
Promotion and Inhibition of Amyloid-β Peptide Aggregation: Molecular Dynamics Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
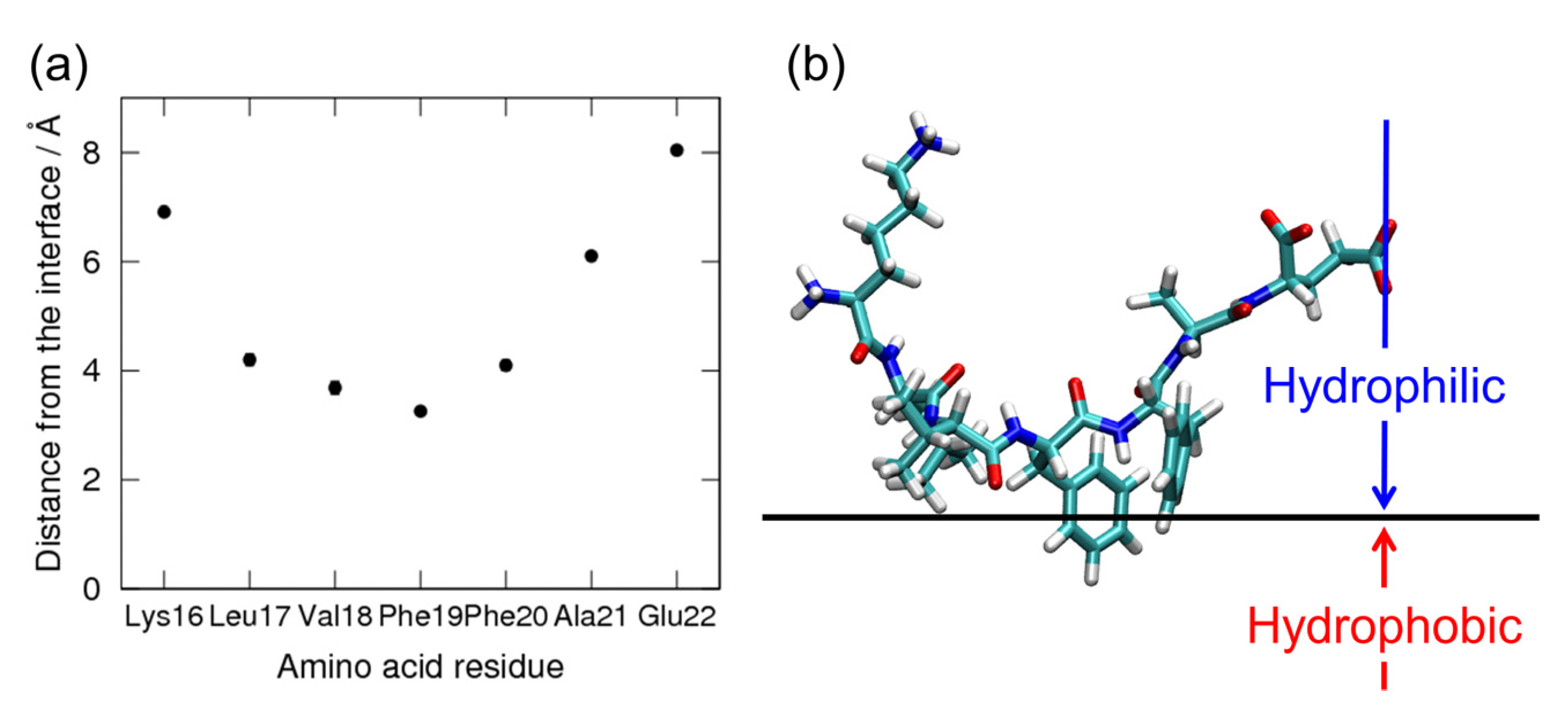
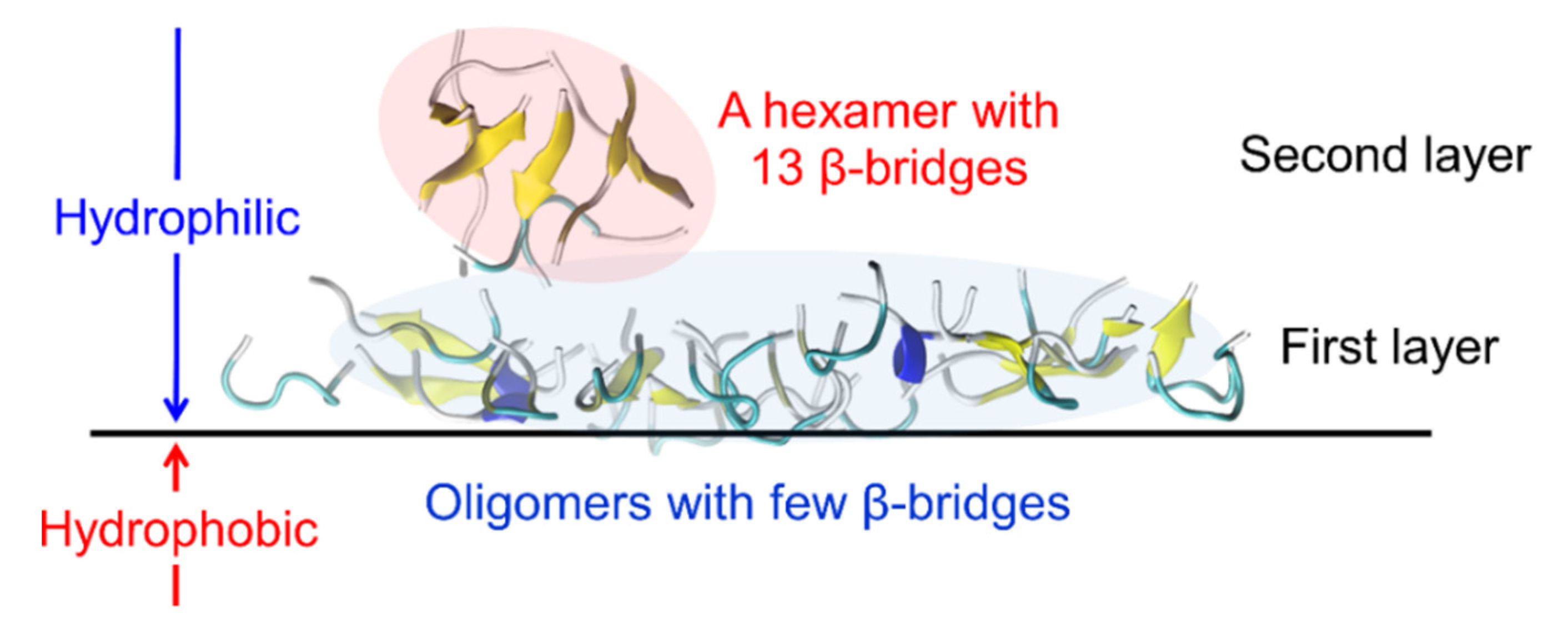
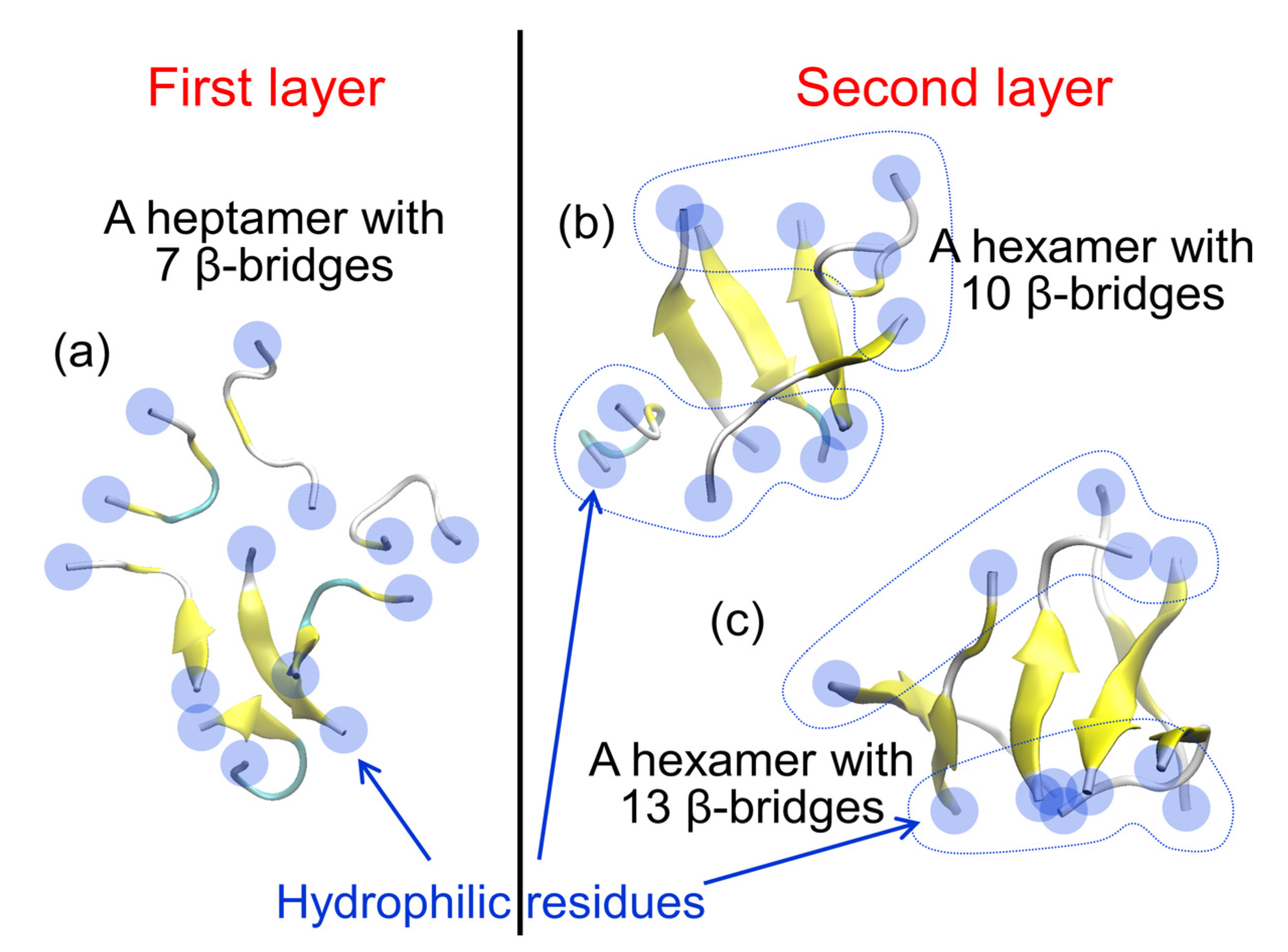
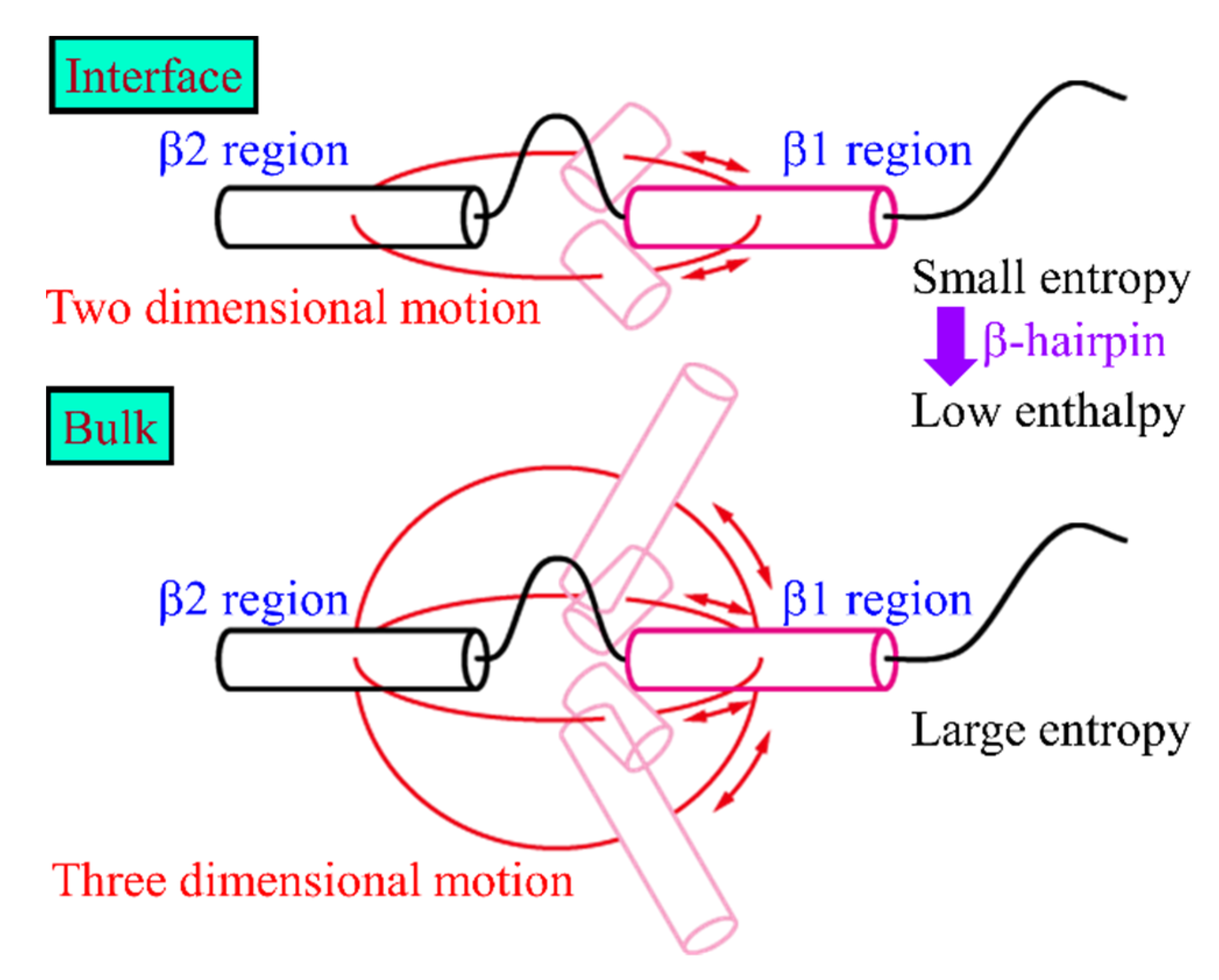
2. Amyloid-β(16–22) Aggregation at Hydrophilic–Hydrophobic Interfaces
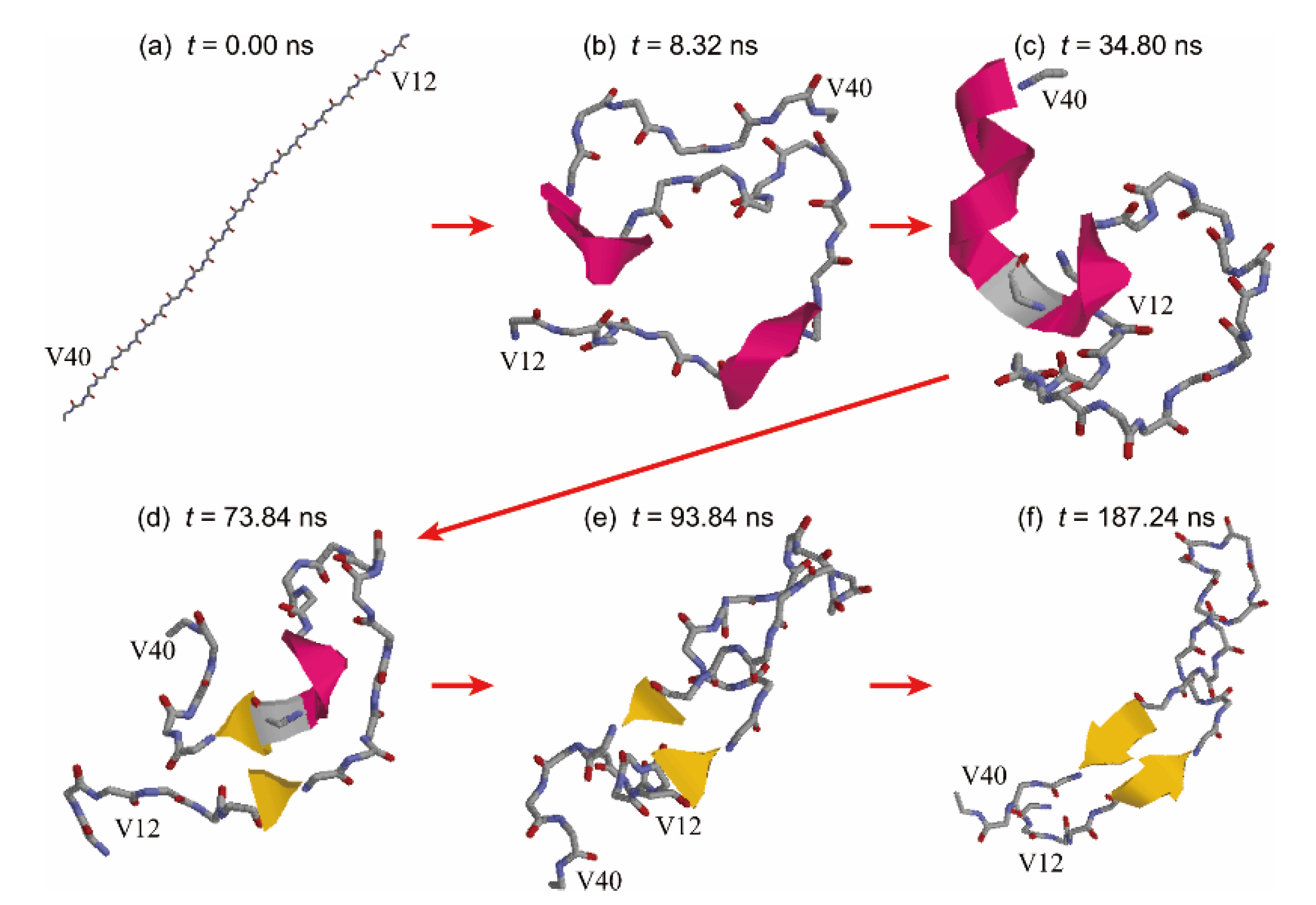
3. Conformation of an Amyloid-β 40 Peptide at a Hydrophilic–Hydrophobic Interface
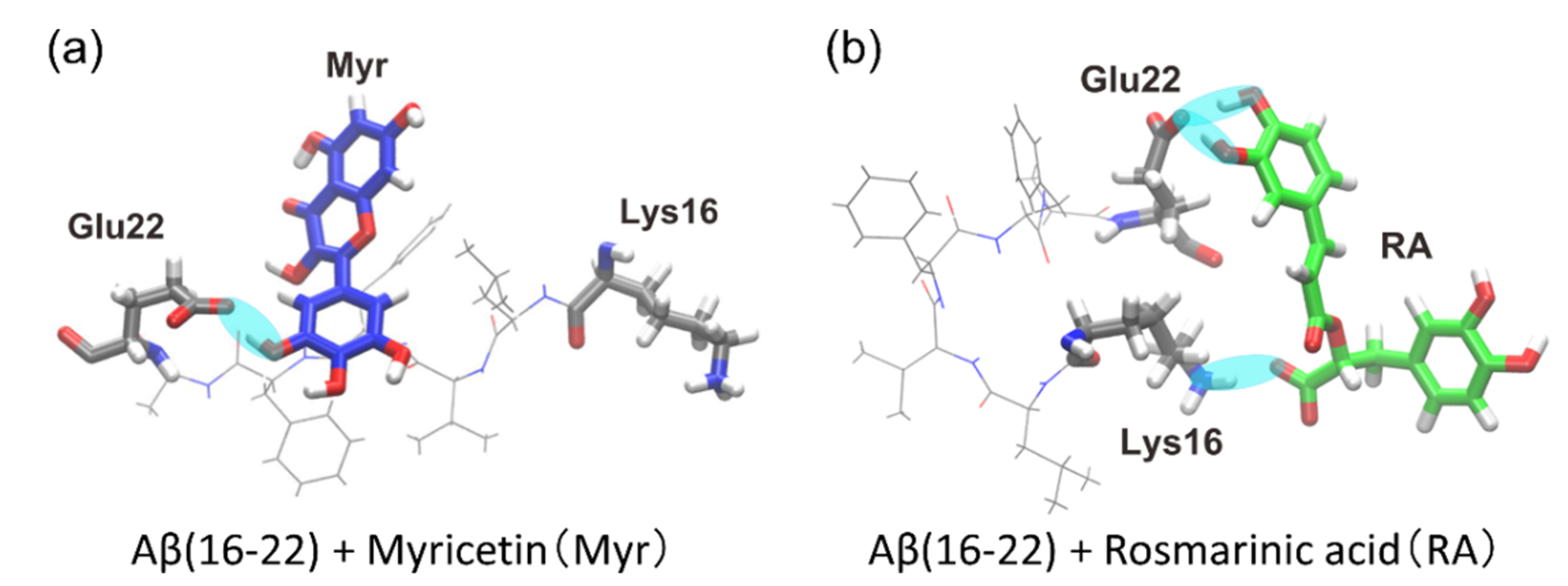
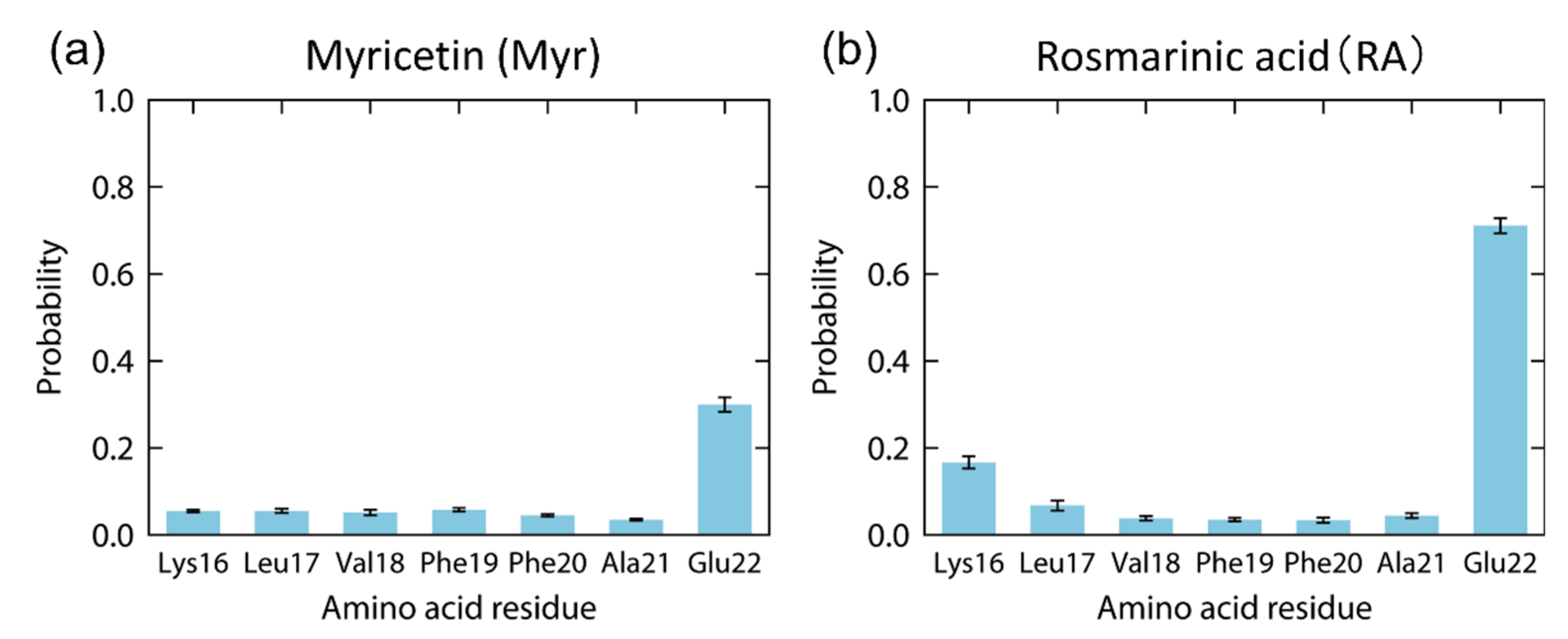
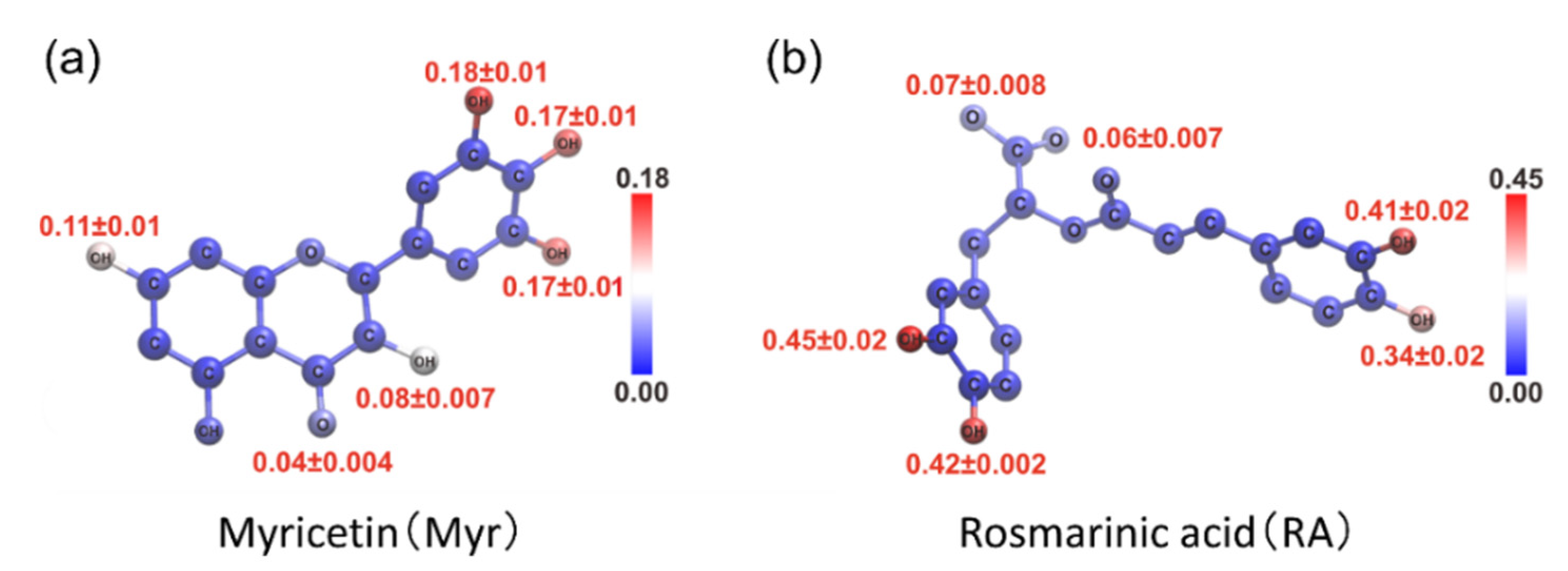
4. Interaction between Amyloid-β(16–22) and Polyphenols
5. Conclusions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sipe, J.D.; Cohen, A.S. Review: History of the Amyloid Fibril. J. Struct. Biol. 2000, 130, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tycko, R. Amyloid polymorphism: Structural basis and neurobiological relevance. Neuron 2015, 86, 632–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C. Common Core Structure of Amyloid Fibrils by Synchrotron X-ray Diffraction. J. Mol. Biol. 1997, 273, 729. [Google Scholar] [CrossRef] [Green Version]
- Petkova, A.T.; Ishii, Y.; Balbach, J.J.; Antzutkin, O.N.; Leapman, R.D.; Delaglio, F.; Tycko, R. A Structural Model for Alzheimer’s β-Amyloid Fibrils Based on Experimental Constraints from Solid State NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 16742. [Google Scholar] [CrossRef] [Green Version]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D Structure of Alzheimer’s Amyloid-β(1–42) Fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342. [Google Scholar] [CrossRef] [Green Version]
- Yagi-Utsumi, M.; Kunihara, T.; Nakamura, T.; Uekusa, Y.; Makabe, K.; Kuwajima, K.; Kato, K. NMR characterization of the interaction of GroEL with amyloid beta as a model ligand. FEBS Lett. 2013, 587, 1605–1609. [Google Scholar] [CrossRef]
- Yagi-Utsumi, M.; Kato, K.; Nishimura, K. Membrane-Induced Dichotomous Conformation of Amyloid beta with the Disordered N-Terminal Segment Followed by the Stable C-Terminal beta Structure. PLoS ONE 2016, 11, e0146405. [Google Scholar] [CrossRef]
- Hoshi, M.; Sato, M.; Matsumoto, S.; Noguchi, A.; Yasutake, K.; Yoshida, N.; Sato, K. Spherical aggregates of β-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3β. Proc. Natl. Acad. Sci. USA 2003, 100, 6370–6375. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, A.; Matsumura, S.; Dezawa, M.; Tada, M.; Yanazawa, M.; Ito, A.; Akioka, M.; Kikuchi, S.; Sato, M.; Ideno, S.; et al. Isolation and Characterization of Patient-derived, Toxic, High Mass Amyloid β-Protein (Aβ) Assembly from Alzheimer Disease Brains*. J. Biol. Chem. 2009, 284, 32895–32905. [Google Scholar] [CrossRef] [Green Version]
- Roychaudhuri, R.; Yang, M.; Hoshi, M.M.; Teplow, D.B. Amyloid β-Protein Assembly and Alzheimer Disease. J. Biol. Chem. 2009, 284, 4749–4753. [Google Scholar] [CrossRef] [Green Version]
