
Preclinical Therapy with Vitamin D3 in Experimental Encephalomyelitis: Efficacy and Comparison with Paricalcitol


 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
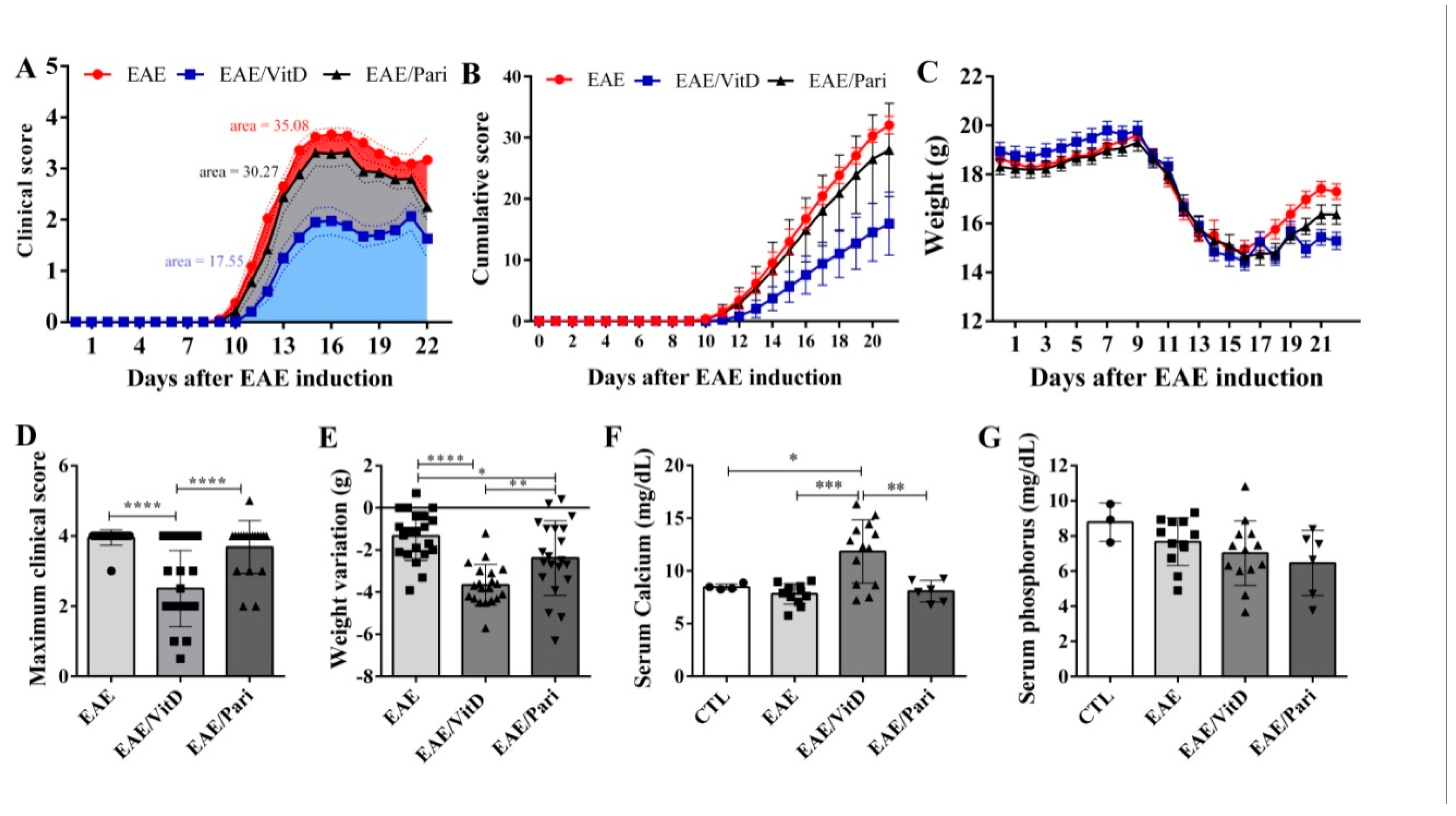
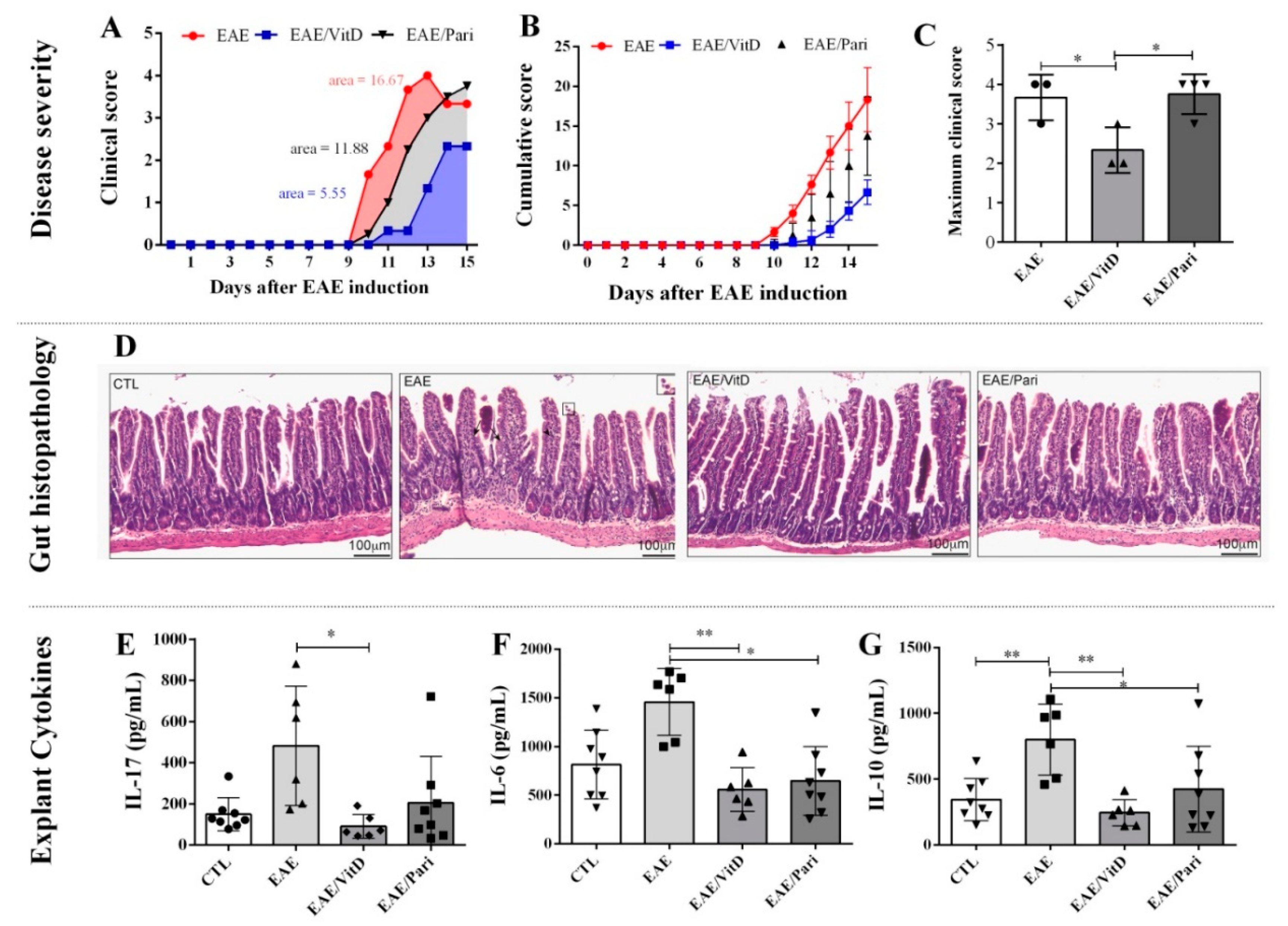
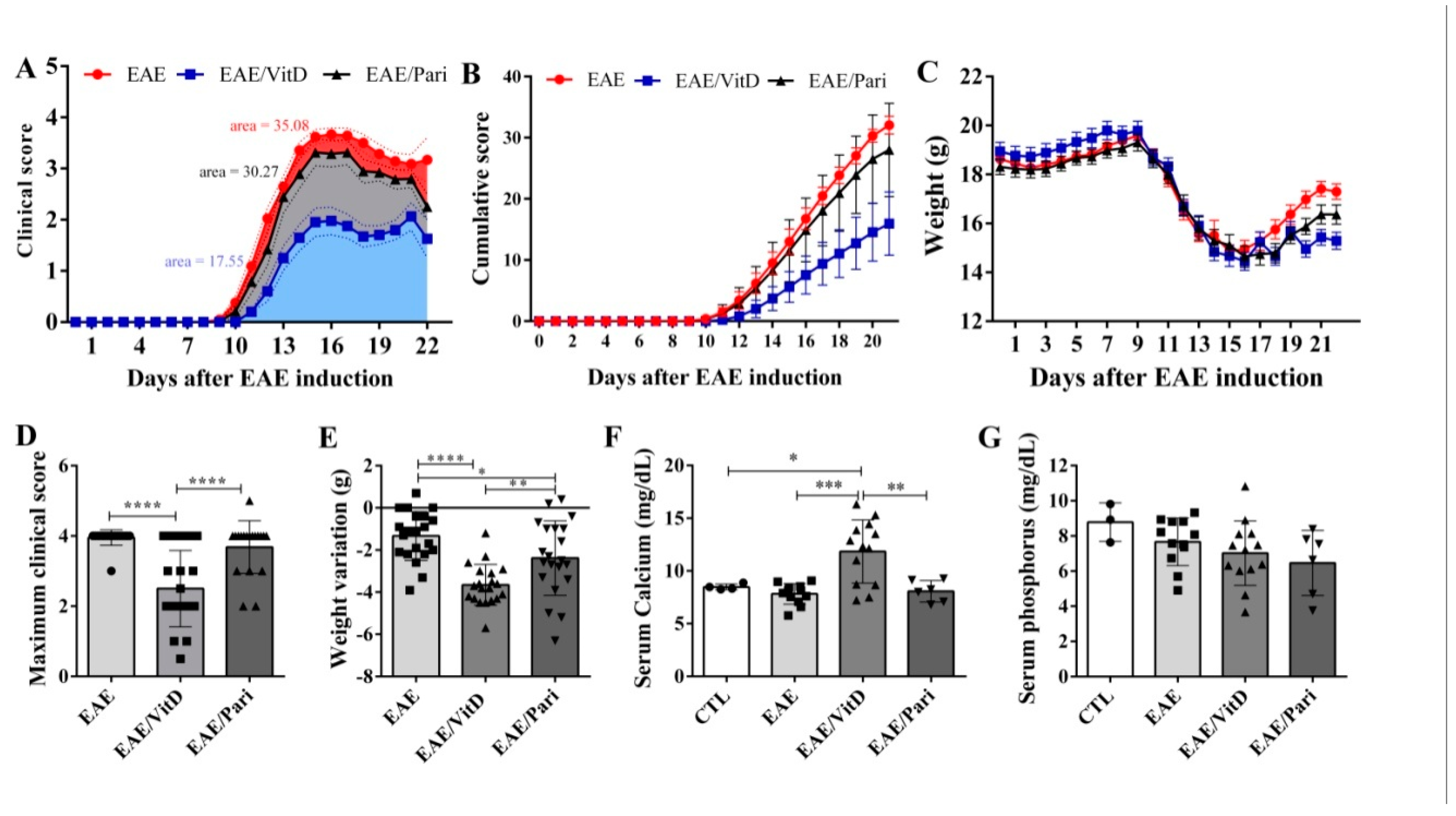
2.1. Preclinical Therapy with Vitamin D3 (VitD), but Not with the Vitamin D Analog Paricalcitol (Pari), Controlled Clinical Experimental Autoimmune Encephalomyelitis (EAE) Severity
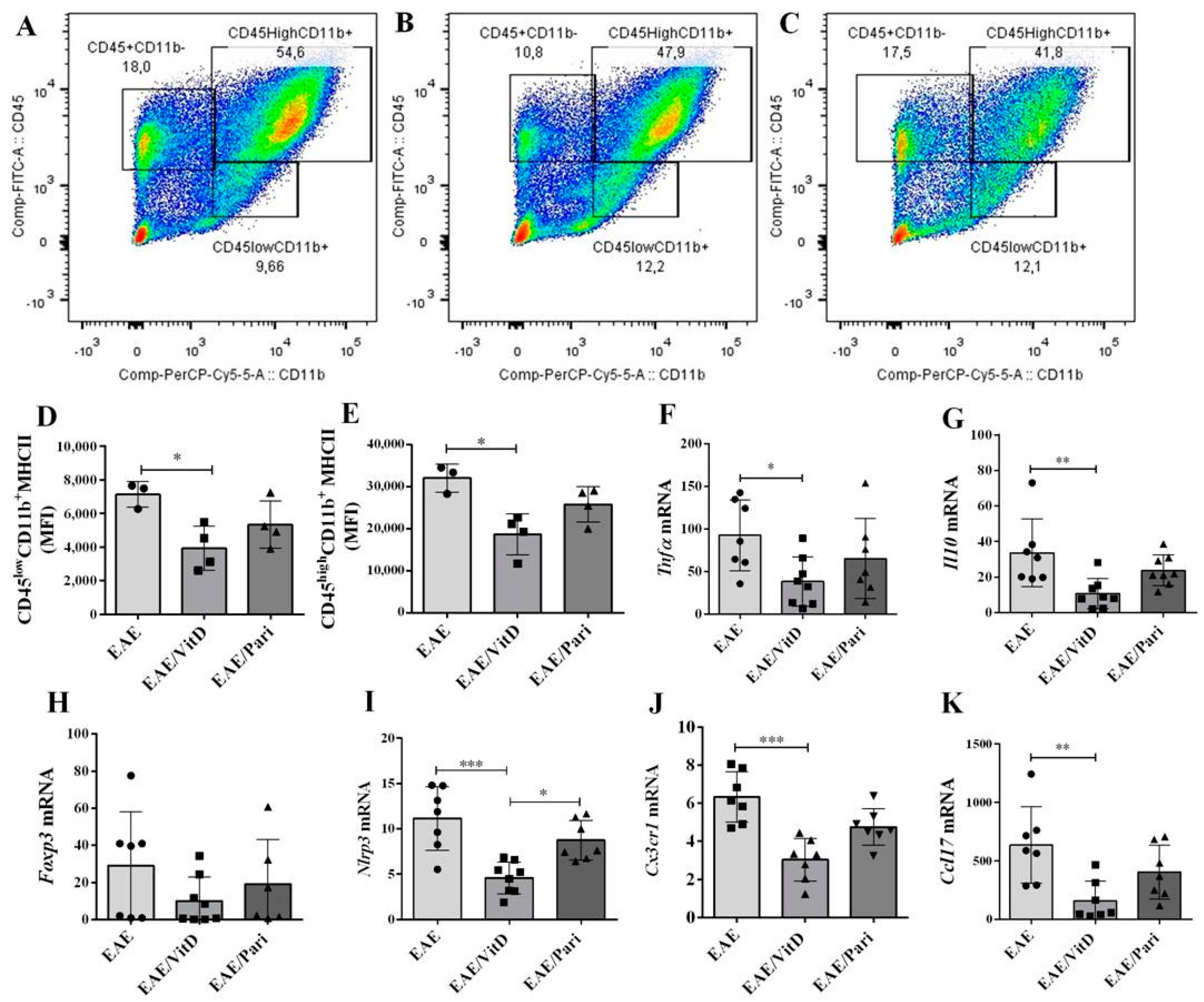
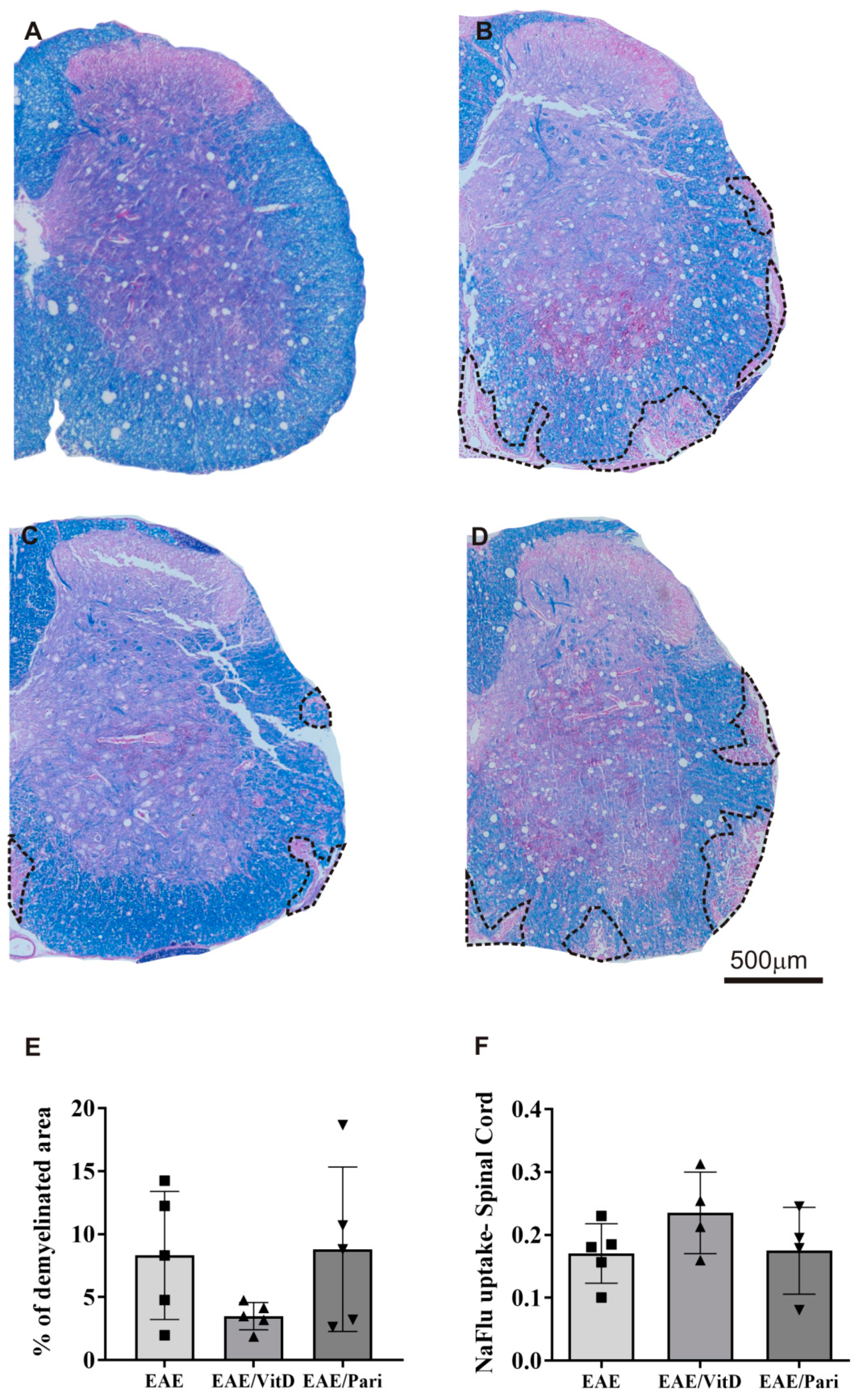
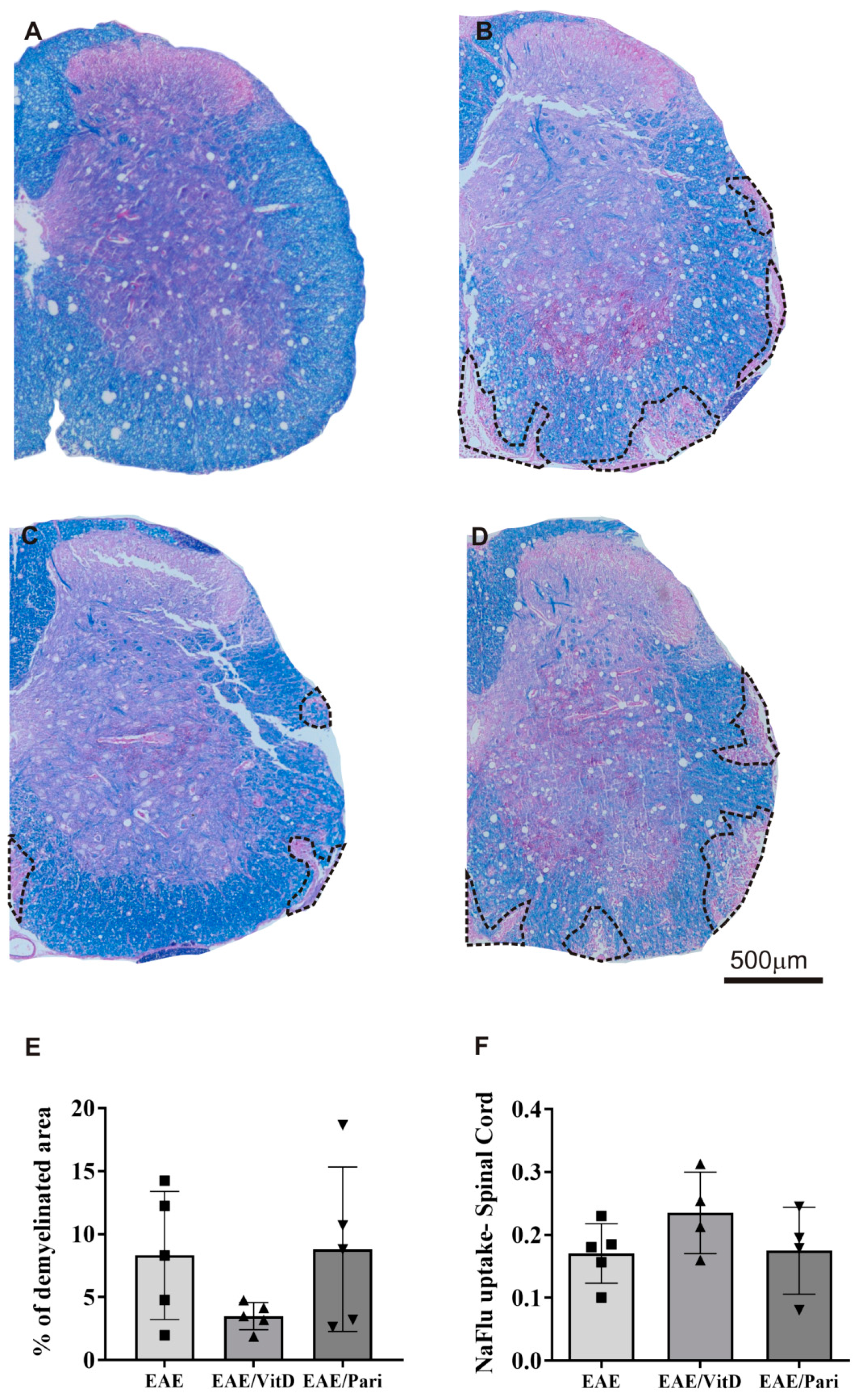
2.2. VitD, but Not Pari, Reduced Inflammation and Demyelination in the Central Nervous System (CNS)
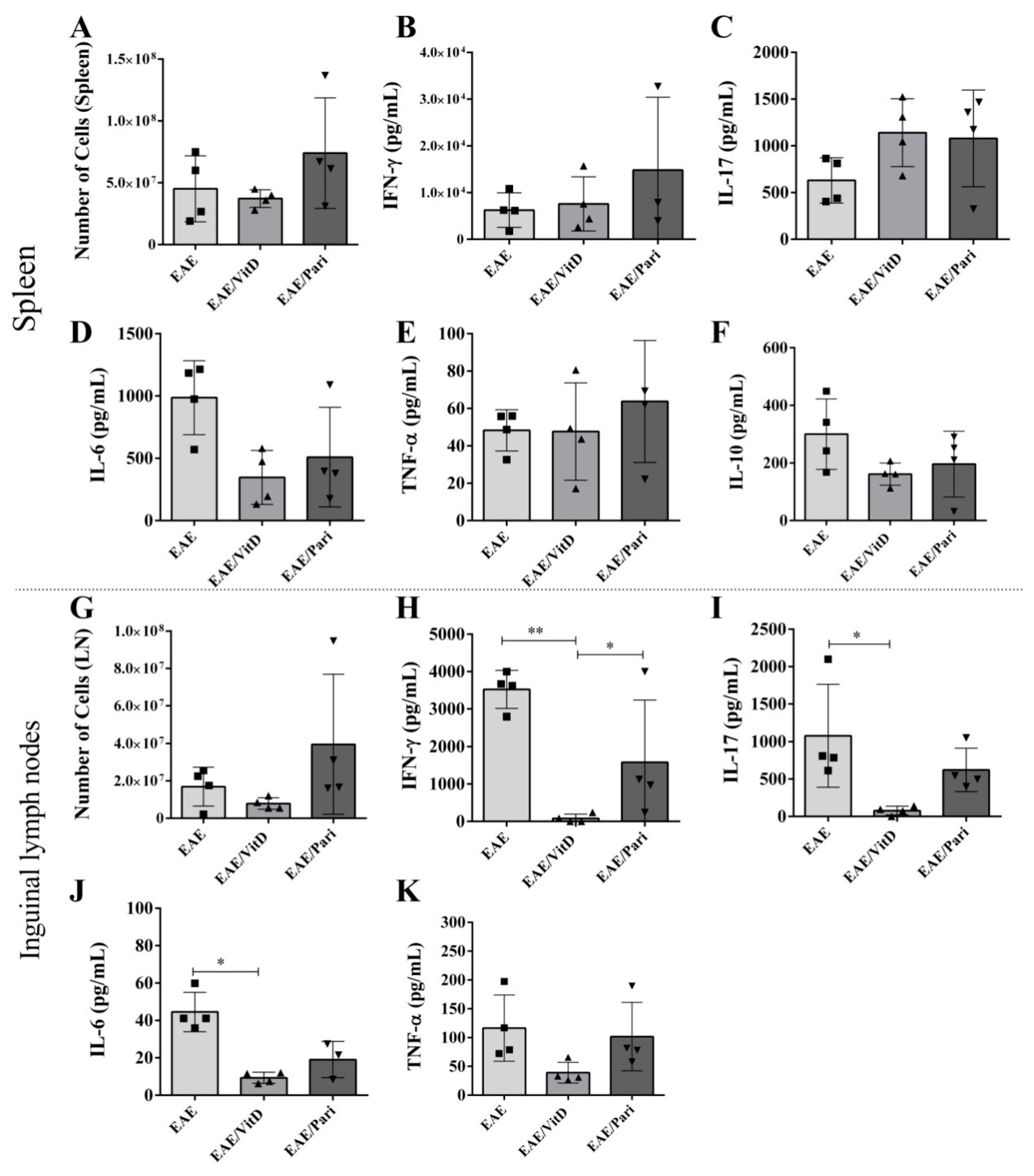
2.3. VitD, but Not Pari, Decreased Cytokine Production by Draining Lymph Nodes Derived Cells
2.4. Gut Inflammation Is Better Controlled by VitD Than by Pari
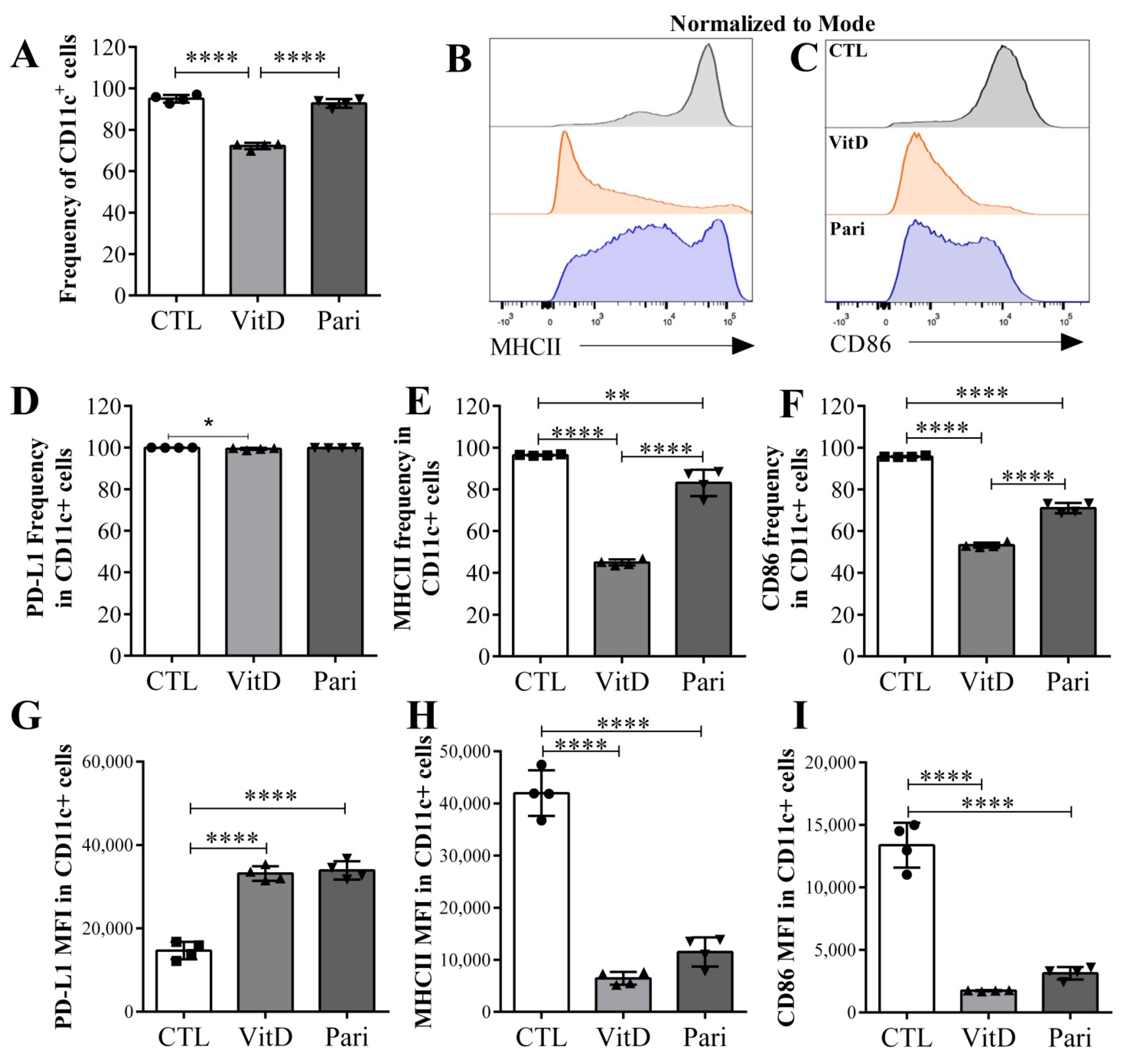
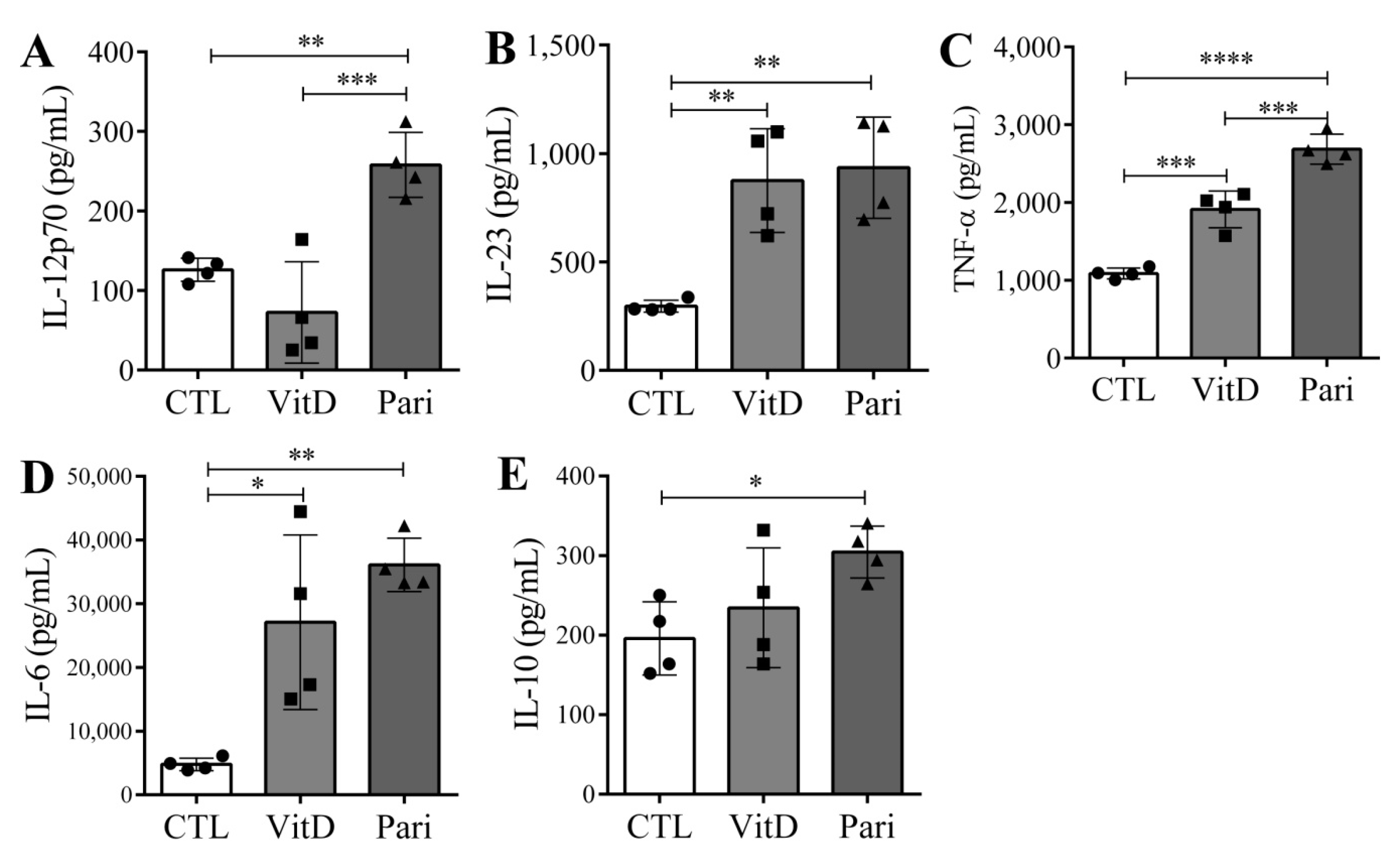
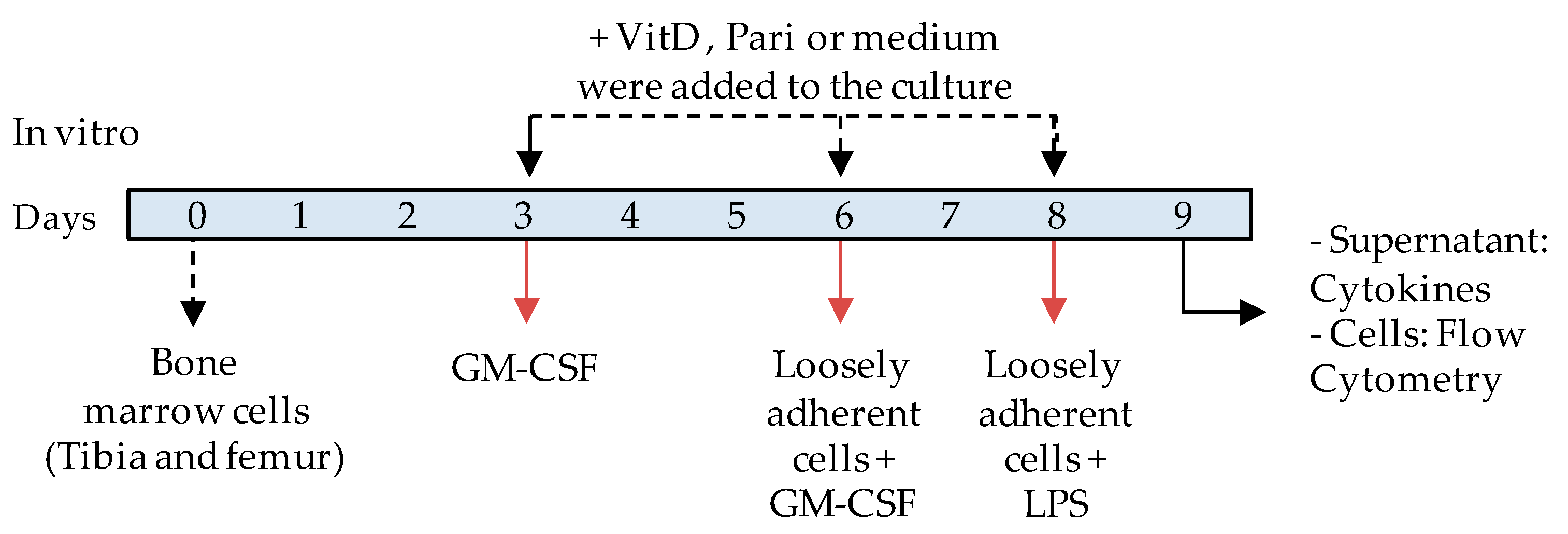
2.5. VitD and Pari Distinctly Affect the Differentiation of Bone Marrow-Derived Dendritic Cells (DCs)
3. Discussion
4. Materials and Methods
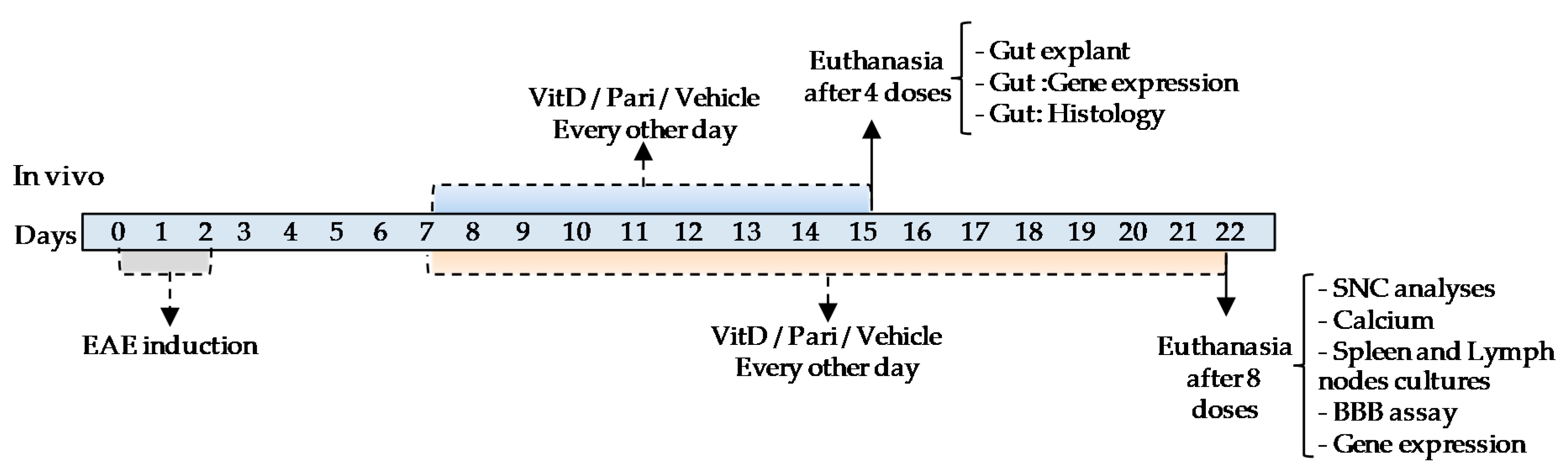
4.1. Experimental Design
4.2. Animals
4.3. Induction and Clinical Assessment of EAE
4.4. Therapy with VitD and Pari
4.5. Serum Calcium and Phosphorus
4.6. Spleen and Lymph Node Cell Cultures
4.7. CNS Mononuclear Cell Isolation
4.8. Blood–Brain Barrier Permeability Assay
4.9. Spinal Cord and Gut Histopathology
4.10. Gut Explant-Immersion Cultures
4.11. In Vitro Generation of Bone Marrow-Derived DCs
4.12. Flow Cytometry
4.13. Cytokine Quantification
4.14. Rt-PCR and qPCR
4.15. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Antigen presenting cells |
| BBB | Blood–brain barrier |
| CTL | Healthy control group |
| CNS | Central nervous system |
| CFA | Complete Freund’s adjuvant |
| DC | Dendritic cell |
| ELISA | Enzyme-linked immunosorbent assay |
| EAE | Experimental autoimmune encephalomyelitis |
| LFB | Luxol fast blue |
| MS | Multiple sclerosis |
| MOG | Myelin oligodendrocyte glycoprotein |
| MHC | Major histocompatibility complex |
| Pari | Paricalcitol, 19-nor-1,25-dihyroxyvitamin D2 |
| VDR | Vitamin D receptor |
| VitD | Vitamin D3, 1,25-dihydroxyvitamin D3, calcitriol |
References
- Goldenberg, M.M. Multiple sclerosis review. Pharmacol. Ther. 2012, 37, 175–184. [Google Scholar]
- Hakim, E.A.; Bakheit, A.M.O.; Bryant, T.N.; Roberts, M.W.H.; Spackman, A.J.; Martin, J.P.; Mclellan, D.L. The social impact of multiple sclerosis—A study of 305 patients and their relatives. Disabil. Rehabil. 2000, 22, 288–293. [Google Scholar] [CrossRef]
