
Dysfunctional Inflammation in Cystic Fibrosis Airways: From Mechanisms to Novel Therapeutic Approaches



{kind=link}
Abstract
:1. Introduction
2. Anti-Inflammatory Therapy in Cystic Fibrosis
Traditional Anti-Inflammatory Treatments
3. Novel Therapeutic Strategies
3.1. Azithromycin
3.2. Compounds Affecting Eicosanoid Pathway
3.3. Cannabinoid-Derived Compounds
3.4. R-Roscovitine
3.5. Thymosin Alpha-1
4. Alternative Strategies
4.1. Inhibitors of Neutrophil Elastase
4.2. Compounds Affecting Lipid Metabolism
4.3. Compounds Targeting Arginine Production and Nitric Oxide
5. New Perspectives
5.1. Stem Cells
5.2. Anticytokines
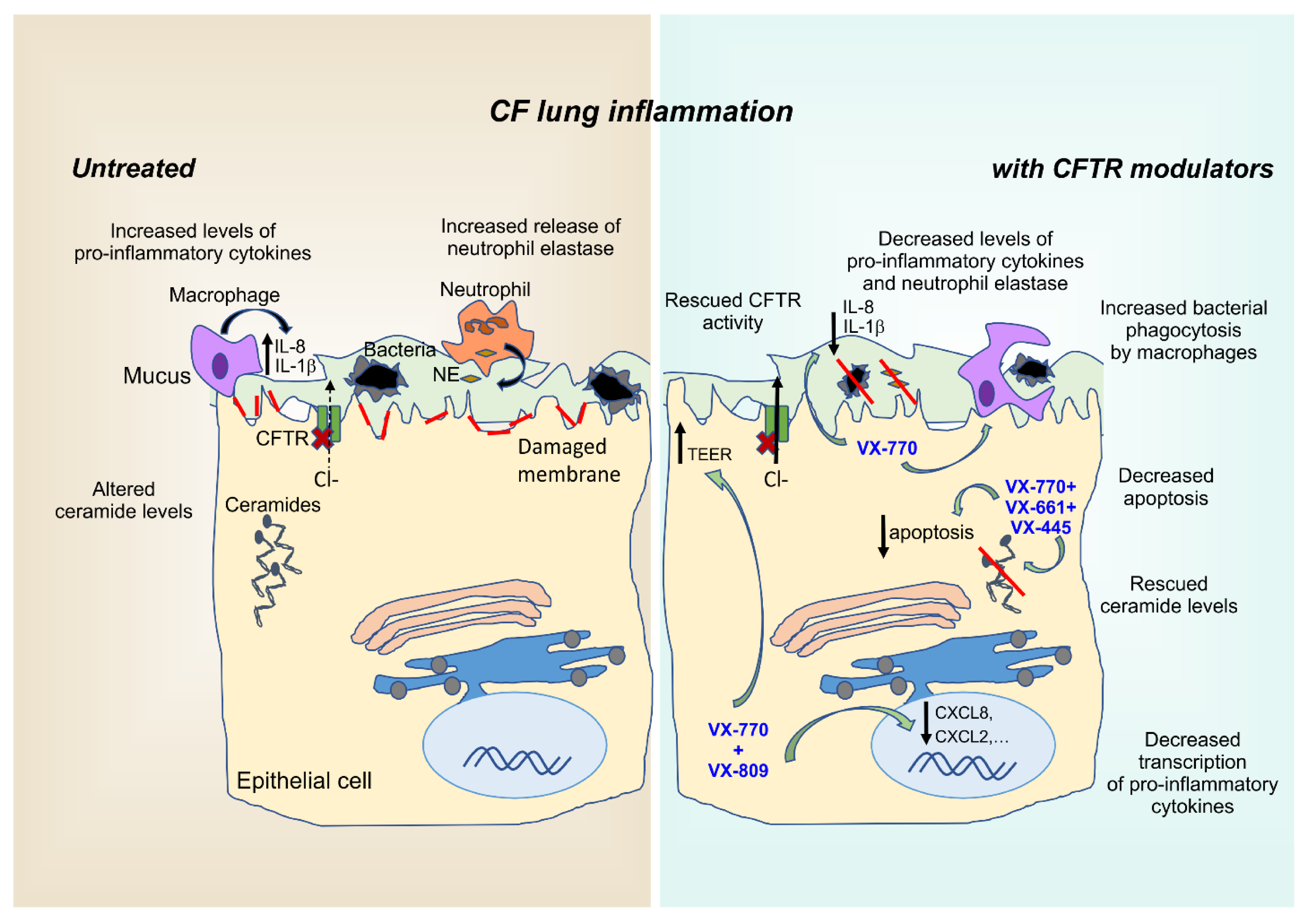
6. Anti-Inflammatory Effects of CFTR Modulators
6.1. Approved CFTR Modulators
6.2. Ivacaftor Monotherapy
6.3. Combination Therapies: Orkambi® and Symdeko®
6.4. Triple-Therapy: Trikafta®
6.5. Iminosugars, TMA Analogs, and Proteostasis Regulators
7. Potential Impact of COVID-19 on Inflammation in CF Patients
8. Conclusions
Funding
Conflicts of Interest
References
- Bergeron, C.; Cantin, A.M. Cystic Fibrosis: Pathophysiology of Lung Disease. Semin. Respir. Crit. Care Med. 2019, 40, 715–726. [Google Scholar] [CrossRef]
- Charles, R.E., Jr.; Muhlebach, M.S.; Ehre, C.; Hill, D.B.; Wolfgang, M.C.; Kesimer, M.; Ramsey, K.A.; Markovetz, M.R.; Garbarine, I.C.; Forest, M.G.; et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci. Transl. Med. 2019, 11, eaav3488. [Google Scholar] [CrossRef] [Green Version]
- Montgomery, S.T.; Mall, M.A.; Kicic, A.; Stick, S.M. Hypoxia and sterile inflammation in cystic fibrosis airways: Mechanisms and potential therapies. Eur. Respir. J. 2016, 49, 1600903. [Google Scholar] [CrossRef] [Green Version]
- Perrem, L.; Ratjen, F. Anti-inflammatories and mucociliary clearance therapies in the age of CFTR modulators. Pediatr. Pulmonol. 2019, 54, S46–S55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [Green Version]
- Robb, C.T.; Regan, K.H.; Dorward, D.A.; Rossi, A.G. Key mechanisms governing resolution of lung inflammation. Semin. Immunopathol. 2016, 38, 425–448. [Google Scholar] [CrossRef] [Green Version]
- Wagner, C.J.; Schultz, C.; Mall, M.A. Neutrophil elastase and matrix metalloproteinase 12 in cystic fibrosis lung disease. Mol. Cell. Pediatr. 2016, 3, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Juncadella, I.J.; Kadl, A.; Sharma, A.K.; Shim, Y.M.; Hochreiter-Hufford, A.; Borish, L.; Ravichandran, K.S. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nat. Cell Biol. 2012, 493, 547–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53, S30–S50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, D.P.; Konstan, M.W.; Chmiel, J.F. Anti-inflammatory Therapies for Cystic Fibrosis-Related Lung Disease. Clin. Rev. Allergy Immunol. 2008, 35, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Tegeder, I.; Pfeilschifter, J.; Geisslinger, G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J. 2001, 15, 2057–2072. [Google Scholar] [CrossRef] [PubMed]
- Konstan, M.W.; Krenicky, J.E.; Finney, M.R.; Kirchner, H.L.; Hilliard, K.A.; Hilliard, J.B.; Davis, P.B.; Hoppel, C.L. Effect of Ibuprofen on Neutrophil Migration in Vivo in Cystic Fibrosis and Healthy Subjects. J. Pharmacol. Exp. Ther. 2003, 306, 1086–1091. [Google Scholar] [CrossRef]
- Tabary, O.; Zahm, J.M.; Hinnrasky, J.; Couetil, J.P.; Cornillet, P.; Guenounou, M.; Gaillard, D.; Puchelle, E.; Jacquot, J. Selective Up-Regulation of Chemokine IL-8 Expression in Cystic Fibrosis Bronchial Gland Cells in Vivo and in Vitro. Am. J. Pathol. 1998, 153, 921–930. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J. Corticosteroid effects on cell signalling. Eur. Respir. J. 2006, 27, 413–426. [Google Scholar] [CrossRef]
- Flume, P.A.; O’Sullivan, B.P.; Robinson, K.A.; Goss, C.H.; Mogayzel, P.J., Jr.; Willey-Courand, D.B.; Bujan, J.; Finder, J.; Lester, M.; Quittell, L.; et al. Cystic fibrosis pulmonary guidelines: Chronic medications for maintenance of lung health. Am. J. Respir. Crit. Care Med. 2007, 176, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Balfour-Lynn, I.M.; Welch, K. Inhaled corticosteroids for cystic fibrosis. Cochrane Database Syst. Rev. 2014, 52, CD001915. [Google Scholar]
