
Metabolic Factors Affecting Tumor Immunogenicity: What Is Happening at the Cellular Level?





Abstract
:1. Introduction
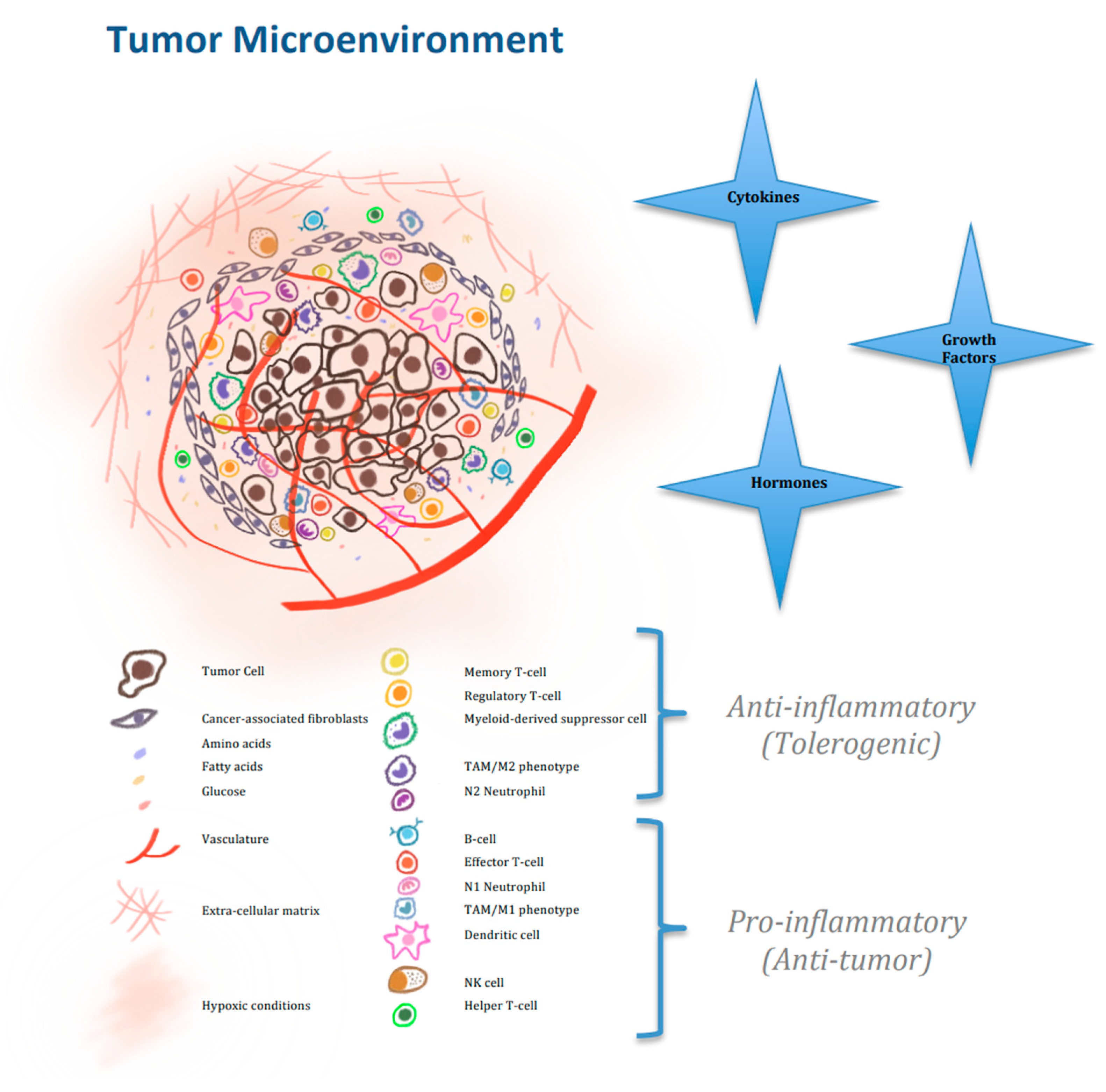
2. Nutrients Affecting the Cellular Activity of Immune Cells in the Tumor Microbiome
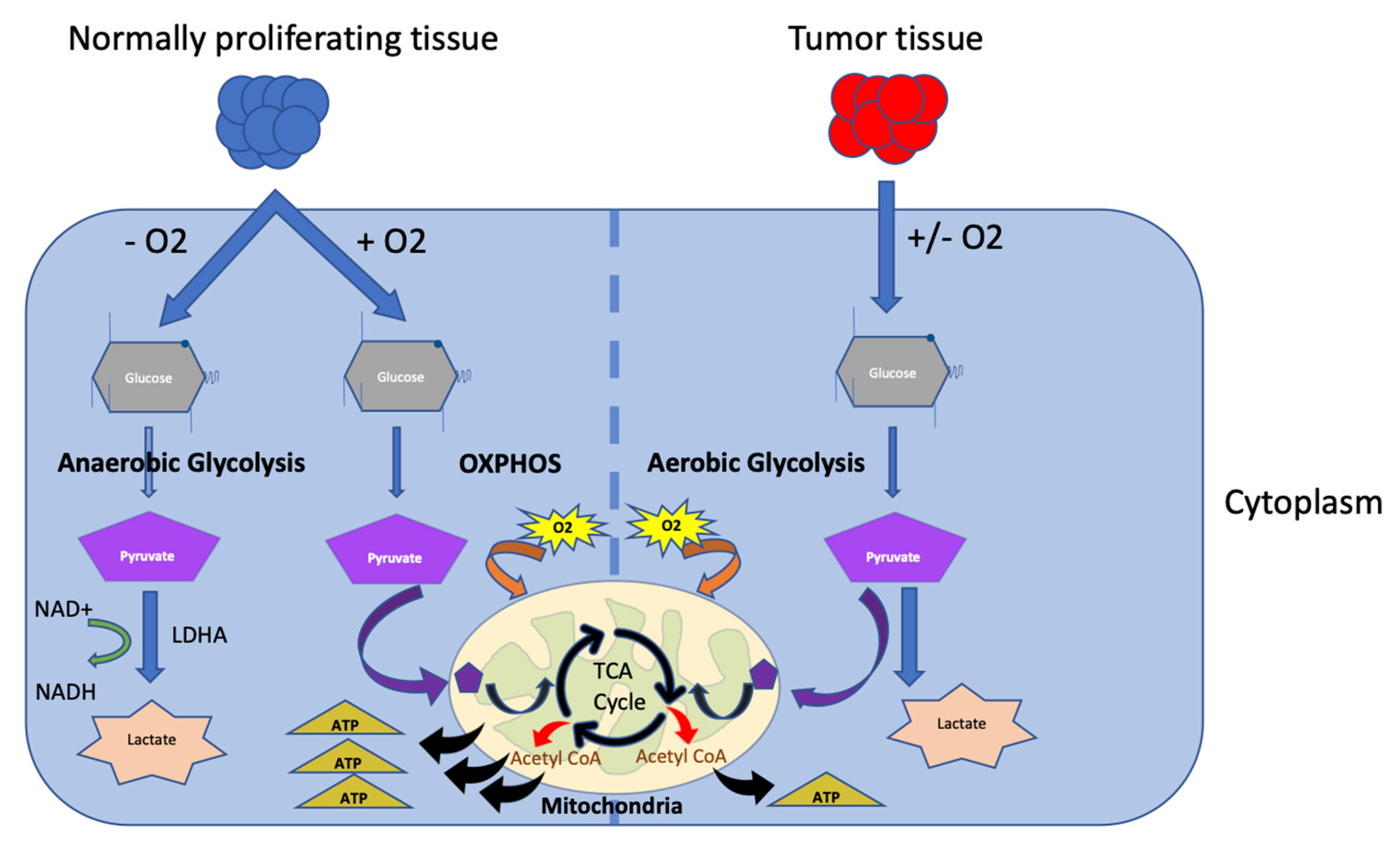
2.1. Glucose Metabolism
2.2. Amino-Acid Metabolism
2.3. Lipid and Fatty Acid Metabolism
3. Mechanisms and Pathways Modulating Metabolism and Affecting Cellular Activity of Immune Cells in TME
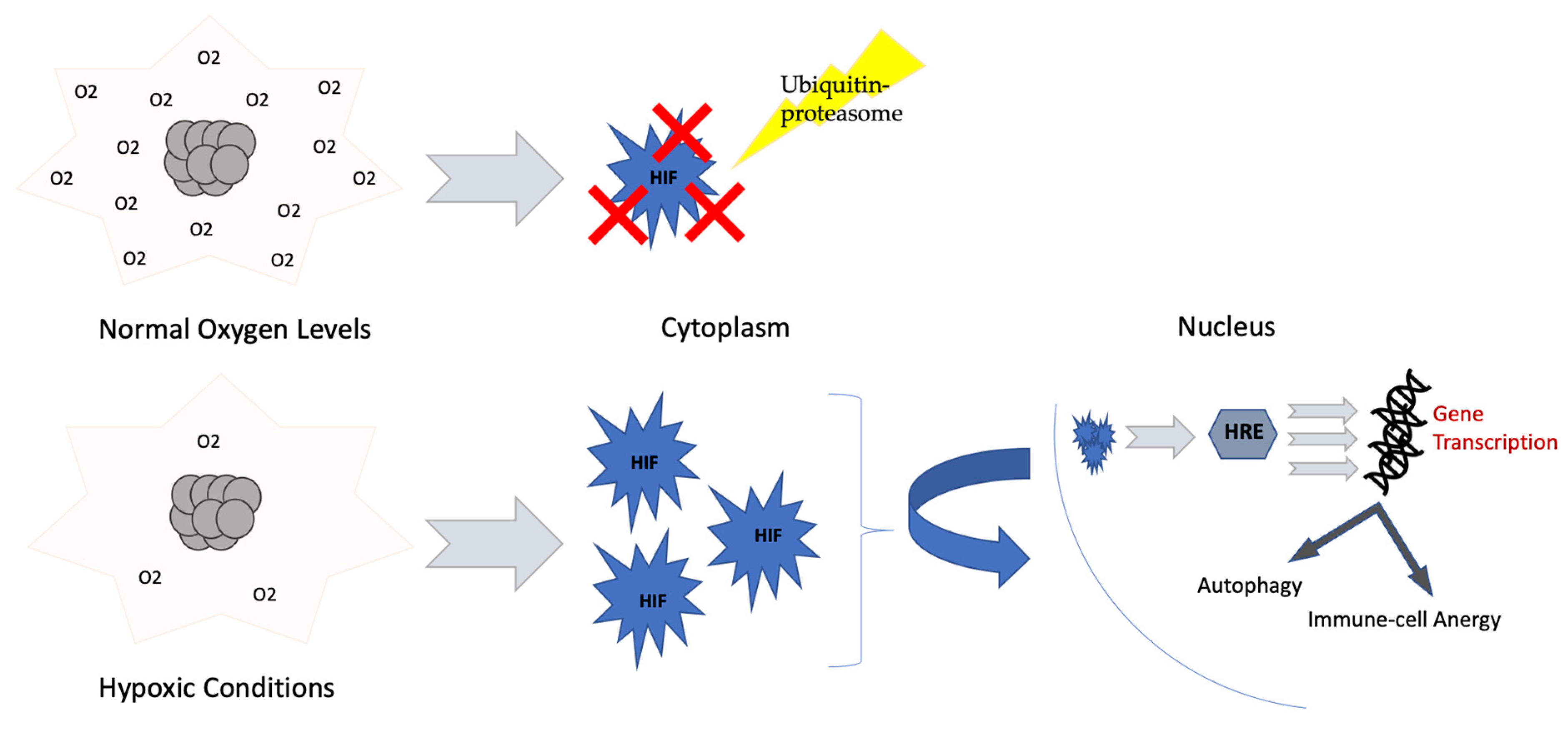
3.1. Hypoxia, HIF-1 α and ROS
3.2. Pyruvate Kinase Muscle Splicing
3.3. Adenosine Pathway (A2A Stimulation)
3.4. Lactic Acidosis
3.5. Concept of Extracellular Vesicles
