Abstract
Hepatic fibrosis occurs when liver tissue becomes scarred from repetitive liver injury and inflammatory responses; it can progress to cirrhosis and eventually to hepatocellular carcinoma. Previously, we reported that neoagarooligosaccharides (NAOs), produced by the hydrolysis of agar by β-agarases, have hepatoprotective effects against acetaminophen overdose-induced acute liver injury. However, the effect of NAOs on chronic liver injury, including hepatic fibrosis, has not yet been elucidated. Therefore, we examined whether NAOs protect against fibrogenesis in vitro and in vivo. NAOs ameliorated PAI-1, α-SMA, CTGF and fibronectin protein expression and decreased mRNA levels of fibrogenic genes in TGF-β-treated LX-2 cells. Furthermore, downstream of TGF-β, the Smad signaling pathway was inhibited by NAOs in LX-2 cells. Treatment with NAOs diminished the severity of hepatic injury, as evidenced by reduction in serum alanine aminotransferase and aspartate aminotransferase levels, in carbon tetrachloride (CCl4)-induced liver fibrosis mouse models. Moreover, NAOs markedly blocked histopathological changes and collagen accumulation, as shown by H&E and Sirius red staining, respectively. Finally, NAOs antagonized the CCl4-induced upregulation of the protein and mRNA levels of fibrogenic genes in the liver. In conclusion, our findings suggest that NAOs may be a promising candidate for the prevention and treatment of chronic liver injury via inhibition of the TGF-β/Smad signaling pathway.
1. Introduction
Liver fibrosis is a highly conserved wound healing response to chronic liver injury [1]. Similar to other tissues, such as the skin, the liver accumulates excessive extracellular matrix (ECM), especially type I and III collagens, as well as proteoglycans, fibronectin, elastin and laminin following injury. In most cases, liver fibrosis is the result of viral and metabolic liver diseases [2,3]. Liver fibrosis may progress to advanced stages of irreversible liver diseases, such as liver cirrhosis and hepatocellular cancer, over time. Currently, liver transplantation is the only treatment available for patients with advanced liver fibrosis. Therefore, new antifibrotic therapies are needed to prevent the late stages of decompensated chronic liver diseases.
During progression of hepatic fibrosis, activation of hepatic stellate cells (HSCs) plays an important role in the excessive production of ECM proteins. Quiescent HSCs store 80% of total liver retinoids (vitamin A) as well as triglycerides, cholesteryl esters, cholesterol, phospholipids and free fatty acids in lipid droplets in the cytoplasm [4]. In contrast, in injured livers, activated HSCs transform into myofibroblasts, with simultaneous loss of their lipid droplets and production of ECM, subsequently leading to hepatic fibrosis. Among various pathogenic factors, transforming growth factor-β (TGF-β) is a key mediator of liver fibrosis via HSC activation. TGF-β forms receptor complexes, with type I receptors in combination with type II receptors. This leads to the activation of kinase domains within the ligand-bound receptors, which triggers phosphorylation cascades involving Smad transcription factors. Activation of Smads regulates the expression of profibrotic genes, including collagens [5], plasminogen activator inhibitor-1 (PAI-1) [6], proteoglycans [7], integrins [8], connective tissue growth factor (CTGF) [9] and matrix metalloproteases (MMPs) [10]. Inhibition of the TGF-β/Smad signaling pathway attenuates the progression of liver fibrosis in vitro and in vivo [11,12].
β-agarase specifically cleaves the β-1,4 glycosidic bond of agarose to produce neoagarooligosaccharides (NAOs)—which have various degrees of polymerization, in contrast to β-agarase—and cleaves the β-1,3 linkage in agarose to produce agarooligosaccharides. Previously, we reported that NAOs inhibit metabolic liver disease and acute liver injury in mice administered a high cholesterol diet or acetaminophen overdose, respectively [13,14]. Moreover, NAOs present no signs of toxicity up to 5,000 mg/kg body weight/day in acute, repeated 14-day and 91-day oral toxicity tests [15]. These results strongly support the potential application of NAOs in dietary supplements and medications. Nevertheless, the therapeutic efficacy and mechanism of NAOs in chronic liver injury have not yet been elucidated.
In this study, we show that NAOs attenuate hepatic fibrosis both in vitro and in vivo. NAOs antagonized TGF-β-induced profibrotic gene expression through inhibition of the Smad signaling pathway in cultured immortalized human semi-activated HSCs. Moreover, NAOs reduced the progression of carbon tetrachloride(CCl4)-induced liver fibrosis as evidenced by serum liver enzymes, histopathological changes, and production of ECM proteins. Therefore, our results indicate that NAOs have hepatoprotective effects in TGF-β-treated HSCs and CCl4-induced liver fibrosis, and have potential applications in clinical interventions.
2. Results
2.1. Suppressive Effect of NAOs on HSCs Activation in Vitro
First, we examined the cytotoxicity of NAOs in LX-2 cells by MTT assay. We found that there were no significant differences between vehicle- and NAOs-treated cells at concentrations up to 1.0 mg/mL (Figure 1A). Therefore, we adopted 0.3–1.0 mg/mL NAOs in following experiments, to demonstrate the effect of NAOs on HSC activation. Treatment of NAOs in LX-2 cells effectively inhibited TGF-β-induced PAI-1, α-SMA, CTGF and fibronectin protein expression—all of which are typical markers of HSC activation (Figure 1B–C, Figures S1 and S2). When we used isolated primary HSC from mice, we also observed increased PAI-1, α-SMA, CTGF and fibronectin expression; TGF- was antagonized by NAOs (Figure 1D). RT-PCR analysis confirmed that TGF-β markedly increased mRNA levels of PAI-1, α-SMA and collagen 1A1 (COL1A1), which were almost completely blocked by pretreatment with NAOs (Figure 1E–G). These findings indicate that NAOs inhibit HSC activation.
