Colorectal Cancer. Cancer Cell 2015, 28, 815–829. [Google Scholar] [CrossRef] [Green Version]
- Marek, L.; Hamacher, A.; Hansen, F.K.; Kuna, K.; Gohlke, H.; Kassack, M.U.; Kurz, T. Histone Deacetylase (HDAC) Inhibitors with a Novel Connecting Unit Linker Region Reveal a Selectivity Profile for HDAC4 and HDAC5 with Improved Activity against Chemoresistant Cancer Cells. J. Med. Chem. 2013, 56, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Milde, T.; Oehme, I.; Korshunov, A.; Kopp-Schneider, A.; Remke, M.; Northcott, P.A.; Deubzer, H.E.; Lodrini, M.; Taylor, M.D.; Von Deimling, A.; et al. HDAC5 and HDAC9 in Medulloblastoma: Novel Markers for Risk Stratification and Role in Tumor Cell Growth. Clin. Cancer Res. 2010, 16, 3240–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Lou, B.; Chen, W.; Zhang, J.; Lin, S.; Lv, F.-F.; Chen, Y. Down-regulation of HDAC5 inhibits growth of human hepatocellular carcinoma by induction of apoptosis and cell cycle arrest. Tumor Biol. 2014, 35, 11523–11532. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Tsai, A.-C.; Chen, M.-C.; Shen, P.-J.; Cheng, Y.-C.; Kuo, C.-C.; Pan, S.-L.; Liu, Y.-M.; Liu, J.-F.; Yeh, T.-K.; et al. Azaindolylsulfonamides, with a More Selective Inhibitory Effect on Histone Deacetylase 6 Activity, Exhibit Antitumor Activity in Colorectal Cancer HCT116 Cells. J. Med. Chem. 2014, 57, 4009–4022. [Google Scholar] [CrossRef] [PubMed]
- Hrckulak, D.; Janeckova, L.; Lanikova, L.; Kriz, V.; Horazna, M.; Babosova, O.; Vojtechova, M.; Galuskova, K.; Sloncova, E.; Korinek, V. Wnt Effector TCF4 Is Dispensable for Wnt Signaling in Human Cancer Cells. Genes 2018, 9, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [Green Version]
- Bienz, M.; Clevers, H. Linking colorectal cancer to Wnt signaling. Cell 2000, 103, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.R.; Myint, T.; Goh, H.S. Over-expression of the c-myc proto-oncogene in colorectal carcinoma. Br. J. Cancer 1993, 68, 407–413. [Google Scholar] [CrossRef] [Green Version]
- Erisman, M.D.; Rothberg, P.G.; Diehl, R.E.; Morse, C.C.; Spandorfer, J.M.; Astrin, S.M. Deregulation of c-myc gene expression in human colon carcinoma is not accompanied by amplification or rearrangement of the gene. Mol. Cell. Biol. 1985, 5, 1969–1976. [Google Scholar] [CrossRef] [Green Version]
- Rochlitz, C.F.; Herrmann, R.; De Kant, E. Overexpression and Amplification of c-myc during Progression of Human Colorectal Cancer. Oncology 1996, 53, 448–454. [Google Scholar] [CrossRef]
- Hatzis, P.; Van Der Flier, L.G.; Van Driel, M.A.; Guryev, V.; Nielsen, F.; Denissov, S.; Nijman, I.J.; Koster, J.; Santo, E.E.; Welboren, W.; et al. Genome-Wide Pattern of TCF7L2/TCF4 Chromatin Occupancy in Colorectal Cancer Cells. Mol. Cell. Biol. 2008, 28, 2732–2744. [Google Scholar] [CrossRef] [Green Version]
- Kriegl, L.; Horst, D.; Reiche, J.A.; Engel, J.; Kirchner, T.; Jung, A. LEF-1 and TCF4 expression correlate inversely with survival in colorectal cancer. J. Transl. Med. 2010, 8, 123. [Google Scholar] [CrossRef] [Green Version]