- Benilova, I.; Karran, E.; De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Ilie, I.M.; Caflisch, A. Simulation Studies of Amyloidogenic Polypeptides and Their Aggregates. Chem. Rev. 2019, 119, 6956–6993. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Derreumaux, P. Structures of the intrinsically disordered Aβ, tau and α-synuclein proteins in aqueous solution from computer simulations. Biophys. Chem. 2020, 264, 106421. [Google Scholar] [CrossRef]
- Allison, J.R.; Varnai, P.; Dobson, C.M.; Vendruscolo, M. Determination of the Free Energy Landscape of α-Synuclein Using Spin Label Nuclear Magnetic Resonance Measurements. J. Am. Chem. Soc. 2009, 131, 18314. [Google Scholar] [CrossRef]
- Sgourakis, N.G.; Merced-Serrano, M.; Boutsidis, C.; Drineas, P.; Du, Z.; Wang, C.; Garcia, A.E. Atomic-Level Characterization of the Ensemble of the Aβ(1–42) Monomer in Water Using Unbiased Molecular Dynamics Simulations and Spectral Algorithms. J. Mol. Biol. 2011, 405, 570. [Google Scholar] [CrossRef] [Green Version]
- Velez-Vega, C.; Escobedo, F.A. Characterizing the Structural Behavior of Selected Aβ-42 Monomers with Different Solubilities. J. Phys. Chem. B 2011, 115, 4900. [Google Scholar] [CrossRef] [PubMed]
- Olubiyi, O.O.; Strodel, B. Structures of the Amyloid β-Peptides. Aβ1–40 and Aβ1–42 as Influenced by pH and a d-Peptide. J. Phys. Chem. B 2012, 116, 3280. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.G.; Okumura, H. Hamiltonian replica-permutation method and its applications to an alanine dipeptide and amyloid-β(29–42) peptides. J. Comput. Chem. 2013, 34, 2493–2497. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.G.; Okumura, H. Coulomb replica-exchange method: Handling electrostatic attractive and repulsive forces for biomolecules. J. Comput. Chem. 2013, 34, 622–639. [Google Scholar] [CrossRef]
- Rosenman, D.J.; Connors, C.R.; Chen, W.; Wang, C.; Garcia, A.E. Aβ Monomers Transiently Sample Oligomer and Fibril-Like Configurations: Ensemble Characterization Using a Combined MD/NMR Approach. J. Mol. Biol. 2013, 425, 3338. [Google Scholar] [CrossRef] [Green Version]
- Rosenman, D.J.; Wang, C.; Garcia, A.E. Characterization of Aβ Monomers through the Convergence of Ensemble Properties among Simulations with Multiple Force Fields. J. Phys. Chem. B 2016, 120, 259. [Google Scholar] [CrossRef]
- Ilie, I.M.; Nayar, D.; den Otter, W.K.; van der Vegt, N.F.A.; Briels, W.J. Intrinsic Conformational Preferences and Interactions in α-Synuclein Fibrils. Insights from Molecular Dynamics simulations. J. Chem. Theory Comput. 2018, 14, 3298. [Google Scholar] [CrossRef]
- Tachi, Y.; Okamoto, Y.; Okumura, H. Conformational Change of Amyloid-β 40 in Association with Binding to GM1-Glycan Cluster. Sci. Rep. 2019, 9, 6853. [Google Scholar] [CrossRef] [Green Version]
