
Analysis of the Conditions That Affect the Selective Processing of Endogenous Notch1 by ADAM10 and ADAM17

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
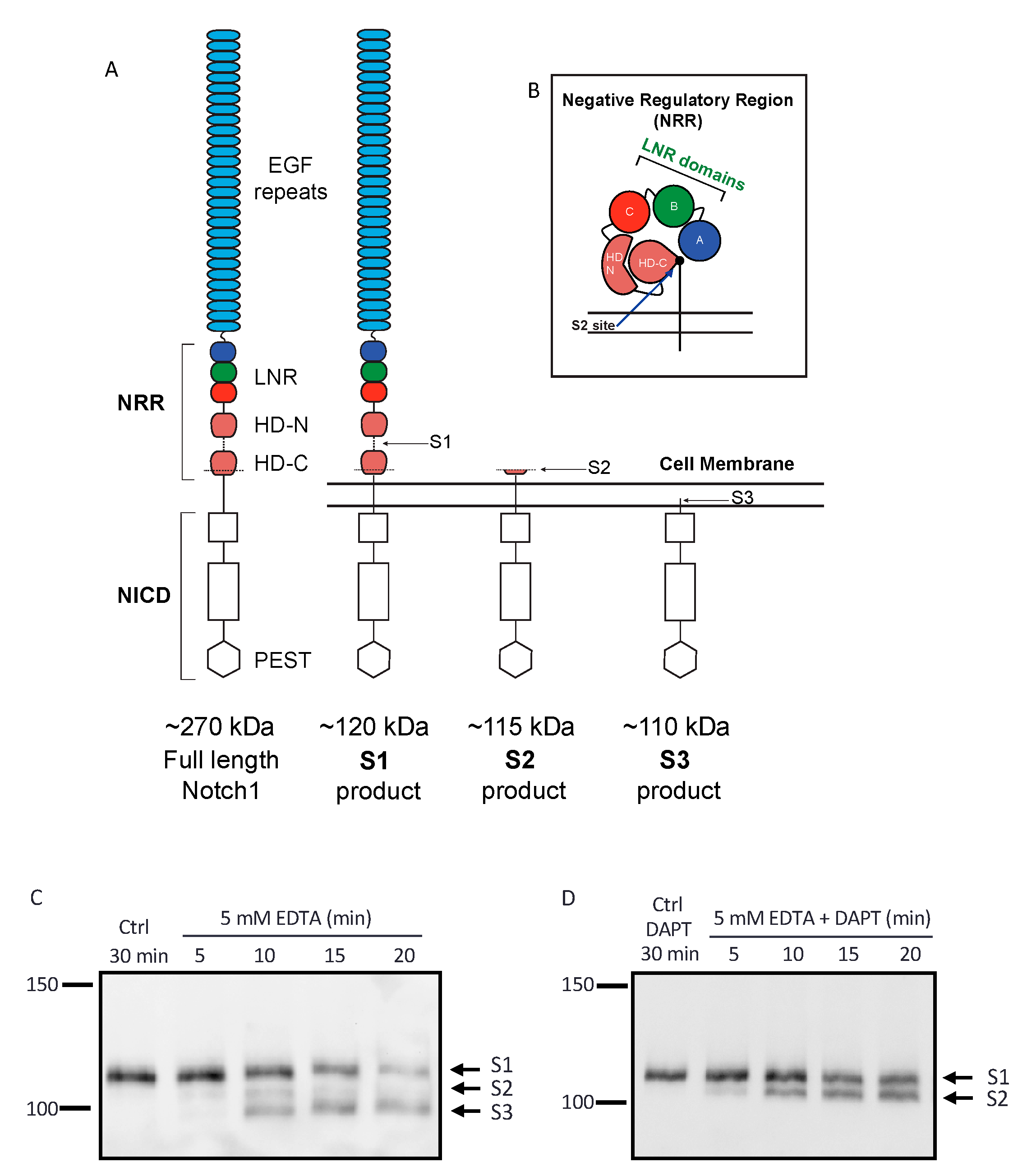
2.1. Establishing an Assay to Evaluate Endogenous Notch1 Processing by ADAM10 and ADAM17
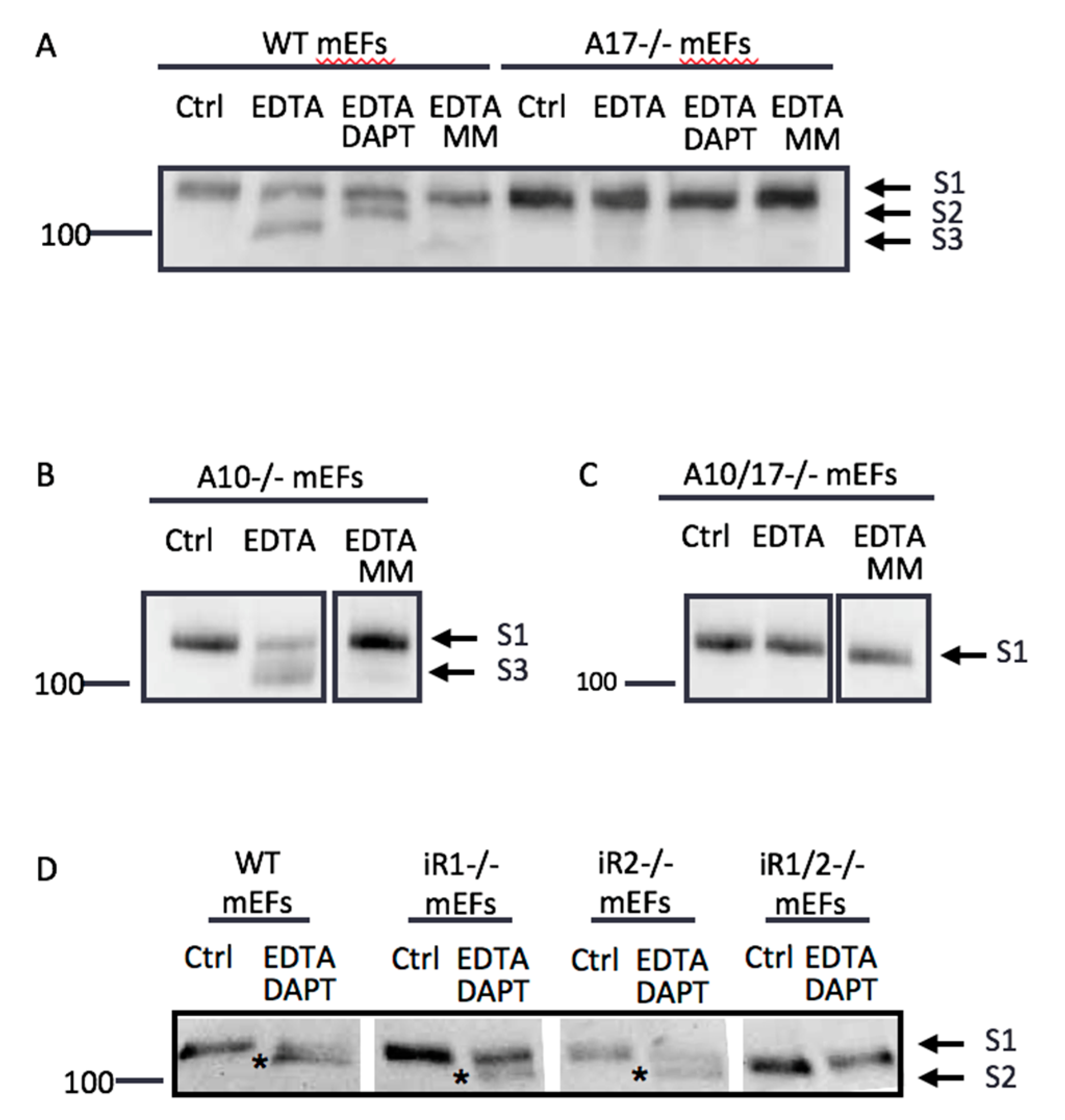
2.2. Endogenous ADAM17 Cleaves at the EDTA-Exposed Notch1 S2 Site
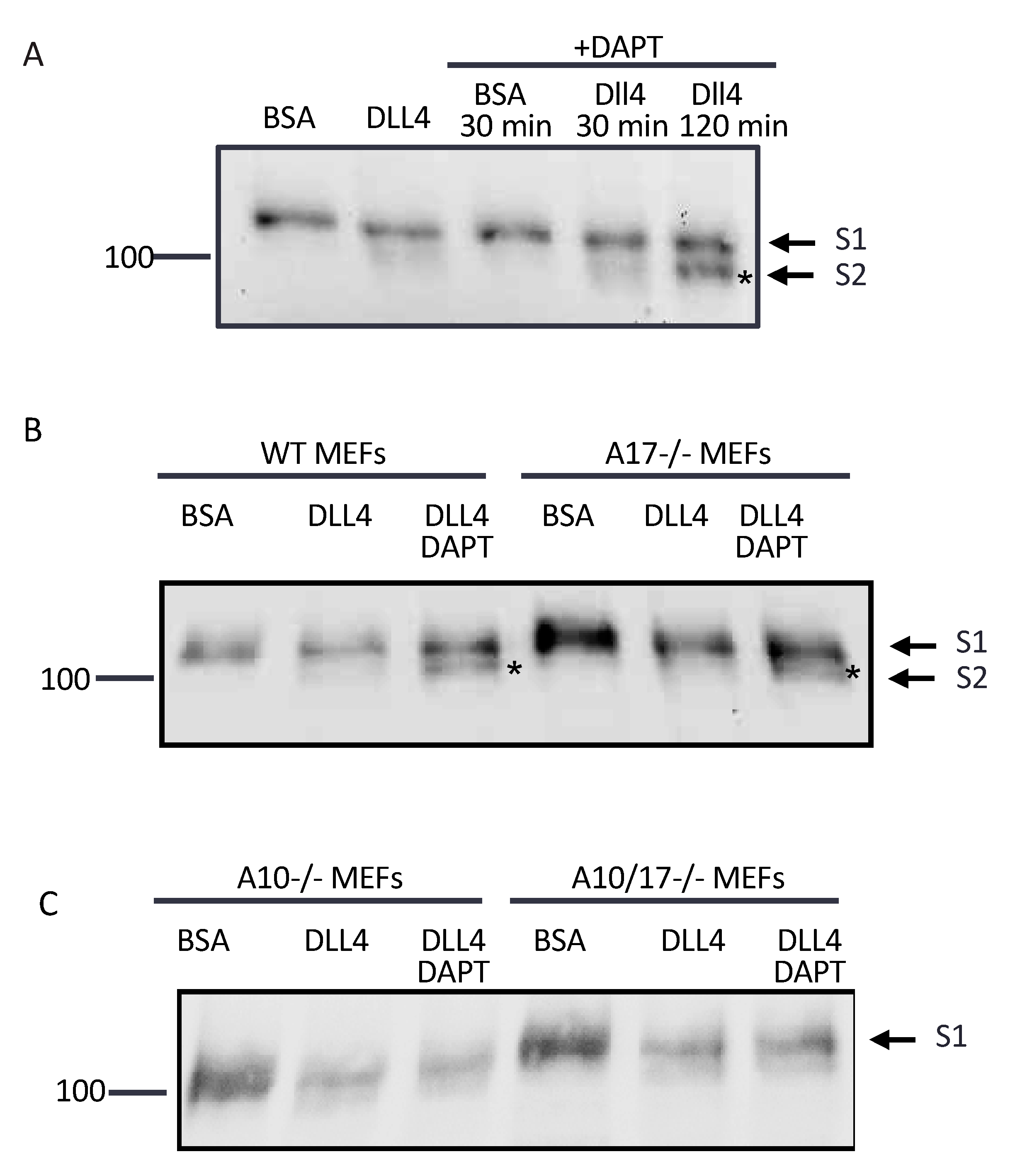
2.3. ADAM10 Cleaves at the S2 Site in Endogenous Notch1 Exposed by Culture of Cells on Immobilized Dll4
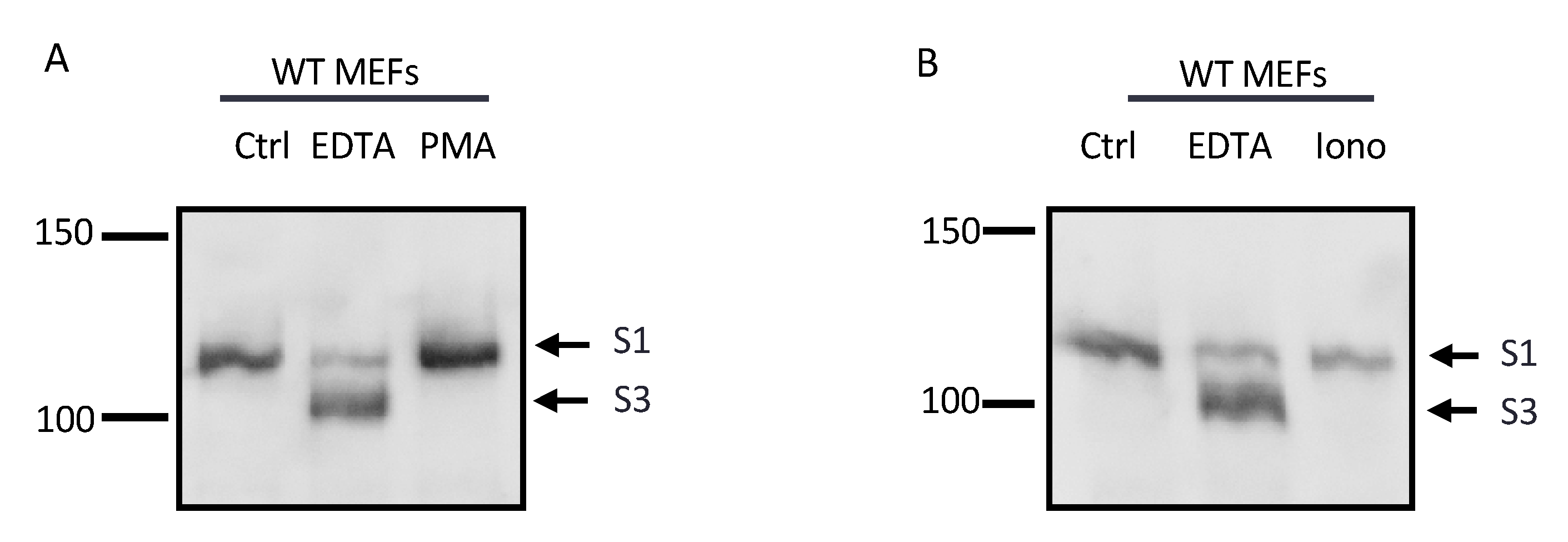
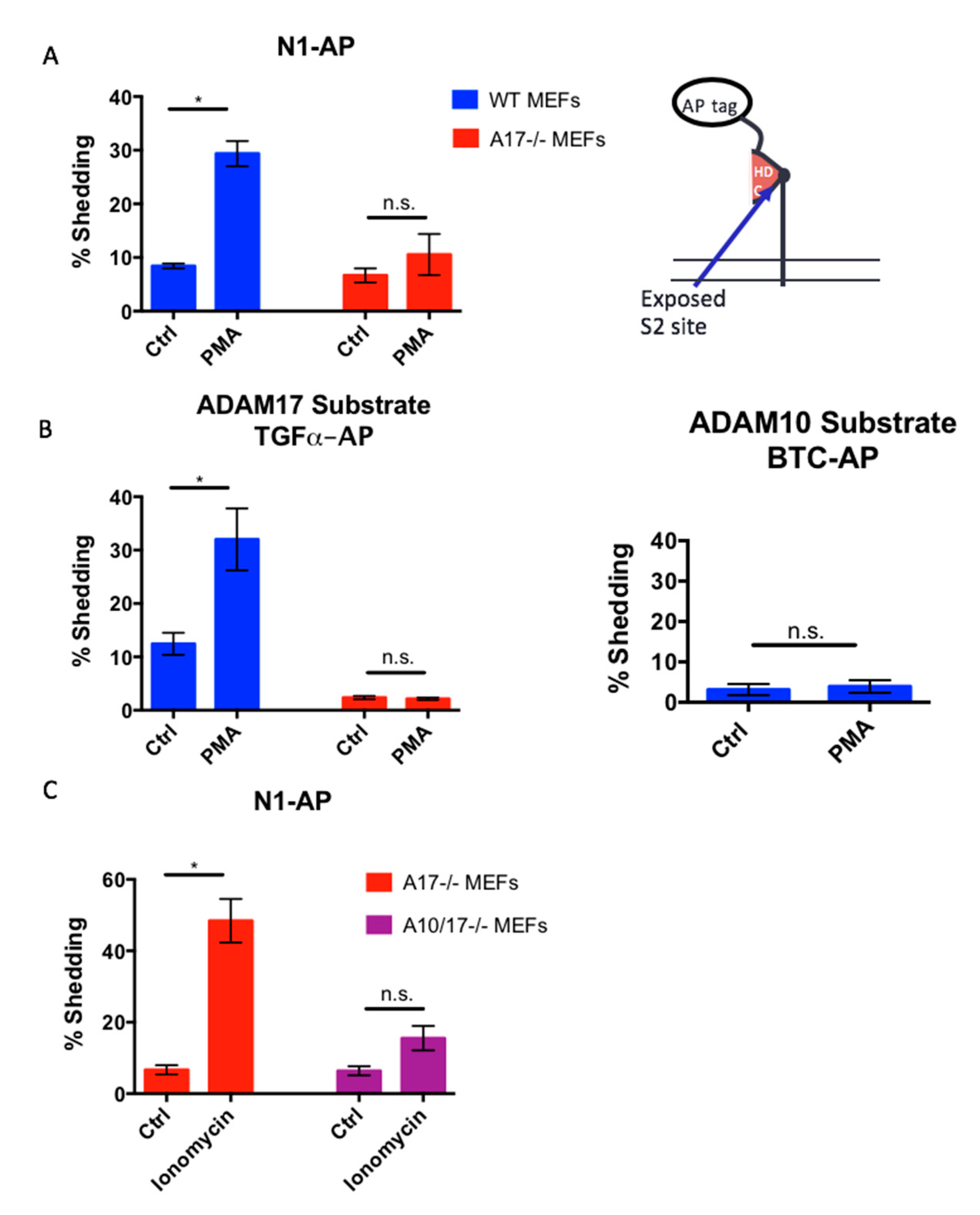
2.4. The Notch1 S2 Site Is Protected from Processing by Stimulated ADAMs
2.5. The Constitutively Exposed S2 Site Behaves Like an ADAM17 Substrate
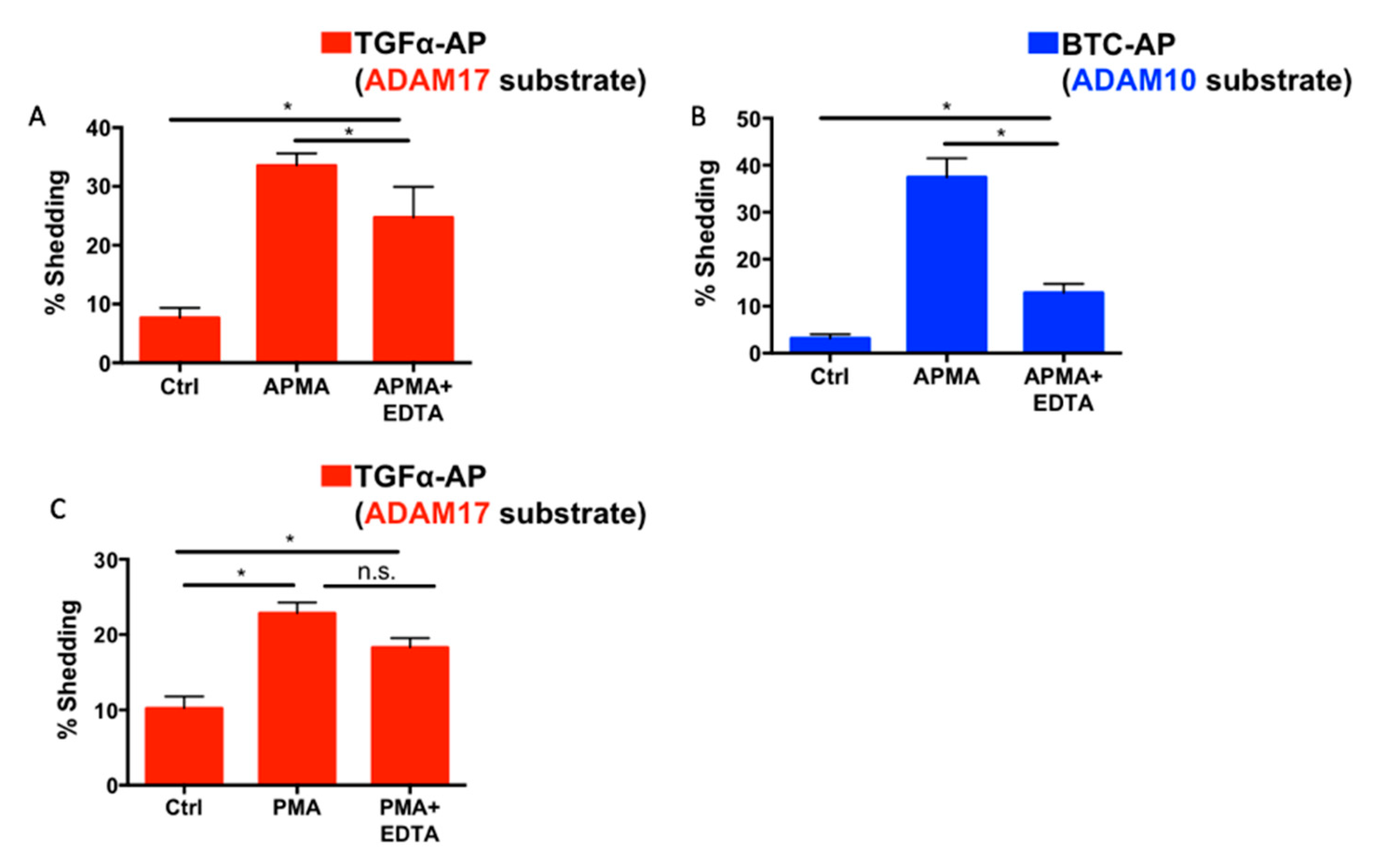
2.6. Differential Effects of EDTA on ADAM10 and ADAM17 Activity
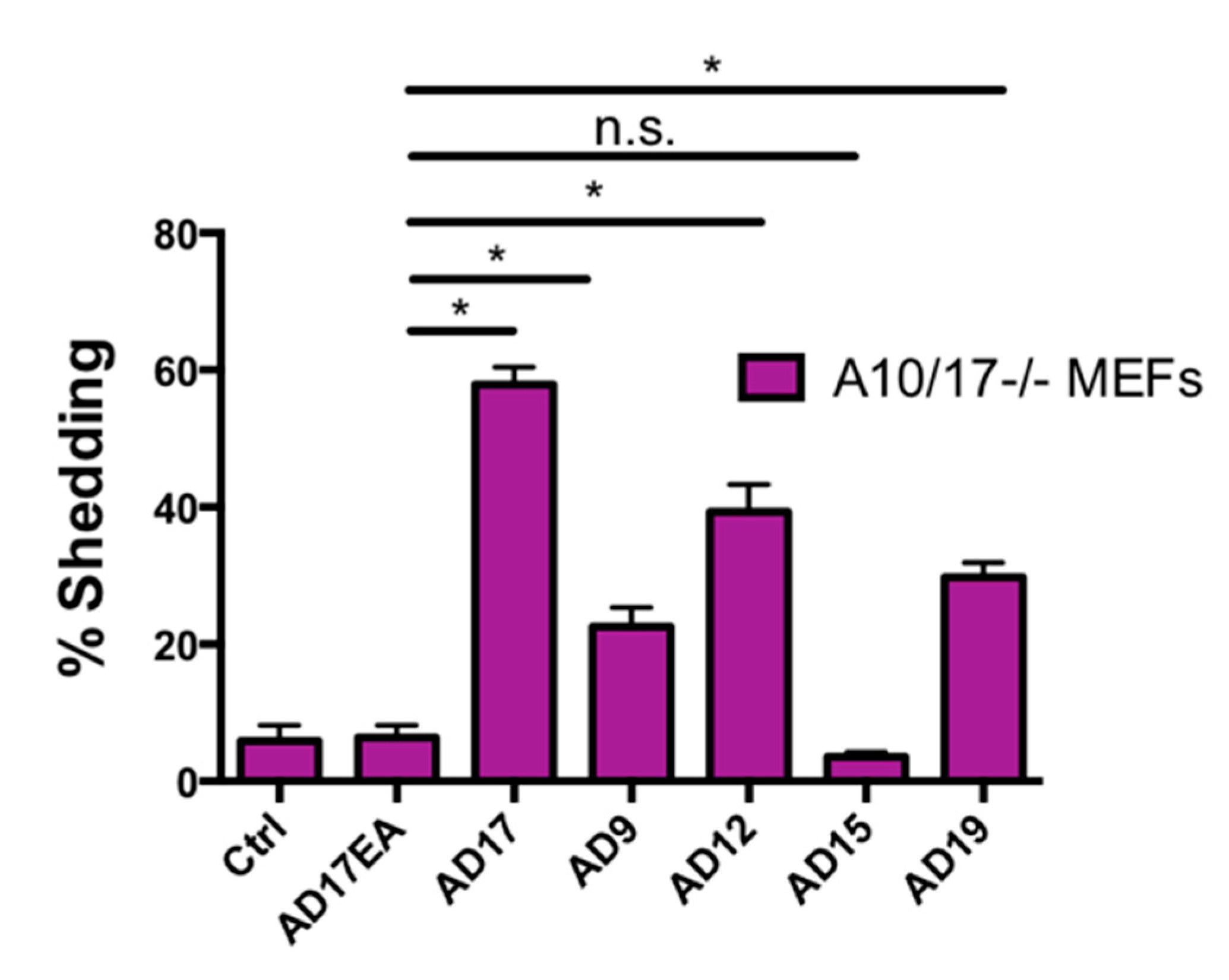
2.7. Other ADAMs Can Cleave the Exposed Notch1 S2 Site
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Reagents and Antibodies
4.2. Plasmids
4.3. Cell Transfection and Alkaline Phosphatase (AP) Ectodomain Shedding Assay
4.4. Western Blot Analysis
4.5. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hori, K.; Sen, A.; Artavanis-Tsakonas, S. Notch Signaling at a Glance. J. Cell Sci. 2013, 126, 2135–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, S.J. Notch Signalling in Context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef] [PubMed]
- Falo-Sanjuan, J.; Bray, S.J. Decoding the Notch Signal. Dev. Growth Differ. 2020, 62, 4–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [Green Version]
- Kovall, R.A.; Gebelein, B.; Sprinzak, D.; Kopan, R. The Canonical Notch Signaling Pathway: Structural and Biochemical Insights into Shape, Sugar, and Force. Dev. Cell 2017, 41, 228–241. [Google Scholar] [CrossRef] [Green Version]
- Blaumueller, C.M.; Qi, H.; Zagouras, P.; Artavanis-Tsakonas, S. Intracellular Cleavage of Notch Leads to a Heterodimeric Receptor on the Plasma Membrane. Cell 1997, 90, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Logeat, F.; Bessia, C.; Brou, C.; LeBail, O.; Jarriault, S.; Seidah, N.G.; Israël, A. The Notch1 Receptor Is Cleaved Constitutively by a Furin-like Convertase. Proc. Natl. Acad. Sci. USA 1998, 95, 8108–8112. [Google Scholar] [CrossRef] [Green Version]
- Gordon, W.R.; Arnett, K.L.; Blacklow, S.C. The Molecular Logic of Notch Signaling—A Structural and Biochemical Perspective. J. Cell Sci. 2008, 121, 3109–3119. [Google Scholar] [CrossRef] [Green Version]
- Kovall, R.A.; Blacklow, S.C. Mechanistic Insights into Notch Receptor Signaling from Structural and Biochemical Studies. Curr. Top. Dev. Biol. 2010, 92, 31–71. [Google Scholar] [CrossRef]
- Kopan, R.; Schroeter, E.H.; Weintraub, H.; Nye, J.S. Signal Transduction by Activated mNotch: Importance of Proteolytic Processing and Its Regulation by the Extracellular Domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1683–1688. [Google Scholar] [CrossRef] [Green Version]