- Granerod, J.; Ambrose, H.E.; Davies, N.W.S.; Clewley, J.P.; Walsh, A.L.; Morgan, D.; Cunningham, R.; Zuckerman, M.; Mutton, K.J.; Solomon, T.; et al. Causes of encephalitis and differences in their clinical presentations in England: A multicentre, population-based prospective study. Lancet Infect. Dis. 2010, 10, 835–844. [Google Scholar] [CrossRef] [Green Version]
- Sassi, F.; Tamone, C.; D’amelio, P. Vitamin D: Nutrient, hormone, and immunomodulator. Nutrients 2018, 10, 1656. [Google Scholar] [CrossRef] [Green Version]
- Hewison, M. Vitamin D and immune function: An overview. Proc. Nutr. Soc. 2012, 71, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Omura, S.; Kawai, E.; Sato, F.; Martinez, N.E.; Minagar, A.; Al-Kofahi, M.; Yun, J.W.; Cvek, U.; Trutschl, M.; Alexander, J.S.; et al. Theiler’s Virus-Mediated Immunopathology in the CNS and Heart: Roles of Organ-Specific Cytokine and Lymphatic Responses. Front. Immunol. 2018, 9, 2870. [Google Scholar] [CrossRef]
- Simpson, S.; Blizzard, L.; Otahal, P.; Van Der Mei, I.; Taylor, B. Latitude is significantly associated with the prevalence of multiple sclerosis: A meta-analysis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1132–1141. [Google Scholar] [CrossRef]
- Di Rosa, M.; Malaguarnera, M.; Nicoletti, F.; Malaguarnera, L. Vitamin D3: A helpful immuno-modulator. Immunology 2011, 134, 123–139. [Google Scholar] [CrossRef]
- Brola, W.; Sobolewski, P.; Szczuchniak, W.; Góral, A.; Fudala, M.; Przybylski, W.; Opara, J. Association of seasonal serum 25-hydroxyvitamin D levels with disability and relapses in relapsing-remitting multiple sclerosis. Eur. J. Clin. Nutr. 2016, 70, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, W.; Li, D.; Yin, X.; Zhang, X.; Olsen, N.; Zheng, S.G. Vitamin D and Chronic Diseases. Aging Dis. 2017, 8, 346–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cyprian, F.; Lefkou, E.; Varoudi, K.; Girardi, G. Immunomodulatory Effects of Vitamin D in Pregnancy and Beyond. Front. Immunol. 2019, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Fernandes de Abreu, D.A.; Eyles, D.; Féron, F. Vitamin D, a neuro-immunomodulator: Implications for neurodegenerative and autoimmune diseases. Psychoneuroendocrinology 2009, 34. [Google Scholar] [CrossRef] [PubMed]
- Bivona, G.; Agnello, L.; Bellia, C.; Iacolino, G.; Scazzone, C.; Lo Sasso, B.; Ciaccio, M. Non-skeletal activities of vitamin d: From physiology to brain pathology. Medicina 2019, 55, 341. [Google Scholar] [CrossRef] [Green Version]
- Dobson, R.; Cock, H.R.; Brex, P.; Giovannoni, G. Vitamin D supplementation. Pract. Neurol. 2018, 18, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotirchos, E.S.; Bhargava, P.; Eckstein, C.; Van Haren, K.; Baynes, M.; Ntranos, A.; Gocke, A.; Steinman, L.; Mowry, E.M.; Calabresi, P.A. Safety and immunologic effects of high- vs low-dose cholecalciferol in multiple sclerosis. Neurology 2016, 86, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Jagannath, V.A.; Fedorowicz, Z.; Asokan, G.V.; Robak, E.W.; Whamond, L. Vitamin D for the management of multiple sclerosis. Cochrane Database Syst. Rev. 2010. [Google Scholar] [CrossRef]