- Itoh, S.G.; Yagi-Utsumi, M.; Kato, K.; Okumura, H. Effects of a Hydrophilic/Hydrophobic Interface on Amyloid-β Peptides Studied by Molecular Dynamics Simulations and NMR Experiments. J. Phys. Chem. B 2019, 123, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Ngoc, L.L.N.; Itoh, S.G.; Sompornpisut, P.; Okumura, H. Replica-permutation molecular dynamics simulations of an amyloid-β(16–22) peptide and polyphenols. Chem. Phys. Lett. 2020, 758, 137913. [Google Scholar] [CrossRef]
- Chebaro, Y.; Mousseau, N.; Derreumaux, P. Structures and Thermodynamics of Alzheimer’s Amyloid-β Aβ(16–35) Monomer and Dimer by Replica Exchange Molecular Dynamics Simulations: Implication for Full-Length Aβ Fibrillation. J. Phys. Chem. B 2009, 113, 7668. [Google Scholar] [CrossRef] [PubMed]
- Cote, S.; Laghaei, R.; Derreumaux, P.; Mousseau, N. Distinct Dimerization for Various Alloforms of the Amyloid-Beta Protein: Aβ1–40, Aβ1–42, and Aβ1–40(D23N). J. Phys. Chem. B 2012, 116, 4043. [Google Scholar] [CrossRef]
- Chiang, H.L.; Chen, C.J.; Okumura, H.; Hu, C.K. Transformation between alpha-helix and beta-sheet structures of one and two polyglutamine peptides in explicit water molecules by replica-exchange molecular dynamics simulations. J. Comput. Chem. 2014, 35, 1430–1437. [Google Scholar] [CrossRef]
- Itoh, S.G.; Okumura, H. Dimerization process of amyloid-β(29–42) studied by the Hamiltonian replica-permutation molecular dynamics simulations. J. Phys. Chem. B 2014, 118, 11428–11436. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Sterpone, F.; Campanera, J.M.; Nasica-Labouze, J.; Derreumaux, P. Impact of the A2V Mutation on the Heterozygous and Homozygous Aβ1–40 Dimer Structures from Atomistic Simulations. ACS Chem. Neurosci. 2016, 7, 823. [Google Scholar] [CrossRef] [PubMed]
- Tarus, B.; Tran, T.T.; Nasica-Labouze, J.; Sterpone, F.; Nguyen, P.H.; Derreumaux, P. Structures of the Alzheimer’s Wild-Type Aβ1–40 Dimer from Atomistic Simulations. J. Phys. Chem. B 2015, 119, 10478. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Sterpone, F.; Pouplana, R.; Derreumaux, P.; Campanera, J.M. Dimerization Mechanism of Alzheimer Aβ40 Peptides: The High Content of Intrapeptide-Stabilized Conformations in A2V and A2T Heterozygous Dimers Retards Amyloid Fibril Formation. J. Phys. Chem. B 2016, 120, 12111. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Chacko, A.R.; Belfort, G. Alzheimer’s Protective Cross-Interaction between Wild-Type and A2T Variants Alters Aβ42 Dimer Structure. ACS Chem. Neurosci. 2017, 8, 606. [Google Scholar] [CrossRef] [PubMed]
- Man, V.H.; Nguyen, P.H.; Derreumaux, P. Conformational Ensembles of the Wild- Type and S8C Aβ1–42 Dimers. J. Phys. Chem. B 2017, 121, 2434. [Google Scholar] [CrossRef] [Green Version]
- Man, V.H.; Nguyen, P.H.; Derreumaux, P. High-Resolution Structures of the Amyloid-β 1–42 Dimers from the Comparison of Four Atomistic Force Fields. J. Phys. Chem. B 2017, 121, 5977. [Google Scholar] [CrossRef] [Green Version]
- Sharma, B.; Ranganathan, S.V.; Belfort, G. Weaker N-Terminal Interactions for the Protective over the Causative Aβ Peptide Dimer Mutants. ACS Chem. Neurosci. 2018, 9, 1247. [Google Scholar] [CrossRef]