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 Signalling Requires Ligand-Induced Proteolytic Release of Intracellular Domain. Nature 1998, 393, 382–386. [Google Scholar] [CrossRef]
- Pan, D.; Rubin, G.M. Kuzbanian Controls Proteolytic Processing of Notch and Mediates Lateral Inhibition during Drosophila and Vertebrate Neurogenesis. Cell 1997, 90, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Sotillos, S.; Roch, F.; Campuzano, S. The Metalloprotease-Disintegrin Kuzbanian Participates in Notch Activation during Growth and Patterning of Drosophila Imaginal Discs. Development 1997, 124, 4769–4779. [Google Scholar] [PubMed]
- Lieber, T.; Kidd, S.; Young, M.W. Kuzbanian-Mediated Cleavage of Drosophila Notch. Genes Dev. 2002, 16, 209–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brou, C.; Logeat, F.; Gupta, N.; Bessia, C.; LeBail, O.; Doedens, J.R.; Cumano, A.; Roux, P.; Black, R.A.; Israël, A. A Novel Proteolytic Cleavage Involved in Notch Signaling: The Role of the Disintegrin-Metalloprotease TACE. Mol. Cell 2000, 5, 207–216. [Google Scholar] [CrossRef]
- Mumm, J.S.; Schroeter, E.H.; Saxena, M.T.; Griesemer, A.; Tian, X.; Pan, D.J.; Ray, W.J.; Kopan, R. A Ligand-Induced Extracellular Cleavage Regulates Gamma-Secretase-like Proteolytic Activation of Notch1. Mol. Cell 2000, 5, 197–206. [Google Scholar] [CrossRef]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A Presenilin-1-Dependent Gamma-Secretase-like Protease Mediates Release of Notch Intracellular Domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef]
- Gordon, W.R.; Roy, M.; Vardar-Ulu, D.; Garfinkel, M.; Mansour, M.R.; Aster, J.C.; Blacklow, S.C. Structure of the Notch1-Negative Regulatory Region: Implications for Normal Activation and Pathogenic Signaling in T-ALL. Blood 2009, 113, 4381–4390. [Google Scholar] [CrossRef] [Green Version]
- Tiyanont, K.; Wales, T.E.; Aste-Amezaga, M.; Aster, J.C.; Engen, J.R.; Blacklow, S.C. Evidence for Increased Exposure of the Notch1 Metalloprotease Cleavage Site upon Conversion to an Activated Conformation. Structure 2011, 19, 546–554. [Google Scholar] [CrossRef] [Green Version]
- Caolo, V.; Debant, M.; Endesh, N.; Futers, T.S.; Lichtenstein, L.; Bartoli, F.; Parsonage, G.; Jones, E.A.; Beech, D.J. Shear Stress Activates ADAM10 Sheddase to Regulate Notch1 via the Piezo1 Force Sensor in Endothelial Cells. Elife 2020, 9. [Google Scholar] [CrossRef]
- Gordon, W.R.; Zimmerman, B.; He, L.; Miles, L.J.; Huang, J.; Tiyanont, K.; McArthur, D.G.; Aster, J.C.; Perrimon, N.; Loparo, J.J.; et al. Mechanical Allostery: Evidence for a Force Requirement in the Proteolytic Activation of Notch. Dev. Cell 2015, 33, 729–736. [Google Scholar] [CrossRef] [Green Version]