- Mitri, C.; Xu, Z.; Bardin, P.; Corvol, H.; Touqui, L.; Tabary, O. Novel Anti-Inflammatory Approaches for Cystic Fibrosis Lung Disease: Identification of Molecular Targets and Design of Innovative Therapies. Front. Pharmacol. 2020, 11, 1096. [Google Scholar] [CrossRef]
- Mogayzel, P.J., Jr.; Naureckas, E.T.; Robinson, K.A.; Mueller, G.; Hadjiliadis, D.; Hoag, J.B.; Lubsch, L.; Hazle, L.; Sabadosa, K.; Marshall, B.; et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am. J. Respir. Crit. Care Med. 2013, 187, 680–689. [Google Scholar] [CrossRef]
- Scheuren, N.; Bang, H.; Munster, T.; Brune, K.; Pahl, A. Modulation of transcription factor NF-kappaB by enantiomers of the nonsteroidal drug ibuprofen. Br. J. Pharmacol. 1998, 123, 645–652. [Google Scholar] [CrossRef]
- Tegeder, I.; Niederberger, E.; Israr, E.; Guhring, H.; Brune, K.; Euchenhofer, C.; Grosch, S.; Geisslinger, G. Inhibition of NF-kappaB and AP-1 activation by R- and S-flurbiprofen. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 2–4. [Google Scholar]
- Jaradat, M.S.; Wongsud, B.; Phornchirasilp, S.; Rangwala, S.M.; Shams, G.; Sutton, M.; Romstedt, K.J.; Noonan, D.J.; Feller, D.R. Activation of peroxisome proliferator-activated receptor isoforms and inhibition of prostaglandin H(2) synthases by ibuprofen, naproxen, and indomethacin. Biochem. Pharmacol. 2001, 62, 1587–1595. [Google Scholar] [CrossRef]
- Rymut, S.M.; Kampman, C.M.; Corey, D.A.; Endres, T.; Cotton, C.U.; Kelley, T.J. Ibuprofen regulation of microtubule dynamics in cystic fibrosis epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2016, 311, L317–L327. [Google Scholar] [CrossRef] [PubMed]
- Devor, D.C.; Schultz, B.D. Ibuprofen inhibits cystic fibrosis transmembrane conductance regulator-mediated Cl- secretion. J. Clin. Investig. 1998, 102, 679–687. [Google Scholar] [CrossRef]
- Carlile, G.W.; Robert, R.; Goepp, J.; Matthes, E.; Liao, J.; Kus, B.; Macknight, S.D.; Rotin, D.; Hanrahan, J.W.; Thomas, D.Y. Ibuprofen rescues mutant cystic fibrosis transmembrane conductance regulator trafficking. J. Cyst. Fibros. 2015, 14, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Xiang, Y.-Y.; Ye, L.; Tsui, L.-C.; Macdonald, J.F.; Hu, J.; Lu, W.-Y. Nonsteroidal anti-inflammatory drugs upregulate function of wild-type and mutant CFTR. Eur. Respir. J. 2008, 32, 334–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruch, B.A.; Singh, S.B.; Ramsey, L.J.; Starner, T.D. Impact of a cystic fibrosis transmembrane conductance regulator (CFTR) modulator on high-dose ibuprofen therapy in pediatric cystic fibrosis patients. Pediatr. Pulmonol. 2018, 53, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Konstan, M.W.; Byard, P.J.; Hoppel, C.L.; Davis, P.B. Effect of High-Dose Ibuprofen in Patients with Cystic Fibrosis. N. Engl. J. Med. 1995, 332, 848–854. [Google Scholar] [CrossRef]
- Lands, L.C.; Milner, R.; Cantin, A.M.; Manson, D.; Corey, M. High-Dose Ibuprofen in Cystic Fibrosis: Canadian Safety and Effectiveness Trial. J. Pediatr. 2007, 151, 249–254. [Google Scholar] [CrossRef]
- Konstan, M.W.; van Devanter, D.R.; Sawicki, G.S.; Pasta, D.J.; Foreman, A.J.; Neiman, E.A.; Morgan, W.J. Association of High-Dose Ibuprofen Use, Lung Function Decline, and Long-Term Survival in Children with Cystic Fibrosis. Ann. Am. Thorac. Soc. 2018, 15, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Lands, L.C.; Stanojevic, S. Oral non-steroidal anti-inflammatory drug therapy for lung disease in cystic fibrosis. Cochrane Database Syst. Rev. 2019, 2019, CD001505. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.P.; Odem-Davis, K.; Cogen, J.D.; Goss, C.H.; Ren, C.L.; Skalland, M.; Somayaji, R.; Heltshe, S.L. Pulmonary Outcomes Associated with Long-Term Azithromycin Therapy in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 430–437. [Google Scholar] [CrossRef]