- Sintzel, M.B.; Rametta, M.; Reder, A.T. Vitamin D and Multiple Sclerosis: A Comprehensive Review. Neurol. Ther. 2017, 7, 59–85. [Google Scholar] [CrossRef] [Green Version]
- Feige, J.; Moser, T.; Bieler, L.; Schwenker, K.; Hauer, L.; Sellner, J. Vitamin D supplementation in multiple sclerosis: A critical analysis of potentials and threats. Nutrients 2020, 12, 783. [Google Scholar] [CrossRef] [Green Version]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2017, 133. [Google Scholar] [CrossRef] [Green Version]
- Bjelobaba, I.; Begovic-Kupresanin, V.; Pekovic, S.; Lavrnja, I. Animal models of multiple sclerosis: Focus on experimental autoimmune encephalomyelitis. J. Neurosci. Res. 2018, 96, 1021–1042. [Google Scholar] [CrossRef] [Green Version]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15. [Google Scholar] [CrossRef]
- Hohlfeld, R.; Dornmair, K.; Meinl, E.; Wekerle, H. The search for the target antigens of multiple sclerosis, part 2: CD8+ T cells, B cells, and antibodies in the focus of reverse-translational research. Lancet Neurol. 2016, 15, 317–331. [Google Scholar] [CrossRef]
- Arneth, B.M. Impact of B cells to the pathophysiology of multiple sclerosis. J. Neuroinflammation 2019, 16, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arellano, G.; Acuña, E.; Reyes, L.I.; Ottum, P.A.; De Sarno, P.; Villarroel, L.; Ciampi, E.; Uribe-San Martín, R.; Cárcamo, C.; Naves, R. Th1 and Th17 cells and associated cytokines discriminate among clinically isolated syndrome and multiple sclerosis phenotypes. Front. Immunol. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human T H 17 lymphocytes promote blood-brain barrier disruption and CNS inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasa, R.; Barcutean, L.; Balasa, A.; Motataianu, A.; Roman-Filip, C.; Manu, D. The action of TH17 cells on blood brain barrier in multiple sclerosis and experimental autoimmune encephalomyelitis. Hum. Immunol. 2020, 81, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Lemire, J.M.; Clay Archer, D. 1,25-dihydroxyvitamin D3 prevents the in vivo induction of murine experimental autoimmune encephalomyelitis. J. Clin. Investig. 1991, 87, 1103–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantorna, M.T.; Hayes, C.E.; Deluca, H.F. 1,25-dihydroxyvitamin D3 reversibly blocks the progression of relapsing encephalomyelitis, a model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 1996, 93, 7861–7864. [Google Scholar] [CrossRef] [Green Version]