4. Other Mechanisms of Adaptation of Immune Response
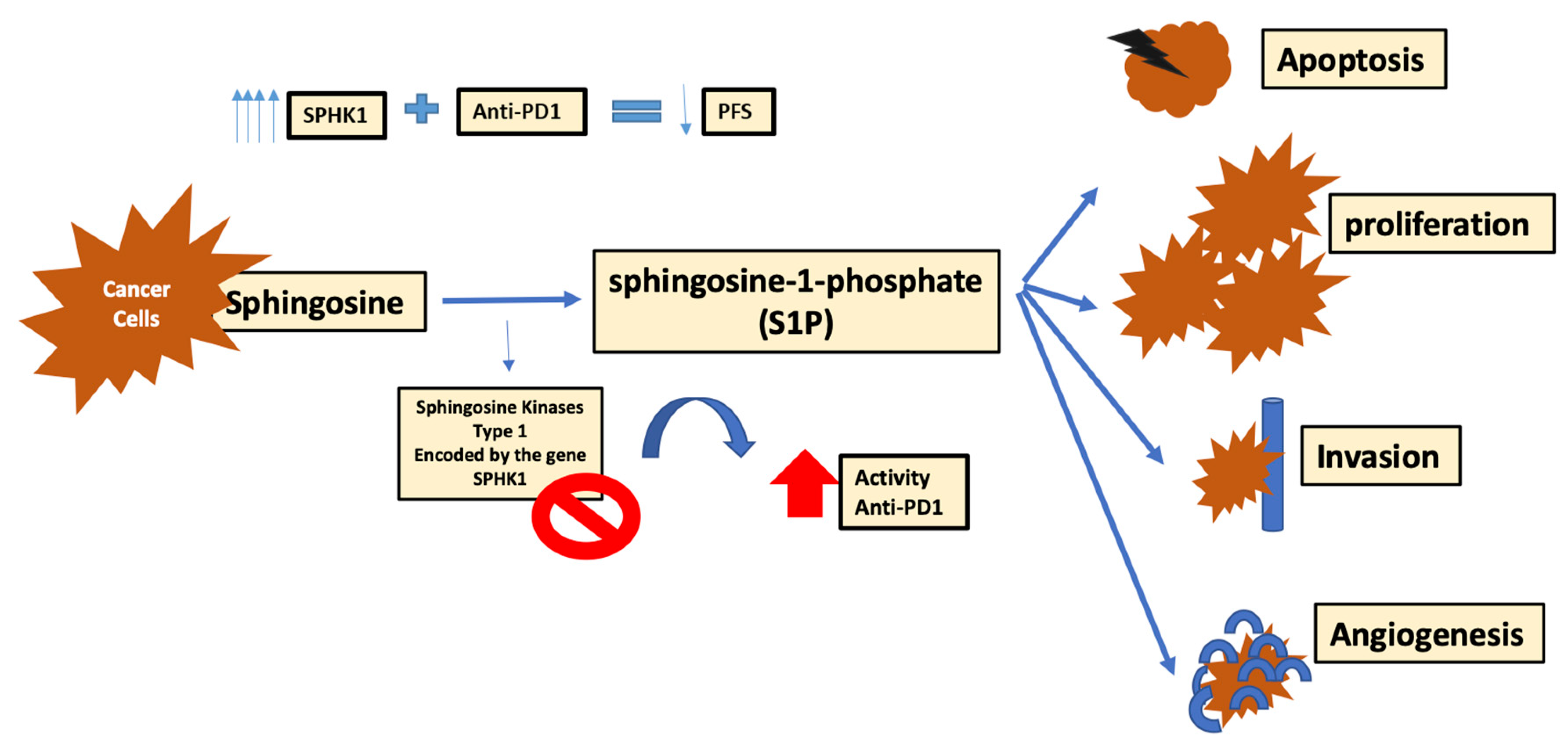
4.1. Role of Sphingosine Kinase-1
4.2. Role of MUC-1 Mucin
4.3. Effect of Acetyl-CoA Carboxylase 1(ACC1), and Mitochondrial Involvement
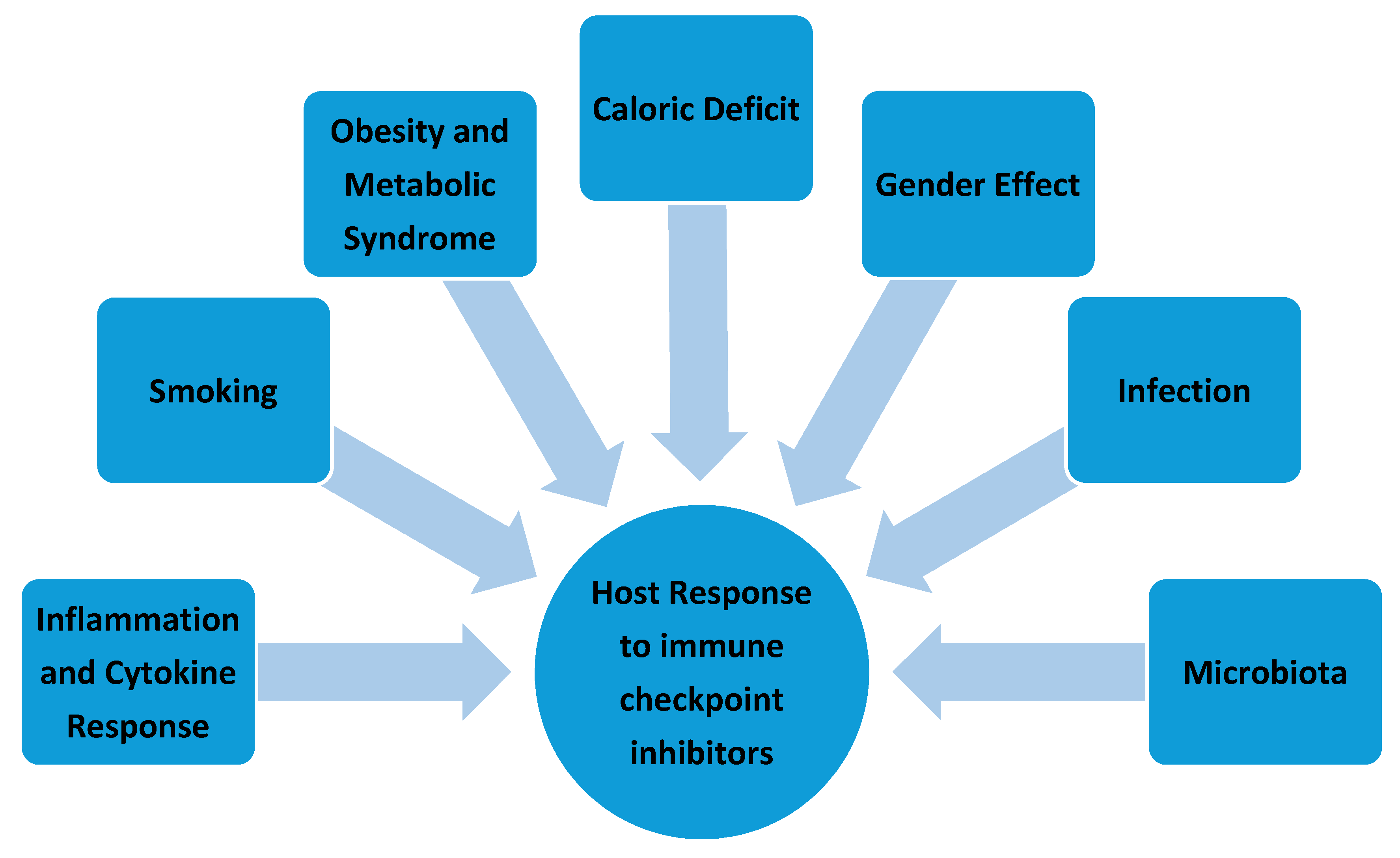
5. Effect of Environmental Factors at the Host Level
5.1. Cytokine Release: Inflammation and Autoimmune Response
5.2. Obesity and Metabolic Syndrome
5.3. Caloric Deficit
5.4. Gender Effect
5.5. Infection
5.6. Microbiota
5.7. Smoking
6. Metabolic Manipulation at the Genetic and Pharmacologic Levels
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACT | Adoptive cell therapy |
| ATP | Adenosine triphosphate |