- Rand, M.D.; Grimm, L.M.; Artavanis-Tsakonas, S.; Patriub, V.; Blacklow, S.C.; Sklar, J.; Aster, J.C. Calcium Depletion Dissociates and Activates Heterodimeric Notch Receptors. Mol. Cell. Biol. 2000, 20, 1825–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbuck, M.P.; Winandy, S. A Review of Notch Processing With New Insights Into Ligand-Independent Notch Signaling in T-Cells. Front. Immunol. 2018, 9, 1230. [Google Scholar] [CrossRef] [Green Version]
- Gordon, W.R.; Vardar-Ulu, D.; Histen, G.; Sanchez-Irizarry, C.; Aster, J.C.; Blacklow, S.C. Structural Basis for Autoinhibition of Notch. Nat. Struct. Mol. Biol. 2007, 14, 295–300. [Google Scholar] [CrossRef]
- Vaccari, T.; Duchi, S.; Cortese, K.; Tacchetti, C.; Bilder, D. The vacuolar ATPase is required for physiological as well as pathological activation of the notch receptor. Development 2010, 137, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Weng, A.P. Activating Mutations of NOTCH1 in Human T Cell Acute Lymphoblastic Leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [Green Version]
- Malecki, M.J.; Sanchez-Irizarry, C.; Mitchell, J.L.; Histen, G.; Xu, M.L.; Aster, J.C.; Blacklow, S.C. Leukemia-Associated Mutations within the NOTCH1 Heterodimerization Domain Fall into at Least Two Distinct Mechanistic Classes. Mol. Cell. Biol. 2006, 26, 4642–4651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Tetering, G.; van Diest, P.; Verlaan, I.; van der Wall, E.; Kopan, R.; Vooijs, M. Metalloprotease ADAM10 Is Required for Notch1 Site 2 Cleavage. J. Biol. Chem. 2009, 284, 31018–31027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, C.; Metzstein, M.M.; Greenwald, I. SUP-17, a Caenorhabditis Elegans ADAM Protein Related to Drosophila KUZBANIAN, and Its Role in LIN-12/NOTCH Signalling. Development 1997, 124, 4759–4767. [Google Scholar]
- Hartmann, D.; de Strooper, B.; Serneels, L.; Craessaerts, K.; Herreman, A.; Annaert, W.; Umans, L.; Lübke, T.; Lena Illert, A.; von Figura, K.; et al. The Disintegrin/Metalloprotease ADAM 10 Is Essential for Notch Signalling but Not for Alpha-Secretase Activity in Fibroblasts. Hum. Mol. Genet. 2002, 11, 2615–2624. [Google Scholar] [CrossRef] [Green Version]
- Glomski, K.; Monette, S.; Manova, K.; De Strooper, B.; Saftig, P.; Blobel, C.P. Deletion of Adam10 in Endothelial Cells Leads to Defects in Organ-Specific Vascular Structures. Blood 2011, 118, 1163–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alabi, R.O.; Glomski, K.; Haxaire, C.; Weskamp, G.; Monette, S.; Blobel, C.P. ADAM10-Dependent Signaling Through Notch1 and Notch4 Controls Development of Organ-Specific Vascular Beds. Circ. Res. 2016, 119, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Alabi, R.O.; Farber, G.; Blobel, C.P. Intriguing Roles for Endothelial ADAM10/Notch Signaling in the Development of Organ-Specific Vascular Beds. Physiol. Rev. 2018, 98, 2025–2061. [Google Scholar] [CrossRef] [PubMed]