- Spach, K.M.; Nashold, F.E.; Dittel, B.N.; Hayes, C.E. IL-10 Signaling Is Essential for 1,25-Dihydroxyvitamin D3-Mediated Inhibition of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2006, 177, 6030–6037. [Google Scholar] [CrossRef] [Green Version]
- Nanduri, R.; Mahajan, S.; Bhagyaraj, E.; Sethi, K.; Kalra, R.; Chandra, V.; Gupta, P. The active form of vitamin D transcriptionally represses Smad7 signaling and activates extracellular signal-regulated kinase (ERK) to inhibit the differentiation of a inflammatory T helper cell subset and suppress experimental autoimmune encephalomyelitis. J. Biol. Chem. 2015, 290, 12222–12236. [Google Scholar] [CrossRef] [Green Version]
- Nataf, S.; Garcion, E.; Darcy, F.; Chabannes, D.; Mulle, J.Y.; Brachet, P. 1,25 Dihydroxyvitamin D3 exerts regional effects in the central nervous system during experimental allergic encephalomyelitis. J. Exp. Neurol. 1996, 55, 904–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, L.R.C.; Mimura, L.A.N.; de Campos Fraga-Silva, T.F.; Ishikawa, L.L.W.; Fernandes, A.A.H.; Zorzella-Pezavento, S.F.G.; Sartori, A. Calcitriol Prevents Neuroinflammation and Reduces Blood-Brain Barrier Disruption and Local Macrophage/Microglia Activation. Front. Pharmacol. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Esplugues, E.; Huber, S.; Gagliani, N.; Hauser, A.E.; Haberman, A.M.; Iwakura, Y.; Kuchroo, V.K.; Kolls, J.K.; Jeffrey, A. Control of TH17 cells occurs in the small intestine. Nature 2011, 475, 514–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosorich, I.; Dalla-Costa, G.; Sorini, C.; Ferrarese, R.; Messina, M.J.; Dolpady, J.; Radice, E.; Mariani, A.; Testoni, P.A.; Canducci, F.; et al. High frequency of intestinal TH17 cells correlates with microbiota alterations and disease activity in multiple sclerosis. Sci. Adv. 2017, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhoury, H.M.A.; Kvietys, P.R.; AlKattan, W.; Al Anouti, F.; Elahi, M.A.; Karras, S.N.; Grant, W.B. Vitamin D and intestinal homeostasis: Barrier, microbiota, and immune modulation. J. Steroid Biochem. Mol. Biol. 2020, 200. [Google Scholar] [CrossRef]
- Abadier, M.; Haghayegh Jahromi, N.; Cardoso Alves, L.; Boscacci, R.; Vestweber, D.; Barnum, S.; Deutsch, U.; Engelhardt, B.; Lyck, R. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood-brain barrier. Eur. J. Immunol. 2015, 45, 1043–1058. [Google Scholar] [CrossRef]
- Lopes Pinheiro, M.A.; Kooij, G.; Mizee, M.R.; Kamermans, A.; Enzmann, G.; Lyck, R.; Schwaninger, M.; Engelhardt, B.; de Vries, H.E. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 461–471. [Google Scholar] [CrossRef]
- Chastain, E.M.L.; Duncan, D.S.; Rodgers, J.M.; Miller, S.D. The Role of Antigen Presenting Cells in Multiple Sclerosis. Biochim. Biophys. Acta 2011, 1812, 265–274. [Google Scholar] [CrossRef]