| ADA | Adenosine deaminase |
| ADK | Adenylate kinase |
| AGK | Acyl glycerol kinase |
| ASS1 | Argininosuccinate Synthase 1 |
| ABHD5 | Abhydrolase-Domain-Containing 5 |
| ACC1 | Acetyl-CoA Carboxylase 1 |
| Bhlhe40 | Bhlhe40: Basic helix-loop-helix-family member e40 |
| BAZ1B | BAZ1B: Bromodomain adjacent to zinc finger domain 1B |
| Bcl-2 | Bcl-2: B-cell lymphoma-2 |
| BMDCs | BMDCs: Bone-marrow-DCs |
| CTL | Cytotoxic T-cells |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| CPI | Checkpoint inhibitors |
| CAF | Cancer-Associated Fibroblasts |
| CRC | Colorectal Cancer |
| CCR-6 | C-C Motif Chemokine Receptor 6 |
| CCL20 | C-C Motif Chemokine Ligand 20 |
| DC | Dendritic Cells |
| ECM | Extracellular matrix |
| ETC | Electron transport chain |
| EZH2 | Zeste methyltransferase enhancer homolog 2 |
| EMT | Epithelial-to-mesenchymal transition |
| EFABP | Epidermal Fatty Acid Binding Protein |
| ENT | Ectonucleotidases |
| EV | Extracellular vesicles |
| EC | Extracellular Domain |
| eATP | Extracellular ATP |
| FAS | Fatty acid biosynthesis |
| FAO | Fatty acid oxidation |
| FASN/SCD1 | Fatty FA synthase and stearoyl-CoA desaturase-1 |
| GSK3 | Glycogen Synthase Kinase-3 |
| Hmgcs1/Acat2 | 3-hydroxy-3-methylglutaryl-CoA synthase/acetyl-CoA acetyl-transferase 2 |
| hnRNP | Heterogeneous nuclear ribonucleoproteins |