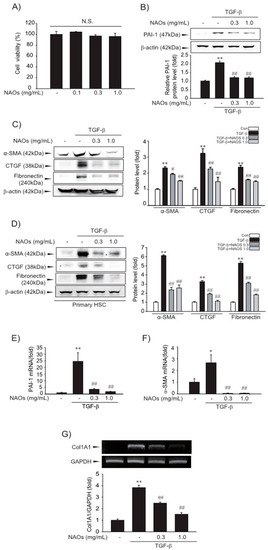
Figure 1.
Inhibition of TGF-β-mediated fibrosis markers by neoagarooligosaccharides (NAOs). (A) MTT assays for cell viability. Effect of NAOs (0.1–1.0 mg/mL, 12 h treatment) on cell viability of LX-2 cells, a well characterized human HSC line evaluated using MTT assays (n = 3). N.S., not significant. (B–D) Effect of varying concentrations of NAOs on TGF-β-mediated fibrogenic protein expression. Cells were treated with 0.3–1.0 mg/mL NAOs and incubated with TGF-β (1 ng/mL) for 6 h. Protein levels in the cell lysates were determined by immunoblotting (n = 3). (E,F) Real-time PCRanalysis. Cells were treated with 0.3–1.0 mg/mL NAOs for 30 min, and then further incubated with TGF-β for 3 h. The α-SMA and PAI-1 transcripts were analyzed by Real-time PCR, with the mRNA level of GAPDH used as a housekeeping gene (n = 3). (G) Collagen (Col) 1A1 transcripts analyzed by RT-PCR assays. Results were confirmed by repeated experiments (n = 3). Data represent the mean ± standard error (S.E.) of three replicates; ** p < 0.01, * p < 0.05, vs. vehicle-treated control; ## p < 0.01, # p < 0.05, vs. TGF-β alone. Minus sign (-) indicates untreated with NAOs.
2.2. Inhibitory Effect of NAOs on TGF-β/Smad Signaling
Next, we investigated the effect of NAOs on the TGF-β-induced Smad pathway, a major mediator of TGF-β signaling. First, to verify the underlying molecular mechanism of inhibition of HSC activation, we performed reporter gene assay of Smad-binding element (SBE)-luciferase activity, which contained nine repeated SBEs. SBE-luciferase activity was significantly increased by TGF-β; however, pretreatment with NAOs (0.3 or 1.0 mg/mL) inhibited SBE-reporter gene activity in LX-2 cells (Figure 2A). Furthermore, Smad3-dependent transcription of SBE reporter activity was inhibited by pretreatment with NAOs (Figure 2B). Consistent with these findings, the increase in TGF-β-induced Smad3 phosphorylation was antagonized by pretreatment with NAOs (Figure 2C). These findings suggest that NAOs inhibit the TGF-β/Smad signaling pathway.
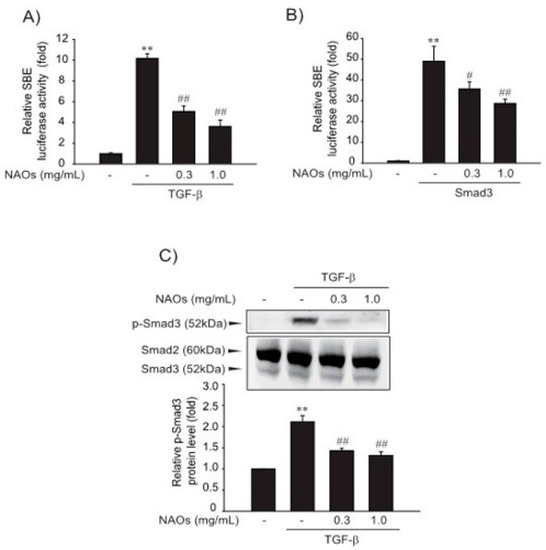
Figure 2.
Inhibition of TGF-β downstream signaling by neoagarooligosaccharides (NAOs). (A) Effect of NAOs on TGF-β-induced Smad pathway in the LX-2 cells, determined by transfecting the cells with pGL-SBE-luciferase construct, followed by treatment with TGF-β (1 ng/mL) and/or NAOs. (B) LX-2 cells co-transfected with Smad3 expression construct and treated with NAOs (12 h). (C) Immunoblotting for Smad3 phosphorylation. Cells treated with 0.3–1.0 mg/mL NAOs for 30 min prior to incubation with TGF-β for 30 min. Cell lysates immunoblotted for phosphorylated Smad3 and results confirmed by repeating the experiments. Data represent the mean ± S.E. of three replicates; ** p < 0.01, vs. vehicle-treated control; ## p < 0.01, # p < 0.05, vs. TGF-β alone. Minus sign (-) indicates untreated with NAOs.