- Huppert, S.S.; Le, A.; Schroeter, E.H.; Mumm, J.S.; Saxena, M.T.; Milner, L.A.; Kopan, R. Embryonic Lethality in Mice Homozygous for a Processing-Deficient Allele of Notch1. Nature 2000, 405, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Peschon, J.J. An Essential Role for Ectodomain Shedding in Mammalian Development. Science 1998, 282, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, E.; Prox, J.; Bernreuther, C.; Weber, S.; Schwanbeck, R.; Serneels, L.; Snellinx, A.; Craessaerts, K.; Thathiah, A.; Tesseur, I.; et al. The Disintegrin/Metalloproteinase ADAM10 Is Essential for the Establishment of the Brain Cortex. J. Neurosci. 2010, 30, 4833–4844. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Niessen, M.T.; Prox, J.; Lüllmann-Rauch, R.; Schmitz, A.; Schwanbeck, R.; Blobel, C.P.; Jorissen, E.; de Strooper, B.; Niessen, C.M.; et al. The Disintegrin/Metalloproteinase Adam10 Is Essential for Epidermal Integrity and Notch-Mediated Signaling. Development 2011, 138, 495–505. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Wu, X.; Chi, C.; Han, M.; Xu, T.; Zhuang, Y. ADAM10 Is Essential for Proteolytic Activation of Notch during Thymocyte Development. Int. Immunol. 2008, 20, 1181–1187. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Tian, L.; Chi, C.; Wu, X.; Yang, X.; Han, M.; Xu, T.; Zhuang, Y.; Deng, K. Adam10 Is Essential for Early Embryonic Cardiovascular Development. Dev. Dyn. 2010, 239, 2594–2602. [Google Scholar] [CrossRef]
- Bozkulak, E.C.; Weinmaster, G. Selective Use of ADAM10 and ADAM17 in Activation of Notch1 Signaling. Mol. Cell. Biol. 2009, 29, 5679–5695. [Google Scholar] [CrossRef] [Green Version]
- Sulis, M.L.; Saftig, P.; Ferrando, A.A. Redundancy and Specificity of the Metalloprotease System Mediating Oncogenic NOTCH1 Activation in T-ALL. Leukemia 2011, 25, 1564–1569. [Google Scholar] [CrossRef] [Green Version]
- Aster, J.C.; Simms, W.B.; Zavala-Ruiz, Z.; Patriub, V.; North, C.L.; Blacklow, S.C. The Folding and Structural Integrity of the First LIN-12 Module of Human Notch1 Are Calcium-Dependent. Biochemistry 1999, 38, 4736–4742. [Google Scholar] [CrossRef] [PubMed]
- Pandiella, A.; Massagué, J. Cleavage of the Membrane Precursor for Transforming Growth Factor Alpha Is a Regulated Process. Proc. Natl. Acad. Sci. USA 1991, 88, 1726–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doedens, J.R.; Mahimkar, R.M.; Black, R.A. TACE/ADAM-17 Enzymatic Activity Is Increased in Response to Cellular Stimulation. Biochem. Biophys. Res. Commun. 2003, 308, 331–338. [Google Scholar] [CrossRef]
- Blobel, C.P. ADAMs: Key Components in EGFR Signalling and Development. Nat. Rev. Mol. Cell Biol. 2005, 6, 32–43. [Google Scholar] [CrossRef]