- Nishizawa, H.; Okumura, H. Classical Molecular Dynamics Simulation to Understand Role of a Zinc Ion for Aggregation of Amyloid-β Peptides. J. Comput. Chem. Jpn. 2018, 17, 76–79. [Google Scholar] [CrossRef] [Green Version]
- Gsponer, J.; Haberthür, U.; Caflisch, A. The Role of Side-chain Interactions in the Early Steps of Aggregation: Molecular Dynamics Simulations of an Amyloid-forming Peptide from the Yeast Prion Sup35. Proc. Natl. Acad. Sci. USA 2003, 100, 5154. [Google Scholar] [CrossRef] [Green Version]
- Urbanc, B.; Betnel, M.; Cruz, L.; Bitan, G.; Teplow, D.B. Elucidation of amyloid beta-protein oligomerization mechanisms: Discrete molecular dynamics study. J. Am. Chem. Soc. 2010, 132, 4266–4280. [Google Scholar] [CrossRef] [Green Version]
- Carballo-Pacheco, M.; Ismail, A.E.; Strodel, B. Oligomer Formation of Toxic and Functional Amyloid Peptides Studied with Atomistic Simulations. J. Phys. Chem. B 2015, 119, 9696. [Google Scholar] [CrossRef]
- Itoh, S.G.; Okumura, H. Oligomer Formation of Amyloid-β(29–42) from Its Monomers Using the Hamiltonian Replica-Permutation Molecular Dynamics Simulation. J. Phys. Chem. B 2016, 120, 6555–6561. [Google Scholar] [CrossRef] [PubMed]
- Barz, B.; Liao, Q.; Strodel, B. Pathways of Amyloid-β Aggregation Depend on Oligomer Shape. J. Am. Chem. Soc. 2018, 140, 319. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ge, X.; Xing, Y.; Wang, B.; Ding, F. β-Barrel Oligomers as Common Intermediates of Peptides Self-Assembling into Cross-β Aggregates. Sci. Rep. 2018, 8, 10353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, H.; Itoh, S.G. Molecular dynamics simulations of amyloid-β(16–22) peptide aggregation at air–water interfaces. J. Chem. Phys. 2020, 152, 095101. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Li, M.S.; Stock, G.; Straub, J.E.; Thirumalai, D. Monomer Adds to Preformed Structured Oligomers of Aβ-Peptides by a Two-stage Dock-Lock Mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, E.P.; Okamoto, Y.; Straub, J.E.; Brooks, B.R.; Thirumalai, D. Thermodynamic Perspective on the Dock-Lock Growth Mechanism of Amyloid Fibrils. J. Phys. Chem. B 2009, 113, 14421. [Google Scholar] [CrossRef] [Green Version]
- Takeda, T.; Klimov, D.K. Probing Energetics of Aβ Fibril Elongation by Molecular Dynamics Simulations. Biophys. J. 2009, 96, 4428. [Google Scholar] [CrossRef] [Green Version]
- Takeda, T.; Klimov, D.K. Replica Exchange Simulations of the Thermodynamics of Aβ Fibril Growth. Biophys. J. 2009, 96, 442. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.S.; Wang, L.; Singh, S.; Ling, Y.L.; Buchanan, L.; Zanni, M.T.; Skinner, J.L.; de Pablo, J.J. Stable and Metastable States of Human Amylin in Solution. Biophys. J. 2010, 99, 2208. [Google Scholar] [CrossRef] [Green Version]