2.3. Suppression of CCl4-Induced Liver Fibrosis by NAOs In Vivo
To study the inhibitory effects of NAOs in vivo, we used the classic animal model of CCl4-induced liver fibrosis. This is the most common adopted method of inducing pernicious effects in the liver by producing highly reactive metabolites, resulting in liver lesions and subsequent fibrosis [16,17]. We pretreated mice with NAOs for 5 days before CCl4 treatment to induce liver fibrosis. CCl4 was administered twice a week for two weeks in concomitant daily treatment with NAOs (Figure 3A). Mice were sacrificed 24 h after the final CCl4 administration. First, we analyzed the levels of the serum biomarkers of liver damage, including alanine aminotransferase (ALT) and aspartate aminotransferase (AST). Injection of CCl4 for 2 weeks significantly increased serum ALT and AST levels; however, these effects were considerably inhibited by NAOs treatment (Figure 3B,C). To determine the hepatoprotective effects of NAOs against CCl4-induced liver damage, we performed histopathological analyses to assess the extent of liver injury. Administration of CCl4 led to degenerative regions, liver centrilobular necrosis, and increased numbers of degenerative hepatocytes and inflammatory cells. However, these CCl4-induced hepatic damages were markedly reduced by NAOs treatment (Figure 4A; Table 1). We stained the mice liver sections with Sirius red to examine the effect of NAOs on collagen accumulation. The results showed that the CCl4-treated mice had large amounts of collagen deposition in the fibrotic septa between nodules. In contrast, treatment with NAOs decreased the collagen accumulation following CCl4 administration (Figure 4B, left; Table 1). HSC activation is a major component of liver fibrosis, and α-SMA is a key marker for HSC activation [18]. α-SMA immunoreactive cells were significantly increased in the centrilobular regions following CCl4 treatment, but this increase in α-SMA immunoreactive cells was markedly inhibited by treatment with NAOs (Figure 4B, right; Table 1). In addition, treatment of NAOs decreased PAI-1, p-Smad3 and TGF-β1 immunoreactive cells by CCl4-treatment (Figure 4C and Table 1). Moreover, the antifibrotic effect of NAOs was confirmed by immunoblotting and real-time PCR analysis of the major markers (PAI-1, α-SMA, and Col1A1) of liver fibrosis taken from three randomly selected mouse samples (Figure 5A–C).
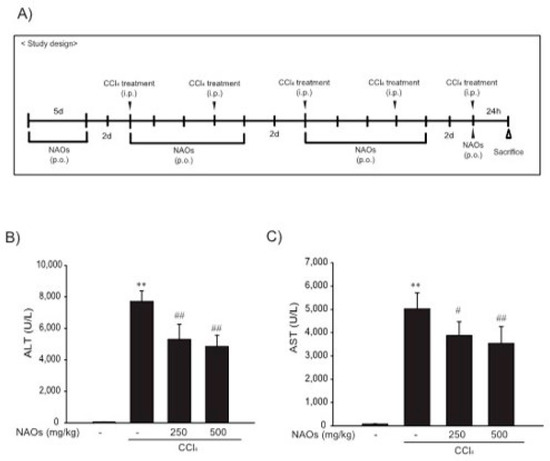
Figure 3.
Inhibition of CCl4-induced liver injury by neoagarooligosaccharides (NAOs). (A) Study design. Prior to CCl4 treatment to induce liver fibrosis, mice are treated with NAOs for 5 days. Liver fibrosis is then induced by intraperitoneal injection of CCl4 (0.5 mg/kg, dissolved in olive oil [10%]) into mice, twice a week for 2 weeks. Mice are also treated with NAOs five times a week for two weeks. Twenty-four hours prior to sacrifice, mice are induced with a final intraperitoneal injection of CCl4. (B,C) Activities of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) analyzed using an automated blood chemistry analyzer. All values are expressed as mean ± S.E. of five mouse serum samples (** p < 0.01, vs. vehicle control; ## p < 0.01, # p < 0.05, significant vs. CCl4).
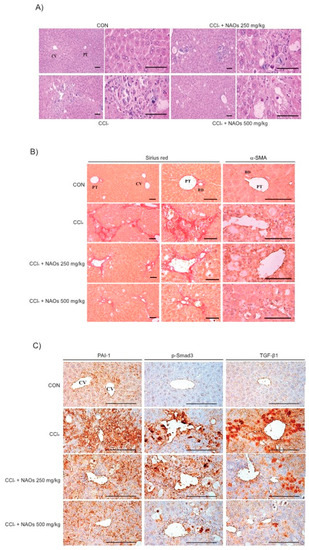
Figure 4.
Inhibition of CCl4-induced liver fibrosis by neoagarooligosaccharides (NAOs). (A) Histopathological changes in mice with CCl4-induced subacute liver damage, showing centrilobular necrosis, including ballooning of hepatocytes and infiltration of inflammatory cells. CCl4+NAOs treated mice show markedly lowered CCl4-induced subacute liver damage as compared to CCl4-treated mice. CCl4: Carbon tetrachloride, CV: Central vein, PT: Portal trial, Scale bar = 120 μm. (B,C) Sirius red and α-SMA, PAI-1, p-Smad3 and TGF-β1 immunohistochemical staining of liver from mice treated with CCl4 or CCl4+NAOs for 2 weeks (Scale bar = 120 μm). CON, vehicle control.

Table 1.
Histomorphometrical analysis of hepatic tissues from vehicle or CCl4-treated mice.