- Sanderson, M.P.; Erickson, S.N.; Gough, P.J.; Garton, K.J.; Wille, P.T.; Raines, E.W.; Dunbar, A.J.; Dempsey, P.J. ADAM10 Mediates Ectodomain Shedding of the Betacellulin Precursor Activated by P-Aminophenylmercuric Acetate and Extracellular Calcium Influx. J. Biol. Chem. 2005, 280, 1826–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horiuchi, K.; Le Gall, S.; Schulte, M.; Yamaguchi, T.; Reiss, K.; Murphy, G.; Toyama, Y.; Hartmann, D.; Saftig, P.; Blobel, C.P. Substrate Selectivity of Epidermal Growth Factor-Receptor Ligand Sheddases and Their Regulation by Phorbol Esters and Calcium Influx. Mol. Biol. Cell 2007, 18, 176–188. [Google Scholar] [CrossRef] [Green Version]
- Le Gall, S.M.; Bobé, P.; Reiss, K.; Horiuchi, K.; Niu, X.-D.; Lundell, D.; Gibb, D.R.; Conrad, D.; Saftig, P.; Blobel, C.P. ADAMs 10 and 17 Represent Differentially Regulated Components of a General Shedding Machinery for Membrane Proteins Such as Transforming Growth Factor Alpha, L-Selectin, and Tumor Necrosis Factor Alpha. Mol. Biol. Cell 2009, 20, 1785–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiss, K.; Maretzky, T.; Ludwig, A.; Tousseyn, T.; de Strooper, B.; Hartmann, D.; Saftig, P. ADAM10 Cleavage of N-Cadherin and Regulation of Cell-Cell Adhesion and Beta-Catenin Nuclear Signalling. EMBO J. 2005, 24, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Maretzky, T.; Reiss, K.; Ludwig, A.; Buchholz, J.; Scholz, F.; Proksch, E.; de Strooper, B.; Hartmann, D.; Saftig, P. ADAM10 Mediates E-Cadherin Shedding and Regulates Epithelial Cell-Cell Adhesion, Migration, and β-Catenin Translocation. Proc. Natl. Acad. Sci. USA 2005, 102, 9182–9187. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; Evers, A.; Le Gall, S.; Alabi, R.O.; Speck, N.; Reiss, K.; Blobel, C.P. The Cytoplasmic Domain of a Disintegrin and Metalloproteinase 10 (ADAM10) Regulates Its Constitutive Activity but Is Dispensable for Stimulated ADAM10-Dependent Shedding. J. Biol. Chem. 2015, 290, 7416–7425. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.-M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct Roles for ADAM10 and ADAM17 in Ectodomain Shedding of Six EGFR Ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Merlos-Suárez, A.; Ruiz-Paz, S.; Baselga, J.; Arribas, J. Metalloprotease-Dependent Protransforming Growth Factor-Alpha Ectodomain Shedding in the Absence of Tumor Necrosis Factor-Alpha-Converting Enzyme. J. Biol. Chem. 2001, 276, 48510–48517. [Google Scholar] [CrossRef] [Green Version]
- Toonen, J.A.; Ronchetti, A.; Sidjanin, D.J. A Disintegrin and Metalloproteinase10 (ADAM10) Regulates NOTCH Signaling during Early Retinal Development. PLoS ONE 2016, 11, e0156184. [Google Scholar] [CrossRef]