- Han, M.; Hansmann, U.H.E. Replica Exchange Molecular Dynamics of the Thermodynamics of Fibril Growth of Alzheimer’s Aβ42 Peptide. J. Chem. Phys. 2011, 135, 065101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straub, J.E.; Thirumalai, D. Toward a Molecular Theory of Early and Late Events in Monomer to Amyloid Fibril Formation. Annu. Rev. Phys. Chem. 2011, 62, 437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurry, T.; Stultz, C.M. Mechanism of Amyloid-β Fibril Elongation. Biochemistry 2014, 53, 6981. [Google Scholar] [CrossRef]
- Han, W.; Schulten, K. Fibril Elongation by Aβ17–42: Kinetic Network Analysis of Hybrid-Resolution Molecular Dynamics Simulations. J. Am. Chem. Soc. 2014, 136, 12450. [Google Scholar] [CrossRef] [Green Version]
- Schwierz, N.; Frost, C.V.; Geissler, P.L.; Zacharias, M. Dynamics of Seeded Aβ40-Fibril Growth from Atomistic Molecular Dynamics Simulations: Kinetic Trapping and Reduced Water Mobility in the Locking Step. J. Am. Chem. Soc. 2016, 138, 527. [Google Scholar] [CrossRef]
- Sasmal, S.; Schwierz, N.; Head-Gordon, T. Mechanism of Nucleation and Growth of Aβ40 Fibrils from All-Atom and Coarse-Grained Simulations. J. Phys. Chem. B 2016, 120, 12088. [Google Scholar] [CrossRef] [Green Version]
- Bacci, M.; Vymetal, J.; Mihajlovic, M.; Caflisch, A.; Vitalis, A. Amyloid β Fibril Elongation by Monomers Involves Disorder at the Tip. J. Chem. Theory Comput. 2017, 13, 5117. [Google Scholar] [CrossRef]
- Ilie, I.M.; den Otter, W.K.; Briels, W.J. The Attachment of α-synuclein to a Fiber: A Coarse Grain Approach. J. Chem. Phys. 2017, 146, 115102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchete, N.V.; Tycko, R.; Hummer, G. Molecular Dynamics Simulations of Alzheimer’s β-Amyloid Protofilaments. J. Mol. Biol. 2005, 353, 804. [Google Scholar] [CrossRef]
- Baumketner, A.; Griffin Krone, M.; Shea, J. Role of the Familial Dutch Mutation E22Q in the Folding and Aggregation of the 15–28 Fragment of the Alzheimer amyloid-β Protein. Proc. Natl. Acad. Sci. USA 2008, 105, 6027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemkul, J.A.; Bevan, D.R. Assessing the Stability of Alzheimer’s Amyloid Protofibrils Using Molecular Dynamics. J. Phys. Chem. B 2010, 114, 1652. [Google Scholar] [CrossRef]
- Okumura, H.; Itoh, S.G. Structural and fluctuational difference between two ends of Abeta amyloid fibril: MD simulations predict only one end has open conformations. Sci. Rep. 2016, 6, 38422. [Google Scholar] [CrossRef]
- Rodriguez, R.A.; Chen, L.Y.; Plascencia-Villa, G.; Perry, G. Thermodynamics of Amyloid-β Fibril Elongation: Atomistic Details of the Transition State. ACS Chem. Neurosci. 2018, 9, 783. [Google Scholar] [CrossRef]
- Davidson, D.S.; Brown, A.M.; Lemkul, J.A. Insights into Stabilizing Forces in Amyloid Fibrils of Differing Sizes from Polarizable Molecular Dynamics Simulations. J. Mol. Biol. 2018, 430, 3819. [Google Scholar] [CrossRef] [PubMed]
- Ilie, I.M.; Caflisch, A. Disorder at the Tips of a Disease-Relevant Aβ42 Amyloid Fibril: A Molecular Dynamics Study. J. Phys. Chem. B 2018, 122, 11072. [Google Scholar] [CrossRef]
- Okumura, H.; Itoh, S.G. Amyloid fibril disruption by ultrasonic cavitation: Nonequilibrium molecular dynamics simulations. J. Am. Chem. Soc. 2014, 136, 10549–10552. [Google Scholar] [CrossRef]