- Mao, C.D.; Byers, S.W. Cell-context dependent TCF/LEF expression and function: Alternative tales of repression, de-repression and activation potentials. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 207–236. [Google Scholar] [CrossRef] [Green Version]
- Hoverter, N.P.; Waterman, M.L. A Wnt-fall for gene regulation: Repression. Sci. Signal. 2008, 1, pe43. [Google Scholar] [CrossRef]
- Arce, L.; Yokoyama, N.N.; Waterman, M.L. Diversity of LEF/TCF action in development and disease. Oncogene 2006, 25, 7492–7504. [Google Scholar] [CrossRef] [Green Version]
- Cadigan, K.M. TCFs and Wnt/beta-catenin signaling: More than one way to throw the switch. Curr. Top Dev. Biol. 2012, 98, 1–34. [Google Scholar]
- Cadigan, K.M.; Waterman, M.L. TCF/LEFs and Wnt Signaling in the Nucleus. Cold Spring Harb. Perspect. Biol. 2012, 4, a007906. [Google Scholar] [CrossRef]
- Brantjes, H.; Roose, J.; Van De Wetering, M.; Clevers, H. All Tcf HMG box transcription factors interact with Groucho-related co-repressors. Nucleic Acids Res. 2001, 29, 1410–1419. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Matise, M.P. Tcf7l2/Tcf4 Transcriptional Repressor Function Requires HDAC Activity in the Developing Vertebrate CNS. PLoS ONE 2016, 11, e0163267. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Schnabel, B. DNA isolation by a rapid method from human blood samples: Effects of MgCl2, EDTA, storage time, and temperature on DNA yield and quality. Biochem. Genet. 1993, 31, 321–328. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.; Consortium, W.G.S.; Wilkie, A.O.; McVean, G.; Lunter, G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Smigielski, E.M.; Sirotkin, K.; Ward, M.; Sherry, S.T. dbSNP: A database of single nucleotide polymorphisms. Nucleic Acids Res. 2000, 28, 352–355. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Steinke, V.; Engel, C.; Buttner, R.; Schackert, H.K.; Schmiegel, W.H.; Propping, P. Hereditary nonpolyposis colorectal cancer (HNPCC)/Lynch syndrome. Dtsch. Arztebl. Int. 2013, 110, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Brandt, A.; Bermejo, J.L.; Sundquist, J.; Hemminki, K. Age of onset in familial cancer. Ann. Oncol. 2008, 19, 2084–2088. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [Green Version]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2009, 20, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Ward, L.D.; Kellis, M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012, 40, D930–D934. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [Green Version]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef]