- Barclay, W.; Shinohara, M.L. Inflammasome activation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Brain Pathol. 2017, 27, 213–219. [Google Scholar] [CrossRef]
- Correale, J.; Farez, M.F. The role of astrocytes in multiple sclerosis progression. Front. Neurol. 2015, 6, 1–12. [Google Scholar] [CrossRef]
- Correale, J.; Gaitán, M.I.; Ysrraelit, M.C.; Fiol, M.P. Progressive multiple sclerosis: From pathogenic mechanisms to treatment. Brain 2017, 140, 527–546. [Google Scholar] [CrossRef] [Green Version]
- Correale, J.; Villa, A. The blood-brain-barrier in multiple sclerosis: Functional roles and therapeutic targeting. Autoimmunity 2007, 40, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Pinke, K.H.; Zorzella-Pezavento, S.F.G.; de Campos Fraga-Silva, T.F.; Mimura, L.A.N.; de Oliveira, L.R.C.; Ishikawa, L.L.W.; Fernandes, A.A.H.; Lara, V.S.; Sartori, A. Calming Down Mast Cells with Ketotifen: A Potential Strategy for Multiple Sclerosis Therapy? Neurotherapeutics 2020, 17. [Google Scholar] [CrossRef] [PubMed]
- Vargas, D.L.; Tyor, W.R. Update on disease-modifying therapies for multiple sclerosis. J. Investig. Med. 2017, 65, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Rafiee Zadeh, A.; Ghadimi, K.; Ataei, A.; Askari, M.; Sheikhinia, N.; Tavoosi, N.; Falahatian, M. Mechanism and adverse effects of multiple sclerosis drugs: A review article. Part 2. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 105–114. [Google Scholar] [PubMed]
- Pfeffer, P.E.; Lu, H.; Mann, E.H.; Chen, Y.H.; Ho, T.R.; Cousins, D.J.; Corrigan, C.; Kelly, F.J.; Mudway, I.S.; Hawrylowicz, C.M. Effects of Vitamin D on inflammatory and oxidative stress responses of human bronchial epithelial cells exposed to particulate matter. PLoS ONE 2018, 13, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Koduah, P.; Paul, F.; Dörr, J.M. Vitamin D in the prevention, prediction and treatment of neurodegenerative and neuroinflammatory diseases. EPMA J. 2017, 8, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Colotta, F.; Jansson, B.; Bonelli, F. Modulation of inflammatory and immune responses by vitamin D. J. Autoimmun. 2017, 85, 78–97. [Google Scholar] [CrossRef] [PubMed]
- Chiuso-Minicucci, F.; Ishikawa, L.L.W.; Mimura, L.A.N.; De Campos Fraga-Silva, T.F.; França, T.G.D.; Zorzella-Pezavento, S.F.G.; Marques, C.; Ikoma, M.R.V.; Sartori, A. Treatment with vitamin D/MOG association suppresses experimental autoimmune encephalomyelitis. PLoS ONE 2015, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mimura, L.A.N.; Chiuso-Minicucci, F.; Fraga-Silva, T.F.C.; Zorzella-Pezavento, S.F.G.; França, T.G.D.; Ishikawa, L.L.W.; Penitenti, M.; Ikoma, M.R.V.; Sartori, A. Association of myelin peptide with vitamin D prevents autoimmune encephalomyelitis development. Neuroscience 2016, 317. [Google Scholar] [CrossRef] [Green Version]