- Shinkai, M.; Foster, G.H.; Rubin, B.K. Macrolide antibiotics modulate ERK phosphorylation and IL-8 and GM-CSF production by human bronchial epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2006, 290, L75–L85. [Google Scholar] [CrossRef] [Green Version]
- Bystrzycka, W.; Manda-Handzlik, A.; Sieczkowska, S.; Moskalik, A.; Demkow, U.; Ciepiela, O. Azithromycin and Chloramphenicol Diminish Neutrophil Extracellular Traps (NETs) Release. Int. J. Mol. Sci. 2017, 18, 2666. [Google Scholar] [CrossRef] [Green Version]
- Haydar, D.; Cory, T.J.; Birket, S.E.; Murphy, B.S.; Pennypacker, K.R.; Sinai, A.P.; Feola, D.J. Azithromycin Polarizes Macrophages to an M2 Phenotype via Inhibition of the STAT1 and NF-kappaB Signaling Pathways. J. Immunol. 2019, 203, 102–1030. [Google Scholar] [CrossRef]
- Saiman, L.; Marshall, B.C.; Mayer-Hamblett, N.; Burns, J.L.; Quittner, A.L.; Cibene, D.A.; Coquillette, S.; Fieberg, A.Y.; Accurso, F.J.; Campbell, P.W., 3rd; et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: A randomized controlled trial. JAMA 2003, 290, 1749–1756. [Google Scholar] [CrossRef] [Green Version]
- Ratjen, F.; Saiman, L.; Mayer-Hamblett, N.; Lands, L.C.; Kloster, M.; Thompson, V.; Emmett, P.; Marshall, B.; Accurso, F.; Sagel, S.; et al. Effect of azithromycin on systemic markers of inflammation in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa. Chest 2012, 142, 1259–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saint-Criq, V.; Rebeyrol, C.; Ruffin, M.; Roque, T.; Guillot, L.; Jacquot, J.; Clement, A.; Tabary, O. Restoration of Chloride Efflux by Azithromycin in Airway Epithelial Cells of Cystic Fibrosis Patients. Antimicrob. Agents Chemother. 2011, 55, 1792–1793. [Google Scholar] [CrossRef] [Green Version]
- Southern, K.W.; Barker, P.M.; Solis-Moya, A.; Patel, L. Macrolide antibiotics for cystic fibrosis. Cochrane Database Syst. Rev. 2012, 11, CD002203. [Google Scholar] [CrossRef]
- Nichols, D.P.; Happoldt, C.L.; Bratcher, P.E.; Caceres, S.M.; Chmiel, J.F.; Malcolm, K.C.; Saavedra, M.T.; Saiman, L.; Taylor-Cousar, J.L.; Nick, J.A. Impact of azithromycin on the clinical and antimicrobial effectiveness of tobramycin in the treatment of cystic fibrosis. J. Cyst. Fibros. 2017, 16, 358–366. [Google Scholar] [CrossRef]
- Konstan, M.; Döring, G.; Heltshe, S.; Lands, L.; Hilliard, K.; Koker, P.; Bhattacharya, S.; Staab, A.; Hamilton, A. A randomized double blind, placebo controlled phase 2 trial of BIIL 284 BS (an LTB4 receptor antagonist) for the treatment of lung disease in children and adults with cystic fibrosis. J. Cyst. Fibros. 2014, 13, 148–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstein, S.H. Ajulemic acid: Potential treatment for chronic inflammation. Pharmacol. Res. Perspect. 2018, 6, e00394. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.P.; Bennett, F.; Norris, P.C.; Maini, A.A.; George, M.J.; Newson, J.; Henderson, A.; Hobbs, A.J.; Tepper, M.; White, B.; et al. Potent Anti-Inflammatory and Pro-Resolving Effects of Anabasum in a Human Model of Self-Resolving Acute Inflammation. Clin. Pharmacol. Ther. 2018, 104, 675–686. [Google Scholar] [CrossRef]
- Tarique, A.A.; Evron, T.; Zhang, G.; Tepper, M.A.; Morshed, M.M.; Andersen, I.S.; Begum, N.; Sly, P.D.; Fantino, E. Anti-inflammatory effects of lenabasum, a cannabinoid receptor type 2 agonist, on macrophages from cystic fibrosis. J. Cyst. Fibros. 2020, 19, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Chmiel, J.F.; Flume, P.; Downey, D.G.; Dozor, A.J.; Colombo, C.; Mazurek, H.; Sapiejka, E.; Rachel, M.; Constantine, S.; Conley, B.; et al. Safety and efficacy of lenabasum in a phase 2 randomized, placebo-controlled trial in adults with cystic fibrosis. J. Cyst. Fibros. 2021, 20, 78–85. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation Website, Drug Development Pipeline—Clinical Trials Tool. Available online: https://www.cff.org/trials/pipeline (accessed on 8 September 2020).