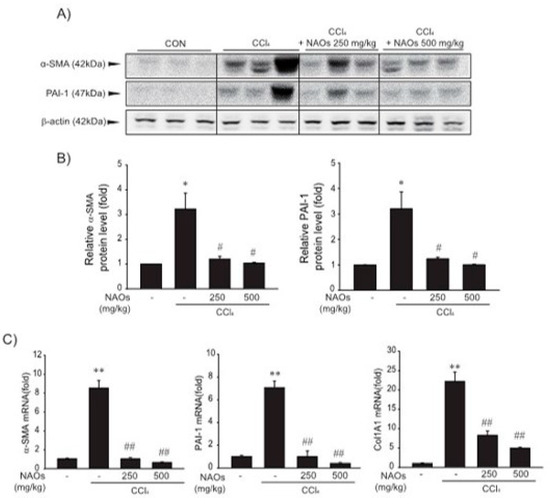
Figure 5.
Inhibition of CCl4-induced expression of fibrogenic genes by neoagarooligosaccharides (NAOs). (A,B) Western blot analysis. α-SMA and PAI-1 protein levels assessed by immunoblotting. (n = 3). Results are presented as means ± S.E. of three replicates. (C) α-SMA, PAI-1, and Col 1A1 transcripts in CCl4-induced mice liver assessed by real-time PCR analysis (n = 3). Results are presented as means ± S.E.; ** p < 0.01, * p < 0.05, vs. vehicle control; ## p < 0.01, # p < 0.05, vs. CCl4; CON, vehicle control.
3. Discussion
Hepatic fibrosis occurs when healthy liver tissues becomes scarred from chronic liver injury and inflammatory responses, and therefore cannot function normally [19,20]. Chronic liver damage leads to liver fibrosis in conjunction with the accumulation of ECM proteins, which is a typical characteristic of most chronic liver diseases [21]. Activated HSCs are major ECM-producing cells in an injured liver; these cells are activated by fibrogenic mediators, such as TGF-β [22]. TGF-β—a key downstream mediator in liver fibrogenesis—exerts its effect via downstream molecular signaling pathways, such as Smad2 and Smad3 [23]. TGF-β bound type II receptor leads to recruitment and phosphorylation of the type I receptor. Ligand-receptor complex formation leads to the activation of kinase domains within the receptors, which triggers phosphorylation of Smads [24]. The Smads then transmit the signals from the receptors of the TGF-β superfamily members to the nucleus, where they initiate transcription of TGF-β target genes [25]. In the present study, treatment with NAOs inhibited TGF-β-induced profibrogenic gene expression in LX-2 cells. Moreover, we found that NAOs blocked the Smad signaling pathway downstream of TGF-β. NAOs might therefore act as an antagonist of TGF-β receptors.
Next, we evaluated the protective effect of NAOs against CCl4-induced hepatic fibrosis in vivo. CCl4, a well-known fibrosis-inducing hepatotoxin, is extensively used in liver-related studies. CCl4-treated rodents exhibited fatty changes along with an increase in inflammatory cell infiltrations, damage to normal hepatocytes, deposition of collagen and formation of fiber segmentation [26]. Chronic liver toxicity induced by CCl4 causes an increase in lipid peroxidation that leads to a decrease in antioxidant enzymes and glutathione activities [27,28]. This CCl4-induced chronic toxicity was suppressed by pretreatment with NAOs. Furthermore, the increase in serum ALT and AST levels induced by administration of CCl4 for 2 weeks was decreased by NAOs treatment (Figure 3). NAOs also dramatically inhibited CCl4-induced hepatic damage, including regions of degeneration, liver centrilobular necrosis, and the increased number of degenerative hepatocytes and inflammatory cells. Treatment with NAOs also reduced CCl4-induced collagen accumulation and synthesis of various ECM components, including PAI-1, α-SMA, and Col1A1 (Figure 4 and Figure 5, Table 1). The in vivo effects of NAOs on liver fibrosis might be due to the protection of hepatocytes and the inhibition of HSC activation. Hepatic regeneration after liver injury is composed not only of fibroblasts, but also of cholangiocytes that orchestrate the deposition of fibrosis by stimulating proliferation and activation of myofibroblasts [29]. Further study of the effect of NAOs on liver regeneration is still required and currently ongoing.
In conclusion, our study demonstrates that NAOs prevent liver fibrosis in vitro and in vivo. Furthermore, NAOs significantly protect hepatocytes from injury and inflammation following CCl4 administration. Collectively, these findings suggest that NAOs are a potential antifibrotic agent for the prevention and treatment of chronic liver diseases via inhibition of HSC activation (Figure 6).
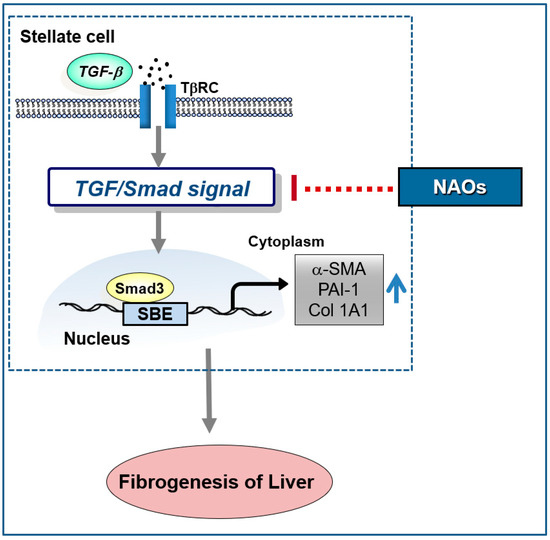
Figure 6.
Schematic diagram demonstrating the mechanism through which neoagarooligosaccharides (NAOs) protect against hepatic fibrosis via inhibition of TGF-β/Smad signaling pathway.