- Murthy, A.; Shao, Y.W.; Narala, S.R.; Molyneux, S.D.; Zúñiga-Pflücker, J.C.; Khokha, R. Notch Activation by the Metalloproteinase ADAM17 Regulates Myeloproliferation and Atopic Barrier Immunity by Suppressing Epithelial Cytokine Synthesis. Immunity 2012, 36, 105–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, D.; Jiang, Q.; Cao, S.; Sun, H.; Chai, Y.; Li, X.; Ren, T.; Yang, R.; Feng, F.; et al. Novel ADAM-17 Inhibitor ZLDI-8 Enhances the In Vitro and In Vivo Chemotherapeutic Effects of Sorafenib on Hepatocellular Carcinoma Cells. Cell Death Dis. 2018, 9, 743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.-D.; Zhao, C.-H.; Ding, H.-W.; Wu, Q.; Ren, T.-S.; Wang, J.; Chen, C.-Q.; Zhao, Q.-C. A Novel Inhibitor of ADAM17 Sensitizes Colorectal Cancer Cells to 5-Fluorouracil by Reversing Notch and Epithelial-Mesenchymal Transition In Vitro and In Vivo. Cell Prolif. 2018, 51, e12480. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Li, Y.; Tsung, A.; Huang, H.; Du, Q.; Yang, M.; Deng, M.; Xiong, S.; Wang, X.; Zhang, L.; et al. INOS Promotes CD24+CD133+ Liver Cancer Stem Cell Phenotype through a TACE/ADAM17-Dependent Notch Signaling Pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E10127–E10136. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wang, D.; Sun, X.; Zhang, Y.; Wang, L.; Suo, J. ADAM17 Promotes Lymph Node Metastasis in Gastric Cancer via Activation of the Notch and Wnt Signaling Pathways. Int. J. Mol. Med. 2019, 43, 914–926. [Google Scholar] [CrossRef]
- Lu, H.-Y.; Zu, Y.-X.; Jiang, X.-W.; Sun, X.-T.; Liu, T.-Y.; Li, R.-L.; Wu, Q.; Zhang, Y.-S.; Zhao, Q.-C. Novel ADAM-17 Inhibitor ZLDI-8 Inhibits the Proliferation and Metastasis of Chemo-Resistant Non-Small-Cell Lung Cancer by Reversing Notch and Epithelial Mesenchymal Transition In Vitro and In Vivo. Pharmacol. Res. 2019, 148, 104406. [Google Scholar] [CrossRef] [PubMed]
- González-Foruria, I.; Santulli, P.; Chouzenoux, S.; Carmona, F.; Chapron, C.; Batteux, F. Dysregulation of the ADAM17/Notch Signalling Pathways in Endometriosis: From Oxidative Stress to Fibrosis. Mol. Hum. Reprod. 2017, 23, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Jin, X.; Jia, H. Inhibition of ADAM-17 More Effectively down-Regulates the Notch Pathway than That of γ-Secretase in Renal Carcinoma. J. Exp. Clin. Cancer Res. 2013, 32, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucher, J.; Linke, D.; Koudelka, T.; Cassidy, L.; Tredup, C.; Wichert, R.; Pietrzik, C.; Becker-Pauly, C.; Tholey, A. LC-MS Based Cleavage Site Profiling of the Proteases ADAM10 and ADAM17 Using Proteome-Derived Peptide Libraries. J. Proteome Res. 2014, 13, 2205–2214. [Google Scholar] [CrossRef]
- Caescu, C.I.; Jeschke, G.R.; Turk, B.E. Active-Site Determinants of Substrate Recognition by the Metalloproteinases TACE and ADAM10. Biochem. J. 2009, 424, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Schlöndorff, J.; Becherer, J.D.; Blobel, C.P. Intracellular Maturation and Localization of the Tumour Necrosis Factor Alpha Convertase (TACE). Biochem. J. 2000, 347 Pt 1, 131–138. [Google Scholar] [CrossRef]
- Lunn, C.A.; Fan, X.; Dalie, B.; Miller, K.; Zavodny, P.J.; Narula, S.K.; Lundell, D. Purification of ADAM 10 from Bovine Spleen as a TNFalpha Convertase. FEBS Lett. 1997, 400, 333–335. [Google Scholar] [CrossRef] [Green Version]