- Meijer, L.; Nelson, D.J.; Riazanski, V.; Gabdoulkhakova, A.G.; Hery-Arnaud, G.; Le Berre, R.; Loaëc, N.; Oumata, N.; Galons, H.; Nowak, E.; et al. Modulating Innate and Adaptive Immunity by (R)-Roscovitine: Potential Therapeutic Opportunity in Cystic Fibrosis. J. Innate Immun. 2016, 8, 330–349. [Google Scholar] [CrossRef] [PubMed]
- Norez, C.; Vandebrouck, C.; Bertrand, J.; Noel, S.; Durieu, E.; Oumata, N.; Galons, H.; Antigny, F.; Chatelier, A.; Bois, P.; et al. Roscovitine is a proteostasis regulator that corrects the trafficking defect of F508del-CFTR by a CDK-independent mechanism. Br. J. Pharmacol. 2014, 171, 4831–4849. [Google Scholar] [CrossRef]
- Yoshida, H.; Kotani, H.; Kondo, T.; Tani, I.; Wei, X.; Tsuruta, S.; Kimura, A.; Asakawa, M.; Ito, M.; Nagai, S.; et al. CDK inhibitors suppress Th17 and promote iTreg differentiation, and ameliorate experimental autoimmune encephalomyelitis in mice. Biochem. Biophys. Res. Commun. 2013, 435, 378–384. [Google Scholar] [CrossRef]
- Moriceau, S.; Lenoir, G.; Witko-Sarsat, V. In Cystic Fibrosis Homozygotes and Heterozygotes, Neutrophil Apoptosis Is Delayed and Modulated by Diamide or Roscovitine: Evidence for an Innate Neutrophil Disturbance. J. Innate Immun. 2010, 2, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Marteyn, B.S.; Burgel, P.-R.; Meijer, L.; Witko-Sarsat, V. Harnessing Neutrophil Survival Mechanisms during Chronic Infection by Pseudomonas aeruginosa: Novel Therapeutic Targets to Dampen Inflammation in Cystic Fibrosis. Front. Cell. Infect. Microbiol. 2017, 7, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riazanski, V.; Gabdoulkhakova, A.G.; Boynton, L.S.; Eguchi, R.R.; Deriy, L.V.; Hogarth, D.K.; Loaëc, N.; Oumata, N.; Galons, H.; Brown, M.E.; et al. TRPC6 channel translocation into phagosomal membrane augments phagosomal function. Proc. Natl. Acad. Sci. USA 2015, 112, E6486–E6495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romani, L.; Oikonomou, V.; Moretti, S.; Iannitti, R.G.; D’Adamo, M.C.; Villella, V.R.; Pariano, M.; Sforna, L.; Borghi, M.; Bellet, M.M.; et al. Thymosin alpha1 represents a potential potent single-molecule-based therapy for cystic fibrosis. Nat. Med. 2017, 23, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, B.K. Unmet needs in cystic fibrosis. Expert Opin. Biol. Ther. 2018, 18, 49–52. [Google Scholar] [CrossRef]
- Armirotti, A.; Tomati, V.; Matthes, E.; Veit, G.; Cholon, D.M.; Phuan, P.-W.; Braccia, C.; Guidone, D.; Gentzsch, M.; Lukacs, G.L.; et al. Bioactive Thymosin Alpha-1 Does Not Influence F508del-CFTR Maturation and Activity. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Matthes, E.; Hanrahan, J.W.; Cantin, A.M. F508del-CFTR is not corrected by thymosin α1. Nat. Med. 2018, 24, 890–891. [Google Scholar] [CrossRef]
- Bellet, M.M.; Borghi, M.; Pariano, M.; Renga, G.; Stincardini, C.; D’Onofrio, F.; Brancorsini, S.; Garaci, E.; Costantini, C.; Romani, L. Thymosin alpha 1 exerts beneficial extrapulmonary effects in cystic fibrosis. Eur. J. Med. Chem. 2021, 209, 112921. [Google Scholar] [CrossRef]
- Causer, A.J.; Shute, J.K.; Cummings, M.H.; Shepherd, A.I.; Gruet, M.; Costello, J.T.; Bailey, S.; Lindley, M.; Pearson, C.; Connett, G.; et al. Circulating biomarkers of antioxidant status and oxidative stress in people with cystic fibrosis: A systematic review and meta-analysis. Redox Biol. 2020, 32, 101436. [Google Scholar] [CrossRef]
- Gaggar, A.; Chen, J.; Chmiel, J.F.; Dorkin, H.L.; Flume, P.A.; Griffin, R.; Nichols, D.P.; Donaldson, S.H. Inhaled alpha 1 -proteinase inhibitor therapy in patients with cystic fibrosis. J. Cyst. Fibros. 2016, 15, 227–233. [Google Scholar] [CrossRef] [Green Version]
- McElvaney, N.G.; Doujaiji, B.; Moan, M.J.; Burnham, M.R.; Wu, M.C.; Crystal, R.G. Pharmacokinetics of Recombinant Secretory Leukoprotease Inhibitor Aerosolized to Normals and Individuals with Cystic Fibrosis. Am. Rev. Respir. Dis. 1993, 148, 1056–1060. [Google Scholar] [CrossRef]
- Quabius, E.S.; Merz, I.; Gorogh, T.; Hedderich, J.; Haag, J.; Rocken, C.; Ambrosch, P.; Hoffmann, M. miRNA-expression in tonsillar squamous cell carcinomas in relation to HPV infection and expression of the antileukoproteinase SLPI. Papillomavirus Res. 2017, 4, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Barth, P.; Bruijnzeel, P.; Wach, A.; Kessler, O.S.; Hooftman, L.; Zimmermann, J.; Naue, N.; Huber, B.; Heimbeck, I.; Kappeler, D.; et al. Single dose escalation studies with inhaled POL6014, a potent novel selective reversible inhibitor of human neutrophil elastase, in healthy volunteers and subjects with cystic fibrosis. J. Cyst. Fibros. 2020, 19, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Hunt, A.M.D.; Glasgow, A.M.A.; Humphreys, H.; Greene, C.M. Alpha-1 Antitrypsin-A Target for MicroRNA-Based Therapeutic Development for Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 836. [Google Scholar] [CrossRef] [Green Version]
- Gregory, S.M.; Nazir, S.A.; Metcalf, J.P. Implications of the innate immune response to adenovirus and adenoviral vectors. Future Virol. 2011, 6, 357–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delacourt, C.; le Bourgeois, M.; D’Ortho, M.P.; Doit, C.; Scheinmann, P.; Navarro, J.; Harf, A.; Hartmann, D.J.; Lafuma, C. Imbalance between 95 kDa type IV collagenase and tissue inhibitor of metalloproteinases in sputum of patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 1995, 152, 765–774. [Google Scholar] [CrossRef]
- Gaggar, A.; Hector, A.; Bratcher, P.E.; Mall, M.A.; Griese, M.; Hartl, D. The role of matrix metalloproteinases in cystic fibrosis lung disease. Eur. Respir. J. 2011, 38, 721–727. [Google Scholar] [CrossRef]