4. Materials and Methods
4.1. Materials
PAI-1 (614024) and fibronectin (610077) antibodies were obtained from BD Biosciences (Franklin Lakes, NJ, USA). Phospho-Smad3 (#9520) and Smad2/3 (#3102) antibodies were provided by Cell Signaling (Danvers, MA, USA). CTGF antibodies (SC-101586) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and TGF-β1 antibody (NBP1-80289) was obtained from Novus Biologicals (Littleton, CO). Horseradish peroxidase-conjugated goat anti-rabbit (G21234) and anti-mouse (G21040) antibodies were purchased from Invitrogen (Carlsbad, CA, USA). α-SMA (A2547) and β-actin (A5441) antibodies, MTT (M2128), and dimethylsulfoxide (D8418) were acquired from Sigma Chemicals (St. Louis, MO, USA). TGF-β (240-B) was purchased from R&D Systems (Minneapolis, MN, USA). CTGF antibody (sc-101586) obtained from Santa Cruz Biotechnology (Santa Cruz, CA). CCl4 (33650-0330) was received from Junsei Chemical (Kyoto, CO).
4.2. Preparation of NAOs
Agar from Gelidium elegans was hydrolyzed to NAOs by β-agarase DagA originating from Streptomyces coelicolor A3(2)_M22-2C43. Enzymatic reactions occurred at 44~46 °C for 16 h. NAOs solution was filtered and concentrated to 10 times its concentration. Freeze-dried NAO was homogenized into 100 mesh-sized powders.
4.3. Cell Culture
Immortalized human semi-activated HSCs and LX-2 cells were kindly donated by Dr. S. L. Friedmann (Mount Sinai School of Medicine, NY, USA), and maintained in Dulbecco’s modified Eagle’s medium (DMEM), containing 10% fetal bovine serum, 50 units/mL penicillin/streptomycin at 37 °C in a humidified 5% CO2 atmosphere. Primary HSCs were isolated from the livers of 8-week-old ICR mice (Oriental Bio, Sungnam, Korea) as previously reported [11,28]. After intubation through the portal vein, the livers were perfused in situ with Ca2+-free Hank’s balanced saline solution at 37 °C for 15 min and then perfused with a solution containing 0.05% collagenase and Ca2+ for 15 min at a flow rate of 10 mL/min. The perfused livers were minced, filtered through a 70 m cell strainer (BD Biosciences), and centrifuged at 50× g for 3 min to separate the supernatant and pellet. The pellet was then discarded. Next, the supernatant was further centrifuged at 500× g for 10 min, resuspended in Ficoll plus Percoll (1:10; GE Healthcare, Chicago, IL, USA), and centrifuged at 1400× g for 17 min. HSCs were collected from the interface.
4.4. Animals
The animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Chosun University (Approval No. CIACUC2015-A0043, 22 December 2015). Male ICR mice (six weeks old) were obtained from Oriental Bio (Sung-nam, Korea) and acclimatized for 1 wk. Mice (n = 5/group) were housed at 20 ± 2 °C with 12 h light/dark cycle and a relative humidity of 50 ± 5% under filtered, pathogen-free air, with food (Purina, Korea) and water available ad libitum.
4.5. CCl4-Induced Hepatic Fibrosis
CCl4-induced hepatic fibrosis model was established as described previously [30]. To induce liver fibrosis, CCl4 dissolved in olive oil (10%) was intraperitoneally injected (0.5 mg/kg) into the mice thrice a week for 2 weeks, and NAOs dissolved in tap water were administered orally for 5 days per week (Figure 3A). The mice were induced by intraperitoneal injection with CCl4 24 h prior to sacrifice.
4.6. Blood Chemistry
Plasma ALT and AST were analyzed using Spectrum®, an automatic blood chemistry analyzer (Abbott Laboratories, Abbott Park, IL, USA).
4.7. Histological Process
Approximately equal regions of individual hepatic samples were crossly trimmed. All crossly trimmed hepatic tissues were refixed in 10% neutral buffered formalin for 24 h, at least in this histopathological observation. After paraffin embedding, 3–4 μm sections were prepared as three serial sections in each liver in paraffin blocks. Representative sections were stained with hematoxylin and eosin (H&E) for general histopathological profiles [31], Sirius red for collagen fiber [32], or Avidin-biotin-peroxidase complex (ABC)-based immunohistochemistry, against a profibrogenic cytokine involved in hepatic fibrosis–TGF-β (1:100) with its target signal molecule–pSmad3 (1:100), against α-SMA (1:100) and PAI-1 (1:100), according to previously established methods. Two histological fields in each hepatic tissue, totalling 10 histological fields in each group, were considered for further statistical analysis in the present histopathological observation. The percentage of degenerative regions (%/mm2) in livers showing centrilobular necrosis, congestion and inflammatory cell infiltrations on hepatic lobules were calculated using a computer-based automated image analyzer (iSolution FL ver 9.1, IMT i-solution Inc., Vancouver, Quebec, Canada) with collagen fiber-occupied region percentages around central veins noted as %/mm2 of hepatic parenchyma under Sirius red staining. The cells occupied by over 20% of immunoreactivities, the density of PAI-1, p-Smad3 and TGF-β1 were regarded as positive, and mean numbers of α-SMA, PAI-1, p-Smad3 and TGF-β1 immunopositive cells were calculated as cells/1000 hepatocytes using a computer-based image analyzer and histological camera system (Nikkon, Tokyo, Japan). The histopathologist was also blind to group distribution when this analysis was made.