- Hoang Viet, M.; Derreumaux, P.; Nguyen, P.H. Nonequilibrium all-atom molecular dynamics simulation of the bubble cavitation and application to dissociate amyloid fibrils. J. Chem. Phys. 2016, 145, 174113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang Viet, M.; Derreumaux, P.; Li, M.S.; Roland, C.; Sagui, C.; Nguyen, P.H. Picosecond dissociation of amyloid fibrils with infrared laser: A nonequilibrium simulation study. J. Chem. Phys. 2015, 143, 155101. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Yahi, N. Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: Common mechanisms in neurodegenerative diseases. Expert Rev. Mol. Med. 2010, 12, e27. [Google Scholar] [CrossRef]
- Morinaga, A.; Hasegawa, K.; Nomura, R.; Ookoshi, T.; Ozawa, D.; Goto, Y.; Yamada, M.; Naiki, H. Critical role of interfaces and agitation on the nucleation of Aβ amyloid fibrils at low concentrations of Aβ monomers. Biochim. Biophys. Acta 2010, 1804, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Jean, L.; Lee, C.F.; Vaux, D.J. Enrichment of Amyloidogenesis at an Air-Water Interface. Biophys. J. 2012, 102, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J. Neurochem. 2003, 87, 172–181. [Google Scholar] [CrossRef]
- Ono, K.; Li, L.; Takamura, Y.; Yoshiike, Y.; Zhu, L.; Han, F.; Mao, X.; Ikeda, T.; Takasaki, J.; Nishijo, H.; et al. Phenolic compounds prevent amyloid beta-protein oligomerization and synaptic dysfunction by site-specific binding. J. Biol. Chem. 2012, 287, 14631–14643. [Google Scholar] [CrossRef] [Green Version]
- Balbach, J.J.; Ishii, Y.; Antzutkin, O.N.; Leapman, R.D.; Rizzo, N.W.; Dyda, F.; Reed, J.; Tycko, R. Amyloid Fibril Formation by Aβ16-22, a Seven-Residue Fragment of the Alzheimer’s β-Amyloid Peptide, and Structural Characterization by Solid State NMR. Biochemistry 2000, 39, 13748–13759. [Google Scholar] [CrossRef]
- Klimov, D.K.; Straub, J.E.; Thirumalai, D. Aqueous urea solution destabilizes Aβ16–22 oligomers. Proc. Natl. Acad. Sci. USA 2004, 101, 14760–14765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, P.H.; Li, M.S.; Derreumaux, P. Effects of all-atom force fields on amyloid oligomerization: Replica exchange molecular dynamics simulations of the Aβ16–22 dimer and trimer. Phys. Chem. Chem. Phys. 2011, 13, 9778–9788. [Google Scholar] [CrossRef]
- Riccardi, L.; Nguyen, P.H.; Stock, G. Construction of the Free Energy Landscape of Peptide Aggregation from Molecular Dynamics Simulations. J. Chem. Theory Comput. 2012, 8, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Barz, B.; Wales, D.J.; Strodel, B. A Kinetic Approach to the Sequence-Aggregation Relationship in Disease-Related Protein Assembly. J. Phys. Chem. B 2014, 118, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Chiricotto, M.; Melchionna, S.; Derreumaux, P.; Sterpone, F. Hydrodynamic effects on β-amyloid (16–22) peptide aggregation. J. Chem. Phys. 2016, 145, 035102. [Google Scholar] [CrossRef] [PubMed]