- Guilbault, C.; de Sanctis, J.B.; Wojewodka, G.; Saeed, Z.; Lachance, C.; Skinner, T.A.A.; Vilela, R.M.; Kubow, S.; Lands, L.C.; Hajduch, M.; et al. Fenretinide Corrects Newly Found Ceramide Deficiency in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2008, 38, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Guilbault, C.; Wojewodka, G.; Saeed, Z.; Hajduch, M.; Matouk, E.; de Sanctis, J.B.; Radzioch, D. Cystic Fibrosis Fatty Acid Imbalance Is Linked to Ceramide Deficiency and Corrected by Fenretinide. Am. J. Respir. Cell Mol. Biol. 2009, 41, 100–106. [Google Scholar] [CrossRef]
- Caretti, A.; Peli, V.; Colombo, M.; Zulueta, A. Lights and Shadows in the Use of Mesenchymal Stem Cells in Lung Inflammation, a Poorly Investigated Topic in Cystic Fibrosis. Cells 2019, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Bonfield, T.L.; Lennon, D.; Ghosh, S.K.; Dimarino, A.M.; Weinberg, A.; Caplan, A.I. Cell based therapy aides in infection and inflammation resolution in the murine model of cystic fibrosis lung disease. Stem Cell Discov. 2013, 3, 139–153. [Google Scholar] [CrossRef] [Green Version]
- Sutton, M.T.; Fletcher, D.; Episalla, N.; Auster, L.; Kaur, S.; Gwin, M.C.; Folz, M.; Velasquez, D.; Roy, V.; van Heeckeren, R.; et al. Mesenchymal Stem Cell Soluble Mediators and Cystic Fibrosis. J. Stem Cell Res. Ther. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zulueta, A.; Colombo, M.; Peli, V.; Falleni, M.; Tosi, D.; Ricciardi, M.; Baisi, A.; Bulfamante, G.; Chiaramonte, R.; Caretti, A. Lung mesenchymal stem cells-derived extracellular vesicles attenuate the inflammatory profile of Cystic Fibrosis epithelial cells. Cell. Signal. 2018, 51, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Alcayaga-Miranda, F.; Cuenca, J.; Khoury, M. Antimicrobial Activity of Mesenchymal Stem Cells: Current Status and New Perspectives of Antimicrobial Peptide-Based Therapies. Front. Immunol. 2017, 8, 339. [Google Scholar] [CrossRef] [PubMed]
- Jacquot, J.; Tabary, O.; Le Rouzic, P.; Clement, A. Airway epithelial cell inflammatory signalling in cystic fibrosis. Int. J. Biochem. Cell Biol. 2008, 40, 1703–1715. [Google Scholar] [CrossRef]
- Moss, R.B.; Mistry, S.J.; Konstan, M.W.; Pilewski, J.M.; Kerem, E.; Tal-Singer, R.; Lazaar, A.L. Safety and early treatment effects of the CXCR2 antagonist SB-656933 in patients with cystic fibrosis. J. Cyst. Fibros. 2013, 12, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Balázs, A.; Mall, M.A. Mucus obstruction and inflammation in early cystic fibrosis lung disease: Emerging role of the IL-1 signaling pathway. Pediatr. Pulmonol. 2019, 54, S5–S12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonneville, F.; Ruffin, M.; Guillot, L.; Rousselet, N.; Le Rouzic, P.; Corvol, H.; Tabary, O. New Insights about miRNAs in Cystic Fibrosis. Am. J. Pathol. 2015, 185, 897–908. [Google Scholar] [CrossRef]
- Galluzzo, M.; Ciraolo, E.; Lucattelli, M.; Hoxha, E.; Ulrich, M.; Campa, C.C.; Lungarella, G.; Doring, G.; Zhou-Suckow, Z.; Mall, M.; et al. Genetic Deletion and Pharmacological Inhibition of PI3K gamma Reduces Neutrophilic Airway Inflammation and Lung Damage in Mice with Cystic Fibrosis-Like Lung Disease. Mediat. Inflamm. 2015, 2015, 545417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine. Front. Pharmacol. 2020, 10, 1662. [Google Scholar] [CrossRef] [Green Version]
- Quon, B.S.; Rowe, S.M. New and emerging targeted therapies for cystic fibrosis. BMJ 2016, 352, i859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Goor, F.; Straley, K.S.; Cao, D.; Gonzalez, J.; Hadida, S.; Hazlewood, A.; Joubran, J.; Knapp, T.; Makings, L.R.; Miller, M.; et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. American journal of physiology. Lung Cell. Mol. Physiol. 2006, 290, L1117–L1130. [Google Scholar] [CrossRef]
- Fukuda, R.; Okiyoneda, T. Peripheral Protein Quality Control as a Novel Drug Target for CFTR Stabilizer. Front. Pharmacol. 2018, 9, 1100. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.P.; Drew, L.; Green, O.; Villella, A.; McEwan, B.; Patel, N.R.; Qiu, D.; Bhalla, A.; Bastos, C.; Parks, D.; et al. CFTR Amplifiers: A New Class of CFTR Modulator that Complements the Substrate Limitations of Other CF Therapeutic Modalities. Am. J. Respir. Crit. Care Med. 2016, 193, A5574. [Google Scholar]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Durmowicz, A.G.; Lim, R.; Rogers, H.; Rosebraugh, C.J.; Chowdhury, B.A. The U.S. Food and Drug Administration’s Experience with Ivacaftor in Cystic Fibrosis. Establishing Efficacy Using In Vitro Data in Lieu of a Clinical Trial. Ann. Am. Thorac. Soc. 2018, 15, 1–2. [Google Scholar] [CrossRef]
- Flume, P.A.; Liou, T.G.; Borowitz, D.S.; Li, H.; Yen, K.; Ordoñez, C.L.; Geller, D.E. Ivacaftor in Subjects with Cystic Fibrosis Who Are Homozygous for the F508del-CFTR Mutation. Chest 2012, 142, 718–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, M.P.; Bell, S.C.; Konstan, M.W.; McColley, S.A.; Rowe, S.M.; Rietschel, E.; Huang, X.; Waltz, D.; Patel, N.R.; Rodman, D. A CFTR corrector (lumacaftor) and a CFTR potentiator (ivacaftor) for treatment of patients with cystic fibrosis who have a phe508del CFTR mutation: A phase 2 randomised controlled trial. Lancet Respir. Med. 2014, 2, 527–538. [Google Scholar] [CrossRef]
- Clancy, J.P.; Rowe, S.M.; Accurso, F.J.; Aitken, M.L.; Amin, R.S.; Ashlock, M.A.; Ballmann, M.; Boyle, M.P.; Bronsveld, I.; Campbell, P.W.; et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012, 67, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, S.H.; Pilewski, J.M.; Griese, M.; Cooke, J.; Viswanathan, L.; Tullis, E.; Davies, J.C.; Lekstrom-Himes, J.A.; Wang, L.T. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. Care Med. 2018, 197, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Symdeko (Tezacaftor/Ivacaftor) [Package Insert]; Vertex Pharmaceuticals Inc.: Boston, MA, USA, 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210491lbl.pdf (accessed on 20 December 2020).