4.8. MTT Assay
To measure cytotoxicity, cells were plated in 96-well plates, treated with the chemicals for 12 or 24 h, and stained with MTT (0.2 mg/mL, 4 h). The medium was then removed from the wells and formazan crystals in the wells were dissolved by adding 200 μL of dimethyl sulfoxide. Absorbance was measured at 540 nm using an enzyme-linked immunosorbent assay microplate reader (Versamax, Molecular Device, Sunnyvale, CA, USA). Cell viability was defined relative to the untreated control (viability [% control] = 100 × [absorbance of treated sample]/[absorbance of control]).
4.9. Immunoblot Analysis
Protein extraction, subcellular fractionation, SDS-polyacrylamide gel electrophoresis and immunoblot analyses were performed as described previously [33]. Briefly, samples were separated by 7.5% or 12% gel electrophoresis and electrophoretically transferred to nitrocellulose membranes. Each nitrocellulose membrane was incubated with the indicated primary antibody (PAI-1, 1:2000; α-SMA, 1:2000; CTGF, 1:200; Phospho-Smad3, 1:1000; Smad2/3, 1:1000 and β-actin, 1:2000) and then with horseradish peroxidase-conjugated secondary antibody (1:500). Immunoreactive proteins were visualized by ECL chemiluminescence (Amersham Biosciences, Buckinghamshire, UK). Equal protein loading was verified using β-actin.
4.10. RNA Isolation and Real-Time RT-PCR Analysis
Total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. To obtain cDNA, total RNA (2 µg) was reverse-transcribed using oligo(dT)16 primer. The cDNA obtained was amplified with a high-capacity cDNA synthesis kit (Bioneer, Daejon, Korea) using a thermal cycler (Bio-Rad, Hercules, CA, USA). Real-time PCR was performed with STEP ONE (Applied Biosystems, Foster City, CA, USA) using SYBR green premix according to the manufacturer’s instructions (Applied Biosystems). Primers were synthesized by Bioneer. The following primer sequences were used: human α-SMA 5’-CGCATCCTCATCCTCCCT-3’ (sense) and 5’-GGCCGTGATCTCCTTCTG-3’ (antisense); human PAI-1 5’-CGCCAGAGCAGGACGAA-3’ (sense) and 5’-CATCTGCATCCTGAAGTTCTCA-3’ (antisense); human Col 1A1 5’-CCTGGGTTTCAGAGACAACTTC-3’ (sense); mouse α-SMA 5’-TCCTCCCTGGAGAAGAGCTAC-3’ (sense) and 5’-TATAGGTGGTTTCGTGGATGC-3’ (antisense); mouse PAI-1 5’-GACACCCTCAGCATGTTCATC-3’ (sense) and 5’-AGGGTTGCACTAAACATGTCAG-3’ (antisense); mouse Col 1A1 5’-ACCTGTGTGTTCCCTACTCA-3’ (sense) and 5’-GACTGTTGCCTTCGCCTCTG-3’ (antisense); and 5’-TCCACATGCTTTATTCCAGCAATC-3’ (antisense). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control for RT-PCR.
4.11. Luciferase Gene Assay
To measure luciferase activity, LX-2 cells were replated in 24-well plates overnight, serum-starved for 6 h, and transiently transfected with SBE-luciferase and pRL-TK plasmids (which encode Renilla luciferase and are used to normalize transfection efficacy) in the presence of Lipofectamine® Reagent (Invitrogen, San Diego, CA, USA) for 3 h. Transfected cells were allowed to recover in DMEM for 3 h and then exposed to 1 μg/mL for 12 h. Firefly and Renilla luciferase activities in cell lysates were measured using the dual luciferase assay system (Promega) according to the manufacturer’s instructions. Relative luciferase activity was calculated by normalizing firefly luciferase activity to that of Renilla luciferase.
4.12. Statistical Analysis
One-way analysis of variance (ANOVA) was used to determine the significance of the differences between the treatment groups. The Newman–Keuls test was used to determine the significance of the differences between multiple group means. Results are expressed as mean ± S.E. p < 0.05 was considered statistically significant.
5. Conclusions
Collectively, the present study clearly shows that NAOs inhibited the expression of profibrotic genes induced by TGF-β in vitro. Furthermore, NAOs antagonized CCl4-induced collagen accumulation and synthesis of various ECM components in vivo. Our data strongly indicate that NAOs may be a promising therapeutic candidate to effectively prevent or treat chronic liver diseases via inhibition of HSC activation.
Supplementary Materials
The following are available online at https://www.mdpi.com/1422-0067/22/4/2041/s1.
Author Contributions
S.H.K., J.H.L. and C.-S.N. conceived and designed the experiments; J.H.Y. performed the in vivo and in vitro evaluation; S.K.K. and I.J.C. performed the histological analysis. S.H.K., C.-S.N. analyzed the data, and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Research Foundation of Korea (NRF) and funded by the Ministry of Science, ICT & Future Planning (No. 2018R1D1A1B07040460 and 2019R1A2C1004636).
Institutional Review Board Statement
The study was conducted according to the National Institute of Health Guidelines for the Care and Use of Laboratory Animals. All experiments were approved by the Institutional Animal Use and Care Committee at Chosun University (CIACUC2015-A0043).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; and in the decision to publish the results.