- Zorzella-Pezavento, S.F.G.; Mimura, L.A.N.; Fraga-Silva, T.F.C.; Ishikawa, L.L.W.; França, T.G.D.; Sartori, A. Experimental autoimmune encephalomyelitis is successfully controlled by epicutaneous administration of MOG plus vitamin D analog. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Renaghan, A.D.; Rosner, M.H. Hypercalcemia: Etiology and management. Nephrol. Dial. Transplant. 2018, 33, 549–551. [Google Scholar] [CrossRef] [Green Version]
- Tebben, P.J.; Singh, R.J.; Kumar, R. Vitamin D-mediated hypercalcemia: Mechanisms, diagnosis, and treatment. Endocr. Rev. 2016, 37, 521–547. [Google Scholar] [CrossRef]
- Trillini, M.; Cortinovis, M.; Ruggenenti, P.; Reyes Loaeza, J.; Courville, K.; Ferrer-Siles, C.; Prandini, S.; Gaspari, F.; Cannata, A.; Villa, A.; et al. Paricalcitol for Secondary Hyperparathyroidism in Renal Transplantation. J. Am. Soc. Nephrol. 2015, 26, 1205–1214. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhu, J.; DeLuca, H.F. Where is the vitamin D receptor? Arch. Biochem. Biophys. 2012, 523, 123–133. [Google Scholar] [CrossRef]
- Yamamoto, E.; Jørgensen, T.N. Immunological effects of vitamin D and their relations to autoimmunity. J. Autoimmun. 2019, 100, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.A.R.; Lopes, M.J.P.; Costa, R.O.; Lima, F.A.V.; Neves, K.R.T.; Calou, I.B.F.; Andrade, G.M.; Viana, G.S.B. Vitamin D protects dopaminergic neurons against neuroinflammation and oxidative stress in hemiparkinsonian rats. J. Neuroinflamm. 2018, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Popescu, D.C.; Huang, H.; Singhal, N.K.; Shriver, L.; McDonough, J.; Clements, R.J.; Freeman, E.J. Vitamin K enhances the production of brain sulfatides during remyelination. PLoS ONE 2018, 13, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; He, L.; Liu, L.; Zhu, J.; Jin, T. The efficacy of vitamin D in multiple sclerosis: A meta-analysis. Mult. Scler. Relat. Disord. 2018, 23, 56–61. [Google Scholar] [CrossRef] [PubMed]
- James, E.; Dobson, R.; Kuhle, J.; Baker, D.; Giovannoni, G.; Ramagopalan, S.V. The effect of vitamin D-related interventions on multiple sclerosis relapses: A meta-analysis. Mult. Scler. J. 2013, 19, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Juedes, A.E.; Ruddle, N.H. Resident and Infiltrating Central Nervous System APCs Regulate the Emergence and Resolution of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2001, 166, 5168–5175. [Google Scholar] [CrossRef]