- Hecht, M.; Bromberg, Y.; Rost, B. Better prediction of functional effects for sequence variants. BMC Genom. 2015, 16 (Suppl. 8), S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betel, D.; Koppal, A.; Agius, P.; Sander, C.; Leslie, C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 2010, 11, R90. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Zhang, S.; Shang, S.; Zhang, B.; Li, S.; Wang, X.; Wang, F.; Su, J.; Wu, Q.; Liu, H.; et al. SEA: A super-enhancer archive. Nucleic Acids Res. 2016, 44, D172–D179. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; Van Der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2019, 48, D87–D92. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [Green Version]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’Ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Gene Name | Variant | Pedigree Segregation | Allele Frequency in NFE | CADD SCORE | Conservational Scores | Intolerance Scores (%) | Deleteriousness Scores * (%) | Amino Acid Change | Snap2 | CGI | Protein Function | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ExAC | gnomAD | GERP | PhyloP | PhastCons | Score | Accuracy (%) | |||||||||
| APCDD1 | 18_10485580_G_A | III7, III8, III10 | 7.37 × 10−5 | 1.4 × 10−4 | 27.5 | 4.94 | 9.26 | 1 | 100 | 66.67 | R299H | 54 | 75 | ND | Inhibition of Wnt signaling, controversial function |
| FLNC | 7_128480709_G_A | III7, III8, III10 | 5.25 × 10−4 | 2.57 × 10−4 | 29.3 | 5.02 | 9.82 | 1 | 100 | 100 | G553S | 33 | 66 | ND | Anchoring of membrane proteins for actin cytoskeleton |
| KCNH6 | 17_61613135_T_G | III7, III8, III10 | 4.51 × 10−5 | 6.16 × 10−5 | 23.4 | 3.39 | 3.96 | 1 | 100 | 91.67 | L403V | 6 | 53 | ND | Regulation of neurotransmitter release, neuronal excitability, epithelial electrolyte transport |
| LSR | 19_35741380_C_A | II7, III7, III8, III10 | . | . | 27.4 | 4.88 | 7.21 | 1 | 75 | 75 | A139D | 80 | 91 | ND | Lipoprotein metabolism |
| MTX1 | 1_155181922_A_G | III7, III8, III10 | 9.25 × 10−5 | 1.52 × 10−4 | 23.3 | 3.64 | 2.89 | 1 | 66.67 | 66.67 | Y228C | 32 | 66 | ND | Mitochondrial protein import |
| SDS | 12_113836573_G_A | III7, III8, III10 | 4.59 × 10−5 | 5.44 × 10−5 | 23.4 | 4 | 5.69 | 1 | 75 | 100 | T91I | 46 | 71 | ND | Serine and glycine metabolism, gluconeogenesis |
| ZW10 | 11_113607367_C_G | III7, III8, III10 | 5.99 × 10−4 | 6.43 × 10−4 | 34 | 6.17 | 7.49 | 1 | 75 | 75 | A732P | 70 | 85 | ND | Chromosome segregation, mitotic checkpoint |
| Gene Name | Variant | Pedigree Segregation | Allele Frequency in NFE | Bedtools Intersect I | CADD v1.4 | SNPnexus | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ExAC | gnomAD NFE | CADD SCORE | Conservational Scores | Chromatin State | TFBS | TFBS Peaks IV | Non Coding Scores V (%) | CpG Island | CpG Ratio VI | TFBS | ||||||||
| PhastCons | PhyloP | GERP | ChromHMM II State | ChromHMM II Score | Segway III | |||||||||||||
| CA4 | 17_58227298_G_C | III7, III8, III10 | . | . | P:58227287 ..58227313,+ | 12.6 | 0.44 | 0.72 | 2.77 | Enh Biv | 0.24 | TF0 | 1 | 4 | 50 | 61 | 0.89 | . |
| CALM3 | 19_47105342_A_G | II7, III7, III8, III10 | . | . | . | 19.8 | 0.87 | 1.94 | 2.49 | TssA | 0.87 | GS | 1 | 1 | 50 | . | . | . |