- Rosendahl, M.S.; Ko, S.C.; Long, D.L.; Brewer, M.T.; Rosenzweig, B.; Hedl, E.; Anderson, L.; Pyle, S.M.; Moreland, J.; Meyers, M.A.; et al. Identification and Characterization of a Pro-Tumor Necrosis Factor-Alpha-Processing Enzyme from the ADAM Family of Zinc Metalloproteases. J. Biol. Chem. 1997, 272, 24588–24593. [Google Scholar] [CrossRef] [Green Version]
- Haining, E.J.; Yang, J.; Bailey, R.L.; Khan, K.; Collier, R.; Tsai, S.; Watson, S.P.; Frampton, J.; Garcia, P.; Tomlinson, M.G. The TspanC8 Subgroup of Tetraspanins Interacts with A Disintegrin and Metalloprotease 10 (ADAM10) and Regulates Its Maturation and Cell Surface Expression. J. Biol. Chem. 2012, 287, 39753–39765. [Google Scholar] [CrossRef] [Green Version]
- Dornier, E.; Coumailleau, F.; Ottavi, J.-F.; Moretti, J.; Boucheix, C.; Mauduit, P.; Schweisguth, F.; Rubinstein, E. TspanC8 Tetraspanins Regulate ADAM10/Kuzbanian Trafficking and Promote Notch Activation in Flies and Mammals. J. Cell Biol. 2012, 199, 481–496. [Google Scholar] [CrossRef] [Green Version]
- Jouannet, S.; Saint-Pol, J.; Fernandez, L.; Nguyen, V.; Charrin, S.; Boucheix, C.; Brou, C.; Milhiet, P.-E.; Rubinstein, E. TspanC8 Tetraspanins Differentially Regulate the Cleavage of ADAM10 Substrates, Notch Activation and ADAM10 Membrane Compartmentalization. Cell Mol. Life Sci. 2016, 73, 1895–1915. [Google Scholar] [CrossRef] [Green Version]
- Koo, C.Z.; Harrison, N.; Noy, P.J.; Szyroka, J.; Matthews, A.L.; Hsia, H.-E.; Müller, S.A.; Tüshaus, J.; Goulding, J.; Willis, K.; et al. The Tetraspanin Tspan15 Is an Essential Subunit of an ADAM10 Scissor Complex. J. Biol. Chem. 2020, 295, 12822–12839. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.C.; Kim, S.; Shepardson, N.; Patel, S.; Hong, S.; Selkoe, D.J. Physical and Functional Interaction between the α- and γ-Secretases: A New Model of Regulated Intramembrane Proteolysis. J. Cell Biol. 2015, 211, 1157–1176. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.R.; Lang, P.A.; Maretzky, T.; Hamada, K.; Ohishi, K.; Maney, S.K.; Berger, T.; Murthy, A.; Duncan, G.; Xu, H.C.; et al. iRhom2 Regulation of TACE Controls TNF-Mediated Protection against Listeria and Responses to LPS. Science 2012, 335, 229–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adrain, C.; Zettl, M.; Christova, Y.; Taylor, N.; Freeman, M. Tumor Necrosis Factor Signaling Requires iRhom2 to Promote Trafficking and Activation of TACE. Science 2012, 335, 225–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Maretzky, T.; Weskamp, G.; Monette, S.; Qing, X.; Issuree, P.D.A.; Crawford, H.C.; McIlwain, D.R.; Mak, T.W.; Salmon, J.E.; et al. IRhoms 1 and 2 Are Essential Upstream Regulators of ADAM17-Dependent EGFR Signaling. Proc. Natl. Acad. Sci. USA. 2015, 112, 6080–6085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Irizarry, C.; Carpenter, A.C.; Weng, A.P.; Pear, W.S.; Aster, J.C.; Blacklow, S.C. Notch Subunit Heterodimerization and Prevention of Ligand-Independent Proteolytic Activation Depend, Respectively, on a Novel Domain and the LNR Repeats. Mol. Cell. Biol. 2004, 24, 9265–9273. [Google Scholar] [CrossRef] [Green Version]