- Nikodemova, M.; Watters, J.J.; Jackson, S.J.; Yang, S.K.; Duncan, I.D. Minocycline down-regulates MHC II expression in microglia and macrophages through inhibition of IRF-1 and protein kinase C (PKC) α/βII. J. Biol. Chem. 2007, 282, 15208–15216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Lu, L.; Guo, T.B.; Qiu, J.; Yang, Y.; Liu, A.; Zhang, J.Z. Novel Immunomodulatory Properties of Berbamine through Selective Down-Regulation of STAT4 and Action of IFN-γ in Experimental Autoimmune Encephalomyelitis. J. Immunol. 2008, 181, 1491–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steeland, S.; Van Ryckeghem, S.; Van Imschoot, G.; De Rycke, R.; Toussaint, W.; Vanhoutte, L.; Vanhove, C.; De Vos, F.; Vandenbroucke, R.E.; Libert, C. TNFR1 inhibition with a Nanobody protects against EAE development in mice. Sci. Rep. 2017, 7, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Becquet, L.; Abad, C.; Leclercq, M.; Miel, C.; Jean, L.; Riou, G.; Couvineau, A.; Boyer, O.; Tan, Y.V. Systemic administration of orexin A ameliorates established experimental autoimmune encephalomyelitis by diminishing neuroinflammation. J. Neuroinflamm. 2019, 16, 1–12. [Google Scholar] [CrossRef]
- Haghmorad, D.; Yazdanpanah, E.; Jadid Tavaf, M.; Zargarani, S.; Soltanmohammadi, A.; Mahmoudi, M.B.; Mahmoudi, M. Prevention and treatment of experimental autoimmune encephalomyelitis induced mice with 1, 25-dihydroxyvitamin D3. Neurol. Res. 2019, 41, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, Z.; He, X.; Yu, H.; Feng, J. Ghrelin attenuates neuroinflammation and demyelination in experimental autoimmune encephalomyelitis involving NLRP3 inflammasome signaling pathway and pyroptosis. Front. Pharmacol. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blauth, K.; Zhang, X.; Chopra, M.; Rogan, S.; Markovic-Plese, S. The role of fractalkine (CX3CL1) in regulation of CD4+ cell migration to the central nervous system in patients with relapsing-remitting multiple sclerosis. Clin. Immunol. 2015, 157, 121–132. [Google Scholar] [CrossRef]
- Ridderstad Wollberg, A.; Ericsson-Dahlstrand, A.; Jureus, A.; Ekerot, P.; Simon, S.; Nilsson, M.; Wiklund, S.-J.; Berg, A.-L.; Ferm, M.; Sunnemark, D.; et al. Pharmacological inhibition of the chemokine receptor CX3CR1 attenuates disease in a chronic-relapsing rat model for multiple sclerosis. Proc. Natl. Acad. Sci. USA 2014, 111, 5409–5414. [Google Scholar] [CrossRef] [Green Version]
- Ruland, C.; Renken, H.; Kuzmanov, I.; Fattahi Mehr, A.; Schwarte, K.; Cerina, M.; Herrmann, A.; Otte, D.M.; Zimmer, A.; Schwab, N.; et al. Chemokine CCL17 is expressed by dendritic cells in the CNS during experimental autoimmune encephalomyelitis and promotes pathogenesis of disease. Brain. Behav. Immun. 2017, 66, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Catherine Lubetzki and Bruno Stankoff Chapter 4—Demyelination in multiple sclerosis. Handb. Clin. Neurol. 2014, 122, 89–99.