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Mall, M.A.; Mayer-Hamblett, N.; Rowe, S.M. Cystic Fibrosis: Emergence of Highly Effective Targeted Therapeutics and Potential Clinical Implications. Am. J. Respir. Crit. Care Med. 2020, 201, 1193–1208. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; van Dalfsen, J.M.; et al. Clinical Mechanism of the Cystic Fibrosis Transmembrane Conductance Regulator Potentiator Ivacaftor in G551D-mediated Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef]
- Heltshe, S.L.; Mayer-Hamblett, N.; Burns, J.L.; Khan, U.; Baines, A.; Ramsey, B.W.; Rowe, S.M. Pseudomonas aeruginosa in Cystic Fibrosis Patients with G551D-CFTR Treated with Ivacaftor. Clin. Infect. Dis. 2015, 60, 703–712. [Google Scholar] [CrossRef]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef]
- Reznikov, L.R.; Alaiwa, M.H.A.; Dohrn, C.L.; Gansemer, N.D.; Diekema, D.J.; Stoltz, D.A.; Welsh, M.J. Antibacterial properties of the CFTR potentiator ivacaftor. J. Cyst. Fibros. 2014, 13, 515–519. [Google Scholar] [CrossRef] [Green Version]
- Millar, B.C.; Rendall, J.C.; Downey, D.G.; Moore, J.E. Does ivacaftor interfere with the antimicrobial activity of commonly used antibiotics against Pseudomonas aeruginosa? Results of an in vitro study. J. Clin. Pharm. Ther. 2018, 43, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.-Y.; Lim, D.J.; Mackey, C.; Skinner, D.; Zhang, S.; McCormick, J.; Woodworth, B.A. Ivacaftor, a Cystic Fibrosis Transmembrane Conductance Regulator Potentiator, Enhances Ciprofloxacin Activity Against Pseudomonas aeruginosa. Am. J. Rhinol. Allergy 2018, 33, 129–136. [Google Scholar] [CrossRef]
- Pohl, K.; Hayes, E.; Keenan, J.; Henry, M.; Meleady, P.; Molloy, K.; Jundi, B.; Bergin, D.A.; McCarthy, C.; McElvaney, O.J.; et al. A neutrophil intrinsic impairment affecting Rab27a and degranulation in cystic fibrosis is corrected by CFTR potentiator therapy. Blood 2014, 124, 999–1009. [Google Scholar] [CrossRef]
- Smyth, A.R.; Bell, S.C.; Bojcin, S.; Bryon, M.; Duff, A.; Flume, P.; Kashirskaya, N.; Munck, A.; Ratjen, F.; Schwarzenberg, S.J.; et al. European Cystic Fibrosis Society Standards of Care: Best Practice guidelines. J. Cyst. Fibros. 2014, 13, S23–S42. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.K.; Wagner, B.D.; Zemanick, E.T.; Robertson, C.E.; Stevens, M.J.; Heltshe, S.L.; Rowe, S.M.; Sagel, S.D. Changes in Airway Microbiome and Inflammation with Ivacaftor Treatment in Patients with Cystic Fibrosis and the G551D Mutation. Ann. Am. Thorac. Soc. 2020, 17, 212–220. [Google Scholar] [CrossRef]
- Ruffin, M.; Roussel, L.; Maillé, E.; Rousseau, S.; Brochiero, E. Vx-809/Vx-770 treatment reduces inflammatory response to Pseudomonas aeruginosa in primary differentiated cystic fibrosis bronchial epithelial cells. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L635–L641. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; de Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, S.; Sly, P.D.; Gangell, C.L.; Sturges, N.; Winfield, K.; Wikström, M.; Gard, S.; Upham, J.W.; Cf, O.B.O.A. Alveolar macrophages and CC chemokines are increased in children with cystic fibrosis. Eur. Respir. J. 2009, 34, 655–661. [Google Scholar] [CrossRef] [Green Version]
- Kopp, B.T.; Abdulrahman, B.A.; Khweek, A.A.; Kumar, S.B.; Akhter, A.; Montione, R.; Tazi, M.F.; Caution, K.; McCoy, K.; Amer, A.O. Exaggerated inflammatory responses mediated by Burkholderia cenocepacia in human macrophages derived from Cystic fibrosis patients. Biochem. Biophys. Res. Commun. 2012, 424, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Shrestha, C.L.; Kopp, B.T. Cystic fibrosis transmembrane conductance regulator (CFTR) modulators have differential effects on cystic fibrosis macrophage function. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Barnaby, R.; Koeppen, K.; Nymon, A.; Hampton, T.H.; Berwin, B.; Ashare, A.; Stanton, B.A. Lumacaftor (VX-809) restores the ability of CF macrophages to phagocytose and kill Pseudomonas aeruginosa. Am. J. Physiol. Cell. Mol. Physiol. 2018, 314, L432–L438. [Google Scholar] [CrossRef] [Green Version]
- Kopp, B.T.; Fitch, J.; Jaramillo, L.; Shrestha, C.L.; Robledo-Avila, F.; Zhang, S.; Palacios, S.; Woodley, F.; Hayes, D.; Partida-Sanchez, S.; et al. Whole-blood transcriptomic responses to lumacaftor/ivacaftor therapy in cystic fibrosis. J. Cyst. Fibros. 2020, 19, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Currie, A.J.; Main, E.T.; Wilson, H.M.; Armstrong-James, D.; Warris, A. CFTR Modulators Dampen Aspergillus-Induced Reactive Oxygen Species Production by Cystic Fibrosis Phagocytes. Front. Cell. Infect. Microbiol. 2020, 10, 372. [Google Scholar] [CrossRef] [PubMed]
- Amin, R.; Dupuis, A.; Aaron, S.D.; Ratjen, F. The Effect of Chronic Infection with Aspergillus fumigatus on Lung Function and Hospitalization in Patients with Cystic Fibrosis. Chest 2010, 137, 171–176. [Google Scholar] [CrossRef]
- Gentzsch, M.; Cholon, D.M.; Quinney, N.L.; Boyles, S.E.; Martino, M.E.B.; Ribeiro, C.M.P. The cystic fibrosis airway milieu enhances rescue of F508del in a pre-clinical model. Eur. Respir. J. 2018, 52, 1801133. [Google Scholar] [CrossRef]
- Pohl, K.; Nichols, D.P.; Taylor-Cousar, J.L.; Saavedra, M.T.; Strand, M.J.; Nick, J.A.; Bratcher, P.E. Corticosteroid use and increased CXCR2 levels on leukocytes are associated with lumacaftor/ivacaftor discontinuation in cystic fibrosis patients homozygous for the F508del CFTR mutation. PLoS ONE 2018, 13, e0209026. [Google Scholar] [CrossRef]
- Khan, T.Z.; Wagener, J.S.; Bost, T.; Martinez, J.; Accurso, F.J.; Riches, D.W.H. Early Pulmonary Inflammation in Infants with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 1995, 151, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Jarosz-Griffiths, H.H.; Scambler, T.; Wong, C.H.; Lara-Reyna, S.; Holbrook, J.; Martinon, F.; Savic, S.; Whitaker, P.; Etherington, C.; Spoletini, G.; et al. Different CFTR modulator combinations downregulate inflammation differently in cystic fibrosis. eLife 2020, 9, e54556. [Google Scholar] [CrossRef]
- Shrestha, C.L.; Zhang, S.; Wisniewski, B.; Häfner, S.; Elie, J.; Meijer, L.; Kopp, B.T. (R)-Roscovitine and CFTR modulators enhance killing of multi-drug resistant Burkholderia cenocepacia by cystic fibrosis macrophages. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Soltis, A.R.; Sukumar, G.; Zhang, X.; Caohuy, H.; Freedy, J.; Dalgard, C.L.; Wilkerson, M.D.; Pollard, H.B.; Pollard, B.S. Gene therapy-emulating small molecule treatments in cystic fibrosis airway epithelial cells and patients. Respir. Res. 2019, 20, 290. [Google Scholar] [CrossRef]
- Trikafta [Package Insert]; Pharmaceuticals Incorporated: Boston, MA, USA, 2019. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212273s000lbl.pdf (accessed on 4 January 2021).