| HDAC5 | 17_42200942_T_G | II7, III7, III8 | . | 6.64 × 10−5 | . | 21.9 | 1.00 | 0.87 | 3.99 | TssA | 0.98 | TSS | 17 | 42 | 50 | 92 | 0.97 | |
| PLAA | 9_26947165_G_A | III7, III8, III10 | . | 6.49 × 10−5 | P:26947129 ..26947212,- | 22 | 0.99 | 0.92 | 4.8 | TssA | 0.94 | TSS | 41 | 61 | 83.3 | 72 | 0.86 | NRF2 |
| PPTC7 | 12_111021082_G_C | III7, III8, III10 | . | 2.69 × 10−4 | SE: hg19_A549_2 12_111015565 | 16.5 | 1.00 | 3.85 | 4.29 | TssA | 0.98 | TSS | 26 | 50 | 50 | 83 | 1.13 | . |
| TMEM 115 | 3_50396814_C_G | III7, III8, III10 | . | 1.69 × 10−3 | . | 18.1 | 1.00 | 1.89 | 4.82 | TssA | 0.82 | GS | 18 | 27 | 66.7 | 64 | 0.67 | . |
| TPM2 | 9_35690678_C_T | III7, III8, III10 | . | 1.36 × 10−3 | . | 17.2 | 1.00 | 0.61 | 3.76 | TssA | 0.95 | GE2 | 6 | 6 | 50 | 109 | 0.76 | . |
| UBE2K | 4_39699921_G_C | III7, III8, III10 | . | 1.16 × 10−4 | . | 17.0 | 1.00 | 1.39 | 4.47 | TssA | 0.93 | TSS | 14 | 25 | 66.7 | 89 | 1.01 | . |
| Gene Name | Variant | Pedigree Segregation | Allele Frequency in NFE | Bedtools intersect I | CADD v1.4 | SNPnexus Non-Coding Scores IV (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ExAC | gnomAD | miRNA Target Sites | Context Score ++ Percentile | Site Type | PHRED | Conservational Scores | Chromatin State | mirSVR-Score | ||||||||
| verPhCons | verPhyloP | GerpN | ChromHMM II State | ChromHMM II Score | Segway III | |||||||||||
| ACTN2 | 1_236927298_T_A | III7, III8, III10 | . | 6.48 × 10−5 | miR-450b-5p miR-891b | 36 71 | 7mer-m8 7mer-1a | 13.84 | 0.84 | 0.60 | 5.70 | Quies | 0.488 | R2 | −0.90 | 66.7 |
| CCNT2 | 2_135714351_C_T | III7, III8, III10 | . | . | miR-4670-3p miR-3606-5p | 93 83 | 7mer-1a 8mer | 15.46 | 1.00 | 3.28 | 5.65 | Tx Wk | 0.551 | GE0 | −1.11 | 83.3 |
| CLK1 | 2_201717953_C_T | III7, III8 | . | 2.01 × 10−3 | miR-4718 let-7c-3p | 98 96 | 7mer-m8 7mer-m8 | 15.57 | 0.68 | 0.38 | 5.34 | Tx | 0.921 | GE0 | −1.21 | 85.7 |
| DDX17 | 22_38881603_ACT_A | II7, III7, III8 | . | 2.84 × 10−3 | miR-550b-3p | 91 | 7mer-m8 | 14.91 | 0.66 | 0.97 | 4.36 | Tx | 0.858 | F1 | −0.35 | 100 |
| FH | 1_241661076_T_C | III7, III8, III10 | . | 3.89 × 10−3 | miR-4294 | 94 | 7mer-m8 | 10.4 | 0.47 | 0.11 | 4.56 | Tx | 0.709 | GE0 | −0.96 | 71.4 |
| LRRC8C | 1_90180607_T_A | III7, III8, III10 | . | 2.59 × 10−4 | miR-499a-5p miR-208-3p miR-4432 miR-8087 | 96 91 81 90 | 8mer 7mer-1a 7mer-1a 7mer-1a | 21.8 | 1.00 | 3.21 | 6.17 | Tx Wk | 0.583 | F1 | −0.76 | 83.3 |
| SEMA4B | 15_90772811_G_A | III7, III8, III10 | . | . | miR-3918 miR-3127-5p miR-506-5p miR-10a-3p | 99 99 92 94 | 8mer 8mer 7mer-m8 7mer-1a | 13.65 | 0.53 | 3.01 | 4.95 | Tx | 0.606 | GE1 | −1.27 | 83.3 |
| TJP1 | 15_29993152_G_A | III7, III8 | . | 6.49 × 10−5 | miR-654-3p | 89 | 8mer | 15.03 | 1.00 | 1.48 | 5.79 | Tx | 0.441 | GE1 | −0.80 | 83.3 |
| Matrix ID | Name | Relative Score | Exclusive for pHDAC5 | Start | End | Strand | Predicted Sequence |
|---|---|---|---|---|---|---|---|
| MA1631.1 | ASCL1(var.2) | 0.859965928 | WT | 710 | 722 | - | cagcacctcctcg |