- Carballo-Pacheco, M.; Ismail, A.E.; Strodel, B. On the Applicability of Force Fields to Study the Aggregation of Amyloidogenic Peptides Using Molecular Dynamics Simulations. J. Chem. Theory Comput. 2018, 14, 6063–6075. [Google Scholar] [CrossRef]
- Okumura, H.; Okamoto, Y. Multibaric-multithermal ensemble molecular dynamics simulations. J. Comput. Chem. 2006, 27, 379–395. [Google Scholar] [CrossRef]
- Okumura, H.; Okamoto, Y. Multibaric-Multithermal Molecular Dynamics Simulation of Alanine Dipeptide in Explicit Water. Bull. Chem. Soc. Jpn. 2007, 80, 1114–1123. [Google Scholar] [CrossRef]
- Okumura, H.; Okamoto, Y. Temperature and pressure dependence of alanine dipeptide studied by multibaric-multithermal molecular dynamics simulations. J. Phys. Chem. B 2008, 112, 12038–12049. [Google Scholar] [CrossRef] [PubMed]
- Okumura, H. Partial multicanonical algorithm for molecular dynamics and Monte Carlo simulations. J. Chem. Phys. 2008, 129, 124116. [Google Scholar] [CrossRef] [PubMed]
- Okumura, H. Optimization of partial multicanonical molecular dynamics simulations applied to an alanine dipeptide in explicit water solvent. Phys. Chem. Chem. Phys. 2011, 13, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Okumura, H. Temperature and pressure denaturation of chignolin: Folding and unfolding simulation by multibaric-multithermal molecular dynamics method. Proteins 2012, 80, 2397–2416. [Google Scholar] [CrossRef] [PubMed]
- Okumura, H.; Itoh, S.G. Transformation of a design peptide between the α-helix and β-hairpin structures using a helix-strand replica-exchange molecular dynamics simulation. Phys. Chem. Chem. Phys. 2013, 15, 13852–13861. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Miller, T.F.; Eleftheriou, M.; Pattnaik, P.; Ndirango, A.; Newns, D.; Martyna, G.J. Symplectic quaternion scheme for biophysical molecular dynamics. J. Chem. Phys. 2002, 116, 8649–8659. [Google Scholar] [CrossRef] [Green Version]
- Okumura, H.; Itoh, S.G.; Okamoto, Y. Explicit symplectic integrators of molecular dynamics algorithms for rigid-body molecules in the canonical, isobaric-isothermal, and related ensembles. J. Chem. Phys. 2007, 126, 084103. [Google Scholar] [CrossRef] [PubMed]
- Petkova, A.T.; Yau, W.M.; Tycko, R. Experimental constraints on quaternary structure in Alzheimer’s beta-amyloid fibrils. Biochemistry 2006, 45, 498–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utsumi, M.; Yamaguchi, Y.; Sasakawa, H.; Yamamoto, N.; Yanagisawa, K.; Kato, K. Up-and-down topological mode of amyloid β-peptide lying on hydrophilic/hydrophobic interface of ganglioside clusters. Glycoconjug. J. 2008, 26, 999. [Google Scholar] [CrossRef] [PubMed]
- Abelein, A.; Abrahams, J.P.; Danielsson, J.; Gräslund, A.; Jarvet, J.; Luo, J.; Tiiman, A.; Wärmländer, S.K. The hairpin conformation of the amyloid β peptide is an important structural motif along the aggregation pathway. J. Biol. Inorg. Chem. 2014, 19, 623–634. [Google Scholar] [CrossRef]