- Cystic Fibrosis Foundation Website. Available online: https://www.cff.org/Research/About-Our-Research/Research-Milestones/ (accessed on 4 January 2021).
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Liessi, N.; Pesce, E.; Braccia, C.; Bertozzi, S.M.; Giraudo, A.; Bandiera, T.; Pedemonte, N.; Armirotti, A. Distinctive lipid signatures of bronchial epithelial cells associated with cystic fibrosis drugs, including Trikafta. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Becker, K.A.; Riethmüller, J.; Seitz, A.P.; Gardner, A.; Boudreau, R.; Kamler, M.; Kleuser, B.; Schuchman, E.; Caldwell, C.C.; Edwards, M.J.; et al. Sphingolipids as targets for inhalation treatment of cystic fibrosis. Adv. Drug Deliv. Rev. 2018, 133, 66–75. [Google Scholar] [CrossRef]
- Chan, C.; Goldkorn, T. Ceramide Path in Human Lung Cell Death. Am. J. Respir. Cell Mol. Biol. 2000, 22, 460–468. [Google Scholar] [CrossRef]
- Lavrentiadou, S.N.; Chan, C.; Kawcak, T.; Ravid, T.; Tsaba, A.; van der Vliet, A.; Rasooly, R.; Goldkorn, T. Ceramide-mediated apoptosis in lung epithelial cells is regulated by glutathione. Am. J. Respir. Cell Mol. Biol. 2001, 25, 676–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauber, K.; Bohn, E.; Kröber, S.M.; Xiao, Y.-J.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic Cells Induce Migration of Phagocytes via Caspase-3-Mediated Release of a Lipid Attraction Signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Asano, N.; Nash, R.J.; Molyneux, R.J.; Fleet, G.W. Sugar-mimic glycosidase inhibitors: Natural occurrence, biological activity and prospects for therapeutic application. Tetrahedron Asymmetry 2000, 11, 1645–1680. [Google Scholar] [CrossRef]
- Lillelund, V.H.; Jensen, H.H.; Liang, X.; Bols, M. Recent developments of transition-state analogue glycosidase inhibitors of non-natural product origin. Chem. Rev. 2002, 102, 515–553. [Google Scholar] [CrossRef]
- Dechecchi, M.C.; Nicolis, E.; Norez, C.; Bezzerri, V.; Borgatti, M.; Mancini, I.; Rizzotti, P.; Ribeiro, C.M.; Gambari, R.; Becq, F.; et al. Anti-inflammatory effect of miglustat in bronchial epithelial cells. J. Cyst. Fibros. 2008, 7, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Norez, C.; Noel, S.; Wilke, M.; Bijvelds, M.; Jorna, H.; Melin, P.; Dejonge, H.; Becq, F. Rescue of functional delF508-CFTR channels in cystic fibrosis epithelial cells by the α-glucosidase inhibitor miglustat. FEBS Lett. 2006, 580, 2081–2086. [Google Scholar] [CrossRef] [Green Version]
- Leonard, A.; Lebecque, P.; Dingemanse, J.; Leal, T. A randomized placebo-controlled trial of miglustat in cystic fibrosis based on nasal potential difference. J. Cyst. Fibros. 2012, 11, 231–236. [Google Scholar] [CrossRef] [Green Version]
- Loberto, N.; Tebon, M.; Lampronti, I.; Marchetti, N.; Aureli, M.; Bassi, R.; Giri, M.G.; Bezzerri, V.; Lovato, V.; Cantu, C.; et al. GBA2-encoded beta-glucosidase activity is involved in the inflammatory response to Pseudomonas aeruginosa. PLoS ONE 2014, 9, e104763. [Google Scholar] [CrossRef] [Green Version]
- Munari, S.; Loberto, N.; Aureli, M.; Vauzeilles, B.; Baron, A.; Guisot, N.; Schiumarini, D.; Bassi, R.; Tironi, M.; Giri, M.; et al. Neoglycoconjugates Derived from Deoxynojirimycin as Possible Therapeutic Agents for Cystic Fibrosis Lung Disease, by Modulation of the Sphingolipid Metabolism. JSM Genet. Genom. 2016, 3, 1015. [Google Scholar]
- Tamanini, A.; Borgatti, M.; Finotti, A.; Piccagli, L.; Bezzerri, V.; Favia, M.; Guerra, L.; Lampronti, I.; Bianchi, N.; Dall’Acqua, F.; et al. Trimethylangelicin reduces IL-8 transcription and potentiates CFTR function. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L380–L390. [Google Scholar] [CrossRef] [Green Version]
- Favia, M.; Mancini, M.T.; Bezzerri, V.; Guerra, L.; LaSelva, O.; Abbattiscianni, A.C.; Debellis, L.; Reshkin, S.J.; Gambari, R.; Cabrini, G.; et al. Trimethylangelicin promotes the functional rescue of mutant F508del CFTR protein in cystic fibrosis airway cells. Am. J. Physiol. Cell. Mol. Physiol. 2014, 307, L48–L61. [Google Scholar] [CrossRef] [Green Version]
- Lampronti, I.; Manzione, M.G.; Sacchetti, G.; Ferrari, D.; Spisani, S.; Bezzerri, V.; Finotti, A.; Borgatti, M.; Dechecchi, M.C.; Miolo, G.; et al. Differential Effects of Angelicin Analogues on NF-kappaB Activity and IL-8 Gene Expression in Cystic Fibrosis IB3-1 Cells. Mediat. Inflamm. 2017, 2017, 2389487. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Venable, J.; LaPointe, P.; Hutt, D.M.; Koulov, A.V.; Coppinger, J.; Gurkan, C.; Kellner, W.; Matteson, J.; Plutner, H.; et al. Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 2006, 127, 803–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.L.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat. Cell Biol. 2010, 12, 863–875. [Google Scholar] [CrossRef]
- Venerando, A.; Franchin, C.; Cant, N.; Cozza, G.; Pagano, M.A.; Tosoni, K.; Al-Zahrani, A.; Arrigoni, G.; Ford, R.C.; Mehta, A.; et al. Detection of Phospho-Sites Generated by Protein Kinase CK2 in CFTR: Mechanistic Aspects of Thr1471 Phosphorylation. PLoS ONE 2013, 8, e74232. [Google Scholar] [CrossRef]
- Tosco, A.; de Gregorio, F.; Esposito, S.; de Stefano, D.; Sana, I.; Ferrari, E.; Sepe, A.; Salvadori, L.; Buonpensiero, P.; di Pasqua, A.; et al. A novel treatment of cystic fibrosis acting on-target: Cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ. 2017, 24, 1305. [Google Scholar] [CrossRef] [Green Version]
- Gahl, W.A. Early oral cysteamine therapy for nephropathic cystinosis. Eur. J. Nucl. Med. Mol. Imaging 2003, 162, S38–S41. [Google Scholar] [CrossRef] [PubMed]