Abbreviations
| α-SMA | smooth muscle actin |
| ALT | Aminotransferase |
| AST | Aspartate aminotransferase |
| CCl4 | Carbon tetrachloride |
| CTGF | Connective tissue growth factor |
| ECM | Extracellular matrix |
| HSCs | Hepatic Stellate Cells |
| NAOs | Neoagarooligosaccharides |
| PAI-1 | Plasminogen activator inhibitor-1 |
| SBEs | Smad-binding elements |
| TGF-β | Transforming growth factor-β |
References
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Zheng, R.-Q.; Wang, Q.-H.; Lu, M.-D.; Xie, S.-B.; Ren, J.; Su, Z.-Z.; Cai, Y.-K.; Yao, J.-L. Liver fibrosis in chronic viral hepatitis: An ultrasonographic study. World J. Gastroenterol. 2003, 9, 2484–2489. [Google Scholar] [CrossRef]
- Hoffmann, C.; Djerir, N.E.H.; Danckaert, A.; Fernandes, J.; Roux, P.; Charrueau, C.; Lachagès, A.-M.; Charlotte, F.; Brocheriou, I.; Clément, K.; et al. Hepatic stellate cell hypertrophy is associated with metabolic liver fibrosis. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Senoo, H.; Mezaki, Y.; Fujiwara, M. The stellate cell system (vitamin A-storing cell system). Anat. Sci. Int. 2017, 92, 387–455. [Google Scholar] [CrossRef]
- Casini, A.; Pinzani, M.; Milani, S.; Grappone, C.; Galli, G.; Jezequel, A.M.; Schuppan, D.; Rotella, C.M.; Surrenti, C. Regulation of extracellular matrix synthesis by transforming growth factor beta 1 in human fat-storing cells. Gastroenterology 1993, 105, 245–253. [Google Scholar] [CrossRef]
- Kawarada, Y.; Inoue, Y.; Kawasaki, F.; Fukuura, K.; Sato, K.; Tanaka, T.; Itoh, Y.; Hayashi, H. TGF-β induces p53/Smads complex formation in the PAI-1 promoter to activate transcription. Sci. Rep. 2016, 6, 35483. [Google Scholar] [CrossRef] [PubMed]
- Kamato, D.; Burch, M.; Zhou, Y.; Mohamed, R.; Stow, J.L.; Osman, N.; Zheng, W.; Little, P.J. Individual Smad2 linker region phosphorylation sites determine the expression of proteoglycan and glycosaminoglycan synthesizing genes. Cell. Signal. 2019, 53, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Katoh, D.; Kozuka, Y.; Noro, A.; Ogawa, T.; Imanaka-Yoshida, K.; Yoshida, T. Tenascin-C Induces Phenotypic Changes in Fibroblasts to Myofibroblasts with High Contractility through the Integrin αvβ1/Transforming Growth Factor β/SMAD Signaling Axis in Human. Breast Cancer Am. J. Pathol. 2020, 190, 2123–2135. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Chen, Y.; Suo, L.; Chen, H.; Zhu, L.; Wan, G.; Han, X. Activin a promotes myofibroblast differentiation of endometrial mesenchymal stem cells via STAT3-dependent Smad/CTGF pathway. Cell Commun. Signal. 2019, 17, 45. [Google Scholar] [CrossRef]
- Gao, W.; Lin, P.; Hwang, E.; Wang, Y.; Yan, Z.; Ngo, H.T.; Yi, T. Pterocarpus santalinus L. Regulated Ultraviolet B Irradiation-induced Procollagen Reduction and Matrix Metalloproteinases Expression Through Activation of TGF-β /Smad and Inhibition of the MAPK/AP-1 Pathway in Normal Human Dermal Fibroblasts. Photochem. Photobiol. 2017, 94, 139–149. [Google Scholar] [CrossRef]
- Yang, J.H.; Kim, S.C.; Kim, K.M.; Jang, C.H.; Cho, S.S.; Kim, S.J.; Ku, S.K.; Cho, I.J.; Ki, S.H. Isorhamnetin attenuates liver fibrosis by inhibiting TGF-β/Smad signaling and relieving oxidative stress. Eur. J. Pharmacol. 2016, 783, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Jia, L.; Xiao, S. Dicliptera Chinensis polysaccharides target TGF-β/Smad pathway and inhibit stellate cells activation in rats with dimethylnitrosamine-induced hepatic fibrosis. Cell Mol. Biol. 2016, 62, 99–103. [Google Scholar] [PubMed]
- Yang, J.H.; Cho, S.S.; Kim, K.M.; Kim, J.Y.; Kim, E.J.; Park, E.Y.; Lee, J.H.; Ki, S.H. Neoagarooligosaccharides enhance the level and efficiency of LDL receptor and improve cholesterol homeostasis. J. Funct. Foods 2017, 38, 529–539. [Google Scholar] [CrossRef]
- Yang, J.H.; Na, C.-S.; Cho, S.S.; Kim, K.M.; Lee, J.H.; Chen, X.-Q.; Ku, S.K.; Cho, I.J.; Kim, E.J.; Lee, J.H.; et al. Hepatoprotective Effect of Neoagarooligosaccharide via Activation of Nrf2 and Enhanced Antioxidant Efficacy. Biol. Pharm. Bull. 2020, 43, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.J.; Lee, J.-H.; Kim, E.J.; Yang, H.J.; Park, J.-S.; Hong, S.-K. Toxicological evaluation of neoagarooligosaccharides prepared by enzymatic hydrolysis of agar. Regul. Toxicol. Pharmacol. 2017, 90, 9–21. [Google Scholar] [CrossRef]