- Issuree, P.D.A.; Maretzky, T.; McIlwain, D.R.; Monette, S.; Qing, X.; Lang, P.A.; Swendeman, S.L.; Park-Min, K.-H.; Binder, N.; Kalliolias, G.D.; et al. IRHOM2 Is a Critical Pathogenic Mediator of Inflammatory Arthritis. J. Clin. Invest. 2013, 123, 928–932. [Google Scholar] [CrossRef] [Green Version]
- Maretzky, T.; Yang, G.; Ouerfelli, O.; Overall, C.M.; Worpenberg, S.; Hassiepen, U.; Eder, J.; Blobel, C.P. Characterization of the Catalytic Activity of the Membrane-Anchored Metalloproteinase ADAM15 in Cell-Based Assays. Biochem. J. 2009, 420, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Saftig, P.; Hartmann, D.; Blobel, C. Evaluation of the Contribution of Different ADAMs to Tumor Necrosis Factor Alpha (TNFalpha) Shedding and of the Function of the TNFalpha Ectodomain in Ensuring Selective Stimulated Shedding by the TNFalpha Convertase (TACE/ADAM17). J. Biol. Chem. 2004, 279, 42898–42906. [Google Scholar] [CrossRef] [Green Version]
- Horiuchi, K.; Zhou, H.-M.; Kelly, K.; Manova, K.; Blobel, C.P. Evaluation of the Contributions of ADAMs 9, 12, 15, 17, and 19 to Heart Development and Ectodomain Shedding of Neuregulins Beta1 and Beta2. Dev. Biol. 2005, 283, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Weskamp, G.; Zheng, Y.; Chesneau, V.; Horiuchi, K.; Blobel, C.P. A Sensitive Method to Monitor Ectodomain Shedding of Ligands of the Epidermal Growth Factor Receptor. Methods Mol. Biol. 2006, 327, 99–113. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alabi, R.O.; Lora, J.; Celen, A.B.; Maretzky, T.; Blobel, C.P. Analysis of the Conditions That Affect the Selective Processing of Endogenous Notch1 by ADAM10 and ADAM17. Int. J. Mol. Sci. 2021, 22, 1846. https://doi.org/10.3390/ijms22041846
Alabi RO, Lora J, Celen AB, Maretzky T, Blobel CP. Analysis of the Conditions That Affect the Selective Processing of Endogenous Notch1 by ADAM10 and ADAM17. International Journal of Molecular Sciences. 2021; 22(4):1846. https://doi.org/10.3390/ijms22041846
Chicago/Turabian StyleAlabi, Rolake O., Jose Lora, Arda B. Celen, Thorsten Maretzky, and Carl P. Blobel. 2021. "Analysis of the Conditions That Affect the Selective Processing of Endogenous Notch1 by ADAM10 and ADAM17" International Journal of Molecular Sciences 22, no. 4: 1846. https://doi.org/10.3390/ijms22041846
APA StyleAlabi, R. O., Lora, J., Celen, A. B., Maretzky, T., & Blobel, C. P. (2021). Analysis of the Conditions That Affect the Selective Processing of Endogenous Notch1 by ADAM10 and ADAM17. International Journal of Molecular Sciences, 22(4), 1846. https://doi.org/10.3390/ijms22041846