- Faraj, J.; Bodas, M.; Pehote, G.; Swanson, D.; Sharma, A.; Vij, N. Novel cystamine-core dendrimer-formulation rescues DeltaF508-CFTR and inhibits Pseudomonas aeruginosa infection by augmenting autophagy. Expert Opin. Drug Deliv. 2019, 16, 177–186. [Google Scholar] [CrossRef]
- Petersen, N.T.; Høiby, N.; Mordhorst, C.H.; Lind, K.; Flensborg, E.W.; Bruun, B. Respiratory infections in cystic fibrosis patients caused by virus, chlamydia and mycoplasma-possible synergism with Pseudomonas aeruginosa. Acta Paediatr. 1981, 70, 623–628. [Google Scholar] [CrossRef]
- Wat, D. Impact of respiratory viral infections on cystic fibrosis. Postgrad. Med. J. 2003, 79, 201–203. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; De, B.P.; Choudhary, S.; Comhair, S.A.; Goggans, T.; Slee, R.; Williams, B.R.; Pilewski, J.; Haque, S.; Erzurum, S.C. Impaired Innate Host Defense Causes Susceptibility to Respiratory Virus Infections in Cystic Fibrosis. Immunity 2003, 18, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Kong, M.; Maeng, P.; Hong, J.; Szczesniak, R.; Sorscher, E.; Sullender, W.; Clancy, J.P. Respiratory Syncytial Virus Infection Disrupts Monolayer Integrity and Function in Cystic Fibrosis Airway Cells. Viruses 2013, 5, 2260–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flight, W.G.; Bright-Thomas, R.J.; Tilston, P.; Mutton, K.J.; Guiver, M.; Morris, J.; Webb, A.K.; Jones, A.M. Incidence and clinical impact of respiratory viruses in adults with cystic fibrosis. Thorax 2013, 69, 247–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, A.R.; Smyth, R.L.; Tong, C.Y.; Hart, C.A.; Heaf, D.P. Effect of respiratory virus infections including rhinovirus on clinical status in cystic fibrosis. Arch. Dis. Child. 1995, 73, 117–120. [Google Scholar] [CrossRef] [Green Version]
- van Ewijk, B.E.; van der Zalm, M.M.; Wolfs, T.F.; Fleer, A.; Kimpen, J.L.; Wilbrink, B.; van der Ent, C.K. Prevalence and impact of respiratory viral infections in young children with cystic fibrosis: Prospective cohort study. Pediatrics 2008, 122, 1171–1176. [Google Scholar] [CrossRef]
- Wat, D.; Gelder, C.; Hibbitts, S.; Cafferty, F.; Bowler, I.; Pierrepoint, M.; Evans, R.; Doull, I. The role of respiratory viruses in cystic fibrosis. J. Cyst. Fibros. 2008, 7, 320–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoek, R.A.S.; Paats, M.S.; Pas, S.D.; Bakker, M.; Hoogsteden, H.C.; Boucher, C.A.B.; van der Eerden, M.M. Incidence of viral respiratory pathogens causing exacerbations in adult cystic fibrosis patients. Scand. J. Infect. Dis. 2012, 45, 65–69. [Google Scholar] [CrossRef]
- Viviani, L.; Assael, B.M.; Kerem, E. Impact of the A (H1N1) pandemic influenza (season 2009–2010) on patients with cystic fibrosis. J. Cyst. Fibros. 2011, 10, 370–376. [Google Scholar] [CrossRef] [Green Version]
- Chan, W.; He, B.; Wang, X.; He, M.-L. Pandemic COVID-19: Current status and challenges of antiviral therapies. Genes Dis. 2020, 7, 502–519. [Google Scholar] [CrossRef] [PubMed]
- Cosgriff, R.; Ahern, S.; Bell, S.C.; Brownlee, K.; Burgel, P.R.; Byrnes, C.; Corvol, H.; Cheng, S.Y.; Elbert, A.; Faro, A.; et al. A multinational report to characterise SARS-CoV-2 infection in people with cystic fibrosis. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2020, 19, 355–358. [Google Scholar] [CrossRef] [PubMed]
- McClenaghan, E.; Cosgriff, R.; Brownlee, K.; Ahern, S.; Burgel, P.R.; Byrnes, C.A.; Colombo, C.; Corvol, H.; Cheng, S.Y.; Daneau, G.; et al. The global impact of SARS-CoV-2 in 181 people with cystic fibrosis. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2020, 19, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Poli, P.; Timpano, S.; Goffredo, M.; Padoan, R.; Badolato, R. Asymptomatic case of COVID-19 in an infant with cystic fibrosis. J. Cyst. Fibros. 2020, 19, e18. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Wang, B.; Mao, J. The pathogenesis and treatment of the ‘Cytokine Storm’ in COVID-19. J. Infect. 2020, 80, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Stanton, B.A.; Hampton, T.H.; Ashare, A. SARS-CoV-2 (COVID-19) and cystic fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L408–L415. [Google Scholar] [CrossRef]
- Ingraham, N.E.; Barakat, A.G.; Reilkoff, R.; Bezdicek, T.; Schacker, T.; Chipman, J.G.; Tignanelli, C.J.; Puskarich, M.A. Understanding the renin-angiotensin-aldosterone-SARS-CoV axis: A comprehensive review. Eur. Respir. J. 2020, 56, 2000912. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghatrif, M.; Cingolani, O.; Lakatta, E.G. The Dilemma of Coronavirus Disease 2019, Aging, and Cardiovascular Disease: Insights from Cardiovascular Aging Science. JAMA Cardiol. 2020, 5, 747–748. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghigo, A.; Prono, G.; Riccardi, E.; De Rose, V. Dysfunctional Inflammation in Cystic Fibrosis Airways: From Mechanisms to Novel Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 1952. https://doi.org/10.3390/ijms22041952
Ghigo A, Prono G, Riccardi E, De Rose V. Dysfunctional Inflammation in Cystic Fibrosis Airways: From Mechanisms to Novel Therapeutic Approaches. International Journal of Molecular Sciences. 2021; 22(4):1952. https://doi.org/10.3390/ijms22041952
Chicago/Turabian StyleGhigo, Alessandra, Giulia Prono, Elisa Riccardi, and Virginia De Rose. 2021. "Dysfunctional Inflammation in Cystic Fibrosis Airways: From Mechanisms to Novel Therapeutic Approaches" International Journal of Molecular Sciences 22, no. 4: 1952. https://doi.org/10.3390/ijms22041952
APA StyleGhigo, A., Prono, G., Riccardi, E., & De Rose, V. (2021). Dysfunctional Inflammation in Cystic Fibrosis Airways: From Mechanisms to Novel Therapeutic Approaches. International Journal of Molecular Sciences, 22(4), 1952. https://doi.org/10.3390/ijms22041952