| MA0598.1 | EHF | 0.869445857 | WT | 711 | 718 | - | acctcctc |
| MA0056.1 | MZF1 | 0.854364496 | WT | 710 | 715 | + | cgagga |
| MA0673.1 | NKX2-8 | 0.857521064 | WT | 708 | 716 | - | ctcctcgac |
| MA1558.1 | SNAI-1 | 0.875024573 | WT | 712 | 721 | + | aggaggtgct |
| MA0745.1 | SNAI-2 | 0.891814443 | WT | 712 | 720 | + | aggaggtgc |
| MA1563.1 | SOX18 | 0.858931866 | MUT | 714 | 721 | - | agcaccGc |
| MA0079.3 | SP1 | 0.871792316 | MUT | 710 | 720 | - | gcaccGcctcg |
| MA0079.3 | SP1 | 0.862428729 | WT | 710 | 720 | - | gcacctcctcg |
| MA0080.2 | SPI1 | 0.865925519 | WT | 712 | 718 | + | aggaggt |
| MA1566.1 | TBX3 | 0.876483102 | WT | 714 | 723 | + | gaggtgctgc |
| MA0806.1 | TBX4 | 0.854062458 | WT | 715 | 722 | + | aggtgctg |
| MA1567.1 | TBX6 | 0.867675674 | WT | 714 | 723 | + | gaggtgctgc |
| MA1648.1 | TCF12(var.2) | 0.868262172 | WT | 711 | 721 | - | agcacctcctc |
| MA0522.3 | TCF3 | 0.875470132 | WT | 711 | 721 | - | agcacctcctc |
| MA0522.2 | TCF3 | 0.869530427 | WT | 712 | 721 | - | agcacctcct |
| MA0830.2 | TCF4 | 0.851528821 | WT | 710 | 722 | - | cagcacctcctcg |
| MA0830.1 | TCF4 | 0.850775152 | WT | 712 | 721 | - | agcacctcct |
| MA0003.1 | TFAP2A | 0.860276583 | MUT | 707 | 715 | - | Gcctcgacg |
| MA0815.1 | TFAP2C(var.3) | 0.858234123 | MUT | 704 | 716 | + | agccgtcgaggCg |
| MA0815.1 | TFAP2C(var.3) | 0.851532375 | MUT | 704 | 716 | - | cGcctcgacggct |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skopelitou, D.; Miao, B.; Srivastava, A.; Kumar, A.; Kuświk, M.; Dymerska, D.; Paramasivam, N.; Schlesner, M.; Lubinski, J.; Hemminki, K.; et al. Whole Exome Sequencing Identifies APCDD1 and HDAC5 Genes as Potentially Cancer Predisposing in Familial Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 1837. https://doi.org/10.3390/ijms22041837
Skopelitou D, Miao B, Srivastava A, Kumar A, Kuświk M, Dymerska D, Paramasivam N, Schlesner M, Lubinski J, Hemminki K, et al. Whole Exome Sequencing Identifies APCDD1 and HDAC5 Genes as Potentially Cancer Predisposing in Familial Colorectal Cancer. International Journal of Molecular Sciences. 2021; 22(4):1837. https://doi.org/10.3390/ijms22041837
Chicago/Turabian StyleSkopelitou, Diamanto, Beiping Miao, Aayushi Srivastava, Abhishek Kumar, Magdalena Kuświk, Dagmara Dymerska, Nagarajan Paramasivam, Matthias Schlesner, Jan Lubinski, Kari Hemminki, and et al. 2021. "Whole Exome Sequencing Identifies APCDD1 and HDAC5 Genes as Potentially Cancer Predisposing in Familial Colorectal Cancer" International Journal of Molecular Sciences 22, no. 4: 1837. https://doi.org/10.3390/ijms22041837
APA StyleSkopelitou, D., Miao, B., Srivastava, A., Kumar, A., Kuświk, M., Dymerska, D., Paramasivam, N., Schlesner, M., Lubinski, J., Hemminki, K., Försti, A., & Bandapalli, O. R. (2021). Whole Exome Sequencing Identifies APCDD1 and HDAC5 Genes as Potentially Cancer Predisposing in Familial Colorectal Cancer. International Journal of Molecular Sciences, 22(4), 1837. https://doi.org/10.3390/ijms22041837