- Scholten, D.; Trebicka, J.; Liedtke, C.; Weiskirchen, R. The carbon tetrachloride model in mice. Lab. Anim. 2015, 49, 4–11. [Google Scholar] [CrossRef]
- Dong, S.; Chen, Q.-L.; Song, Y.-N.; Sun, Y.; Wei, B.; Li, X.-Y.; Hu, Y.-Y.; Liu, P.; Su, S.-B. Mechanisms of CCl4-induced liver fibrosis with combined transcriptomic and proteomic analysis. J. Toxicol. Sci. 2016, 41, 561–572. [Google Scholar] [CrossRef]
- Carpino, G.; Morini, S.; Corradini, S.G.; Franchitto, A.; Merli, M.; Siciliano, M.; Gentili, F.; Muda, A.O.; Berloco, P.B.; Rossi, M. Alpha-SMA expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig. Liver Dis. 2005, 37, 349–356. [Google Scholar] [CrossRef]
- Cordero-Espinoza, L.; Huch, M. The balancing act of the liver: Tissue regeneration versus fibrosis. J. Clin. Investig. 2018, 128, 85–96. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic fibrosis—Overview. Toxicology 2008, 254, 120–129. [Google Scholar] [CrossRef]
- Geervliet, E.; Bansal, R. Matrix Metalloproteinases as Potential Biomarkers and Therapeutic Targets in Liver Diseases. Cells 2020, 9, 1212. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-β in Hepatic Stellate Cell Activation and Liver Fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.S.; Mokhtari, R.B.; El Hout, Y.; Azadi, M.A.; Alauddin, M.; Yeger, H.; Farhat, W.A. TGF-β1 induces EMT reprogramming of porcine bladder urothelial cells into collagen producing fibroblasts-like cells in a Smad2/Smad3-dependent manner. J. Cell Commun. Signal. 2013, 8, 39–58. [Google Scholar] [CrossRef]
- Huang, F.; Chen, Y.-G. Regulation of TGF-β receptor activity. Cell Biosci. 2012, 2, 9. [Google Scholar] [CrossRef]
- Frederick, J.P.; Liberati, N.T.; Waddell, D.S.; Shi, Y.; Wang, X.-F. Transforming Growth Factor β-Mediated Transcriptional Repression of c-myc Is Dependent on Direct Binding of Smad3 to a Novel Repressive Smad Binding Element. Mol. Cell. Biol. 2004, 24, 2546–2559. [Google Scholar] [CrossRef] [PubMed]
- Al-Tamimi, J.; Alhazza, I.M.; Al-Khalifa, M.; Metwalli, A.; Rady, A.; Ebaid, H. Potential effects of samsum ant, Brachyponera sennaarensis, venom on TNF-α/NF-κB mediated inflammation in CCl4-toxicity in vivo. Lipids Health Dis. 2016, 15, 198. [Google Scholar] [CrossRef]
- Goodla, L.; Manubolu, M.; Pathakoti, K.; Jayakumar, T.; Sheu, J.-R.; Fraker, M.; Tchounwou, P.B.; Poondamalli, P.R. Protective Effects of Ammannia baccifera against CCl4-Induced Oxidative Stress in Rats. Int. J. Environ. Res. Public Health 2019, 16, 1440. [Google Scholar] [CrossRef]
- Yang, J.H.; Kim, K.M.; Cho, S.S.; Shin, S.M.; Ka, S.O.; Na, C.-S.; Park, B.H.; Jegal, K.H.; Kim, J.K.; Ku, S.K.; et al. Inhibitory Effect of Sestrin 2 on Hepatic Stellate Cell Activation and Liver Fibrosis. Antioxid. Redox Signal. 2019, 31, 243–259. [Google Scholar] [CrossRef]
- Raven, A.; Lu, W.-Y.; Man, T.Y.; Ferreira-Gonzalez, S.; O’Duibhir, E.; Dwyer, B.J.; Thomson, J.P.; Meehan, R.R.; Bogorad, R.; Koteliansky, V.; et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 2017, 547, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Ki, S.H.; Yang, J.H.; Ku, S.K.; Kim, S.C.; Kim, Y.W.; Cho, I.J. Red ginseng extract protects against carbon tetrachloride-induced liver fibrosis. J. Ginseng Res. 2013, 37, 45–53. [Google Scholar] [CrossRef]
- Knockaert, L.; Berson, A.; Ribault, C.; Prost, P.-E.; Fautrel, A.; Pajaud, J.; Lepage, S.; Lucas-Clerc, C.; Bégué, J.-M.; Fromenty, B.; et al. Carbon tetrachloride-mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Lab. Investig. 2011, 92, 396–410. [Google Scholar] [CrossRef]
- Tipoe, G.L.; Leung, T.M.; Liong, E.C.; Lau, T.Y.H.; Fung, M.L.; Nanji, A.A. Epigallocatechin-3-gallate (EGCG) reduces liver inflammation, oxidative stress and fibrosis in carbon tetrachloride (CCl4)-induced liver injury in mice. Toxicology 2010, 273, 45–52. [Google Scholar] [CrossRef]
- Shin, B.Y.; Jin, S.H.; Cho, I.J.; Ki, S.H. Nrf2-ARE pathway regulates induction of Sestrin-2 expression. Free. Radic. Biol. Med. 2012, 53, 834–841. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).