- Yang, J.; Lindsberg, P.J.; Hukkanen, V.; Seljelid, R.; Gahmberg, C.G.; Meri, S. Differential expression of cytokines (IL-2, IFN-γ, IL-10) and adhesion molecules (VCAM-1, LFA-1, CD44) between spleen and lymph nodes associates with remission in chronic relapsing experimental autoimmune encephalomyelitis. Scand. J. Immunol. 2002, 56, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Odoardi, F.; Sie, C.; Streyl, K.; Ulaganathan, V.K.; Schläger, C.; Lodygin, D.; Heckelsmiller, K.; Nietfeld, W.; Ellwart, J.; Klinkert, W.E.F.; et al. T cells become licensed in the lung to enter the central nervous system. Nature 2012, 488, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Wandu, W.S.; Lee, R.S.; Hinshaw, S.H.; Klinman, D.M.; Wawrousek, E.; Gery, I. SHEDDING NEW LIGHT ON THE PROCESS OF “LICENSING” FOR PATHOGENICITY BY Th-LYMPHOCYTES. Nature 2017, 198, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanayama, M.; Danzaki, K.; He, Y.W.; Shinohara, M.L. Lung inflammation stalls Th17-cell migration en route to the central nervous system during the development of experimental autoimmune encephalomyelitis. Int. Immunol. 2016, 28, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Duc, D.; Vigne, S.; Bernier-Latmani, J.; Yersin, Y.; Ruiz, F.; Gaïa, N.; Leo, S.; Lazarevic, V.; Schrenzel, J.; Petrova, T.V.; et al. Disrupting Myelin-Specific Th17 Cell Gut Homing Confers Protection in an Adoptive Transfer Experimental Autoimmune Encephalomyelitis. Cell Rep. 2019, 29, 378–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Shih, D.Q.; Zhang, X. Mechanisms underlying effects of 1,25-dihydroxyvitamin D 3 on the Th17 cells. Eur. J. Microbiol. Immunol. 2013, 3, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Barragan, M.; Good, M.; Kolls, J.K. Regulation of dendritic cell function by vitamin D. Nutrients 2015, 7, 8127–8151. [Google Scholar] [CrossRef]
- Szymczak, I.; Pawliczak, R. The Active Metabolite of Vitamin D3 as a Potential Immunomodulator. Scand. J. Immunol. 2016, 83, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Dáňová, K.; Klapetková, A.; Kayserová, J.; Šedivá, A.; Špíšek, R.; Jelínková, L.P. NF-kappa;B, p38 MAPK, ERK1/2, mTOR, STAT3 and increased glycolysis regulate stability of paricalcitol/dexamethasone-generated tolerogenic dendritic cells in the inflammatory environment. Oncotarget 2015, 6, 14123–14138. [Google Scholar] [CrossRef]
- Sochorová, K.; Budinský, V.; Rožková, D.; Tobiasová, Z.; Dusilová-Sulková, S.; Špíšek, R.; Bartůňková, J. Paricalcitol (19-nor-1,25-dihydroxyvitamin D2) and calcitriol (1,25-dihydroxyvitamin D3) exert potent immunomodulatory effects on dendritic cells and inhibit induction of antigen-specific T cells. Clin. Immunol. 2009, 133, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Mattner, F.; Smiroldo, S.; Galbiati, F.; Muller, M.; Di Lucia, P.; Poliani, P.L.; Martino, G.; Panina-Bordignon, P.; Adorini, L. Inhibition of Th1 development and treatment of chronic-relapsing experimental allergic encephalomyelitis by a non-hypercalcemic analogue of 1,25-dihydroxyvitamin D3. Eur. J. Immunol. 2000, 30, 498–508. [Google Scholar] [CrossRef]
- Na, S.; Ma, Y.; Zhao, J.; Schmidt, C.; Zeng, Q.Q.; Chandrasekhar, S.; Chin, W.W.; Nagpal, S. A nonsecosteroidal vitamin D receptor modulator ameliorates experimental autoimmune encephalomyelitis without causing hypercalcemia. Autoimmune Dis. 2011, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Qiao, L.; Fu, T. Paricalcitol improves experimental autoimmune encephalomyelitis (EAE) by suppressing inflammation via NF-κB signaling. Biomed. Pharmacother. 2020, 125, 109528. [Google Scholar] [CrossRef]
- Garcion, E.; Sindji, L.; Nataf, S.; Brachet, P.; Darcy, F.; Montero-Menei, C.N. Treatment of experimental autoimmune encephalomyelitis in rat by 1,25-dihydroxyvitamin D3 lead to early effects within the central nervous system. Acta Neuropathol. 2003, 105, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Mazzaferro, S.; Goldsmith, D.; Larsson, T.E.; Massy, Z.A.; Cozzolino, M. Vitamin D metabolites and/or analogs: Which D for which patient? Curr. Vasc. Pharmacol. 2014, 12, 339–349. [Google Scholar] [CrossRef]
- Wasnik, S.; Sharma, I.; Baylink, D.J.; Tang, X. Vitamin D as a potential therapy for multiple sclerosis: Where are we? Int. J. Mol. Sci. 2020, 21, 3102. [Google Scholar] [CrossRef]
- Desena, A.D.; Graves, D.C.; Greenberg, B.M. Do Low Vitamin D-25-OH Levels Predict a Higher Likelihood of Relapse for Acute Disseminated Encephalomyelitis: A Report of 5 Pediatric Cases; 2011; Volume 408, Available online: https://cmsc.confex.com/cmsc/2013/webprogram/Paper1631.html (accessed on 16 December 2020).
- Pellegrino, P.; Radice, S.; Clementi, E. Geoepidemiology of acute disseminated encephalomyelitis. Epidemiology 2014, 25, 928–929. [Google Scholar] [CrossRef]
- Christy, A.L.; Walker, M.E.; Hessner, M.J.; Brown, M.A. Mast cell activation and neutrophil recruitment promotes early and robust inflammation in the meninges in EAE. J. Autoimmun. 2013, 42, 50–61. [Google Scholar] [CrossRef]
- Bareiss, P.M.; Metzger, M.; Sohn, K.; Rupp, S.; Frick, J.S.; Autenrieth, I.B.; Lang, F.; Schwarz, H.; Skutella, T.; Just, L. Organotypical tissue cultures from adult murine colon as an in vitro model of intestinal mucosa. Histochem. Cell Biol. 2008, 129, 795–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randall, K.J.; Turton, J.; Foster, J.R. Explant culture of gastrointestinal tissue: A review of methods and applications. Cell Biol. Toxicol. 2011, 27, 267–284. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mimura, L.A.N.; Fraga-Silva, T.F.d.C.; Oliveira, L.R.C.d.; Ishikawa, L.L.W.; Borim, P.A.; Machado, C.d.M.; Júnior, J.d.A.d.C.e.H.; Fonseca, D.M.d.; Sartori, A. Preclinical Therapy with Vitamin D3 in Experimental Encephalomyelitis: Efficacy and Comparison with Paricalcitol. Int. J. Mol. Sci. 2021, 22, 1914. https://doi.org/10.3390/ijms22041914
Mimura LAN, Fraga-Silva TFdC, Oliveira LRCd, Ishikawa LLW, Borim PA, Machado CdM, Júnior JdAdCeH, Fonseca DMd, Sartori A. Preclinical Therapy with Vitamin D3 in Experimental Encephalomyelitis: Efficacy and Comparison with Paricalcitol. International Journal of Molecular Sciences. 2021; 22(4):1914. https://doi.org/10.3390/ijms22041914
Chicago/Turabian StyleMimura, Luiza Ayumi Nishiyama, Thais Fernanda de Campos Fraga-Silva, Larissa Ragozzo Cardoso de Oliveira, Larissa Lumi Watanabe Ishikawa, Patrícia Aparecida Borim, Carla de Moraes Machado, José de Anchieta de Castro e Horta Júnior, Denise Morais da Fonseca, and Alexandrina Sartori. 2021. "Preclinical Therapy with Vitamin D3 in Experimental Encephalomyelitis: Efficacy and Comparison with Paricalcitol" International Journal of Molecular Sciences 22, no. 4: 1914. https://doi.org/10.3390/ijms22041914
APA StyleMimura, L. A. N., Fraga-Silva, T. F. d. C., Oliveira, L. R. C. d., Ishikawa, L. L. W., Borim, P. A., Machado, C. d. M., Júnior, J. d. A. d. C. e. H., Fonseca, D. M. d., & Sartori, A. (2021). Preclinical Therapy with Vitamin D3 in Experimental Encephalomyelitis: Efficacy and Comparison with Paricalcitol. International Journal of Molecular Sciences, 22(4), 1914. https://doi.org/10.3390/ijms22041914