- Maity, S.; Hashemi, M.; Lyubchenko, Y.L. Nano-assembly of amyloid β peptide: Role of the hairpin fold. Sci. Rep. 2017, 7, 2344. [Google Scholar] [CrossRef] [PubMed]
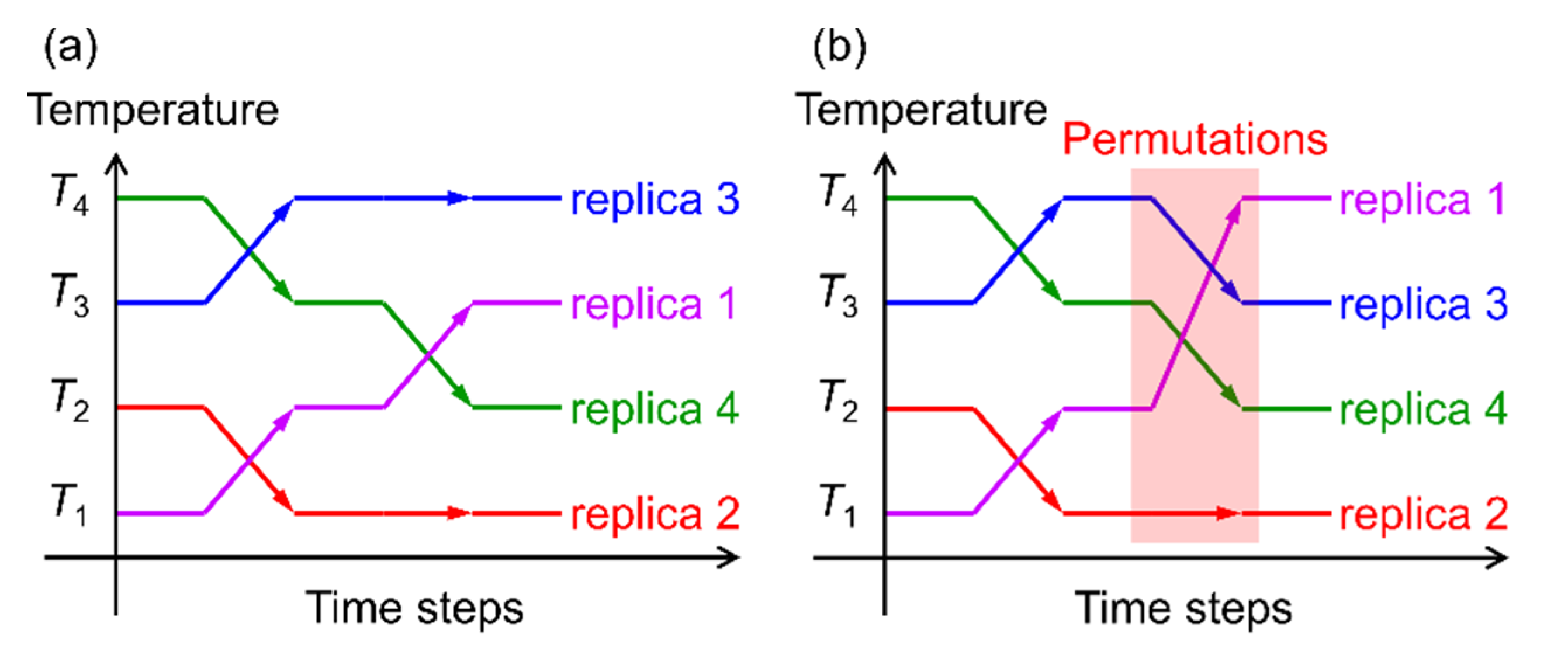
- Itoh, S.G.; Okumura, H. Replica-Permutation Method with the Suwa-Todo Algorithm beyond the Replica-Exchange Method. J. Chem. Theory Comput. 2013, 9, 570–581. [Google Scholar] [CrossRef] [Green Version]
- Mitsutake, A.; Sugita, Y.; Okamoto, Y. Generalized-ensemble algorithms for molecular simulations of biopolymers. Biopolymers 2001, 60, 96–123. [Google Scholar] [CrossRef]
- Itoh, S.G.; Okumura, H.; Okamoto, Y. Generalized-ensemble algorithms for molecular dynamics simulations. Mol. Simul. 2007, 33, 47–56. [Google Scholar] [CrossRef]
- Yamauchi, M.; Mori, Y.; Okumura, H. Molecular simulations by generalized-ensemble algorithms in isothermal-isobaric ensemble. Biophys. Rev. 2019, 11, 457–469. [Google Scholar] [CrossRef]
- Hukushima, K.; Nemoto, K. Exchange Monte Carlo method and application to spin glass simulations. J. Phys. Soc. Jpn. 1996, 65, 1604–1608. [Google Scholar] [CrossRef] [Green Version]
- Sugita, Y.; Okamoto, Y. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Suwa, H.; Todo, S. Markov chain Monte Carlo method without detailed balance. Phys. Rev. Lett. 2010, 105, 120603. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, M.; Okumura, H. Replica sub-permutation method for molecular dynamics and monte carlo simulations. J. Comput. Chem. 2019, 40, 2694–2711. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Okumura, H. Development of isothermal-isobaric replica-permutation method for molecular dynamics and Monte Carlo simulations and its application to reveal temperature and pressure dependence of folded, misfolded, and unfolded states of chignolin. J. Chem. Phys. 2017, 147, 184107. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, H.; Okumura, H. Comparison of Replica-Permutation Molecular Dynamics Simulations with and without Detailed Balance Condition. J. Phys. Soc. Jpn. 2015, 84, 074801. [Google Scholar] [CrossRef]
- Mori, Y.; Okumura, H. Simulated tempering based on global balance or detailed balance conditions: Suwa–Todo, heat bath, and Metropolis algorithms. J. Comput. Chem. 2015, 36, 2344–2349. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Itoh, S.G.; Okumura, H. Promotion and Inhibition of Amyloid-β Peptide Aggregation: Molecular Dynamics Studies. Int. J. Mol. Sci. 2021, 22, 1859. https://doi.org/10.3390/ijms22041859
Itoh SG, Okumura H. Promotion and Inhibition of Amyloid-β Peptide Aggregation: Molecular Dynamics Studies. International Journal of Molecular Sciences. 2021; 22(4):1859. https://doi.org/10.3390/ijms22041859
Chicago/Turabian StyleItoh, Satoru G., and Hisashi Okumura. 2021. "Promotion and Inhibition of Amyloid-β Peptide Aggregation: Molecular Dynamics Studies" International Journal of Molecular Sciences 22, no. 4: 1859. https://doi.org/10.3390/ijms22041859