| HIF-1α | Hypoxia-inducible factor 1α |
| HRE | Hypoxia-Responsive Element |
| Hh | Hedgehog |
| ITZ | Itraconazole |
| IDA | Indoleamine 2,3-dioxygenase |
| iNOSs | inducible NO synthase |
| IFNAR1 | Interferon-alpha/beta receptor alpha chain |
| Kif7 | Kinesin protein |
| LDHA | Lactate Dehydrogenase A |
| 14LDM | Lanosterol 14-α-Demethylase |
| MDSCs | Myeloid-derived suppressor cells |
| NSCLC | Non-small cell lung cancer |
| NO | Nitric oxide |
| NK | Natural Killer |
| OXPHOS | Oxidative phosphorylation |
| OTC | Ornithine transcarbamylase |
| PK | Pyruvate kinase |
| PKA | Protein kinase A |
| PMN-MDSCs | Polymorphonuclear MDSCs |
| PTB | Polypyrimidine tract binding protein |
| PEP | Phosphoenolpyruvate |
| PD-1 | Programmed cell death protein-1 |
| PDL-1 | Programmed cell-death protein ligand-1 |
| PCK1 | Phosphoenolpyruvate carboxykinase 1 |
| PSIP1 | PC4 and SFRS1 interacting protein 1 |
| PGC-1β | Peroxisome proliferator-activated receptor Gamma Coactivator-1 beta |
| PEM | Polymorphic Epithelial Mucin |
| RCC | Renal cell carcinoma |
| ROS | Reactive Oxygen Species |
| SUFU | Suppressor of Fused |
| SRC | Spare Respiratory Capacity |
| S1P | Sphingosine-1-Phosphate |
| SK | Sphingosine kinases |
| SR-A1 | Scavenger Receptor A |
| SERCA | Sarco/ER Ca2+-ATPase |
| SREBPs | Sterol regulatory element binding proteins |
| SMO | Smoothened receptor |
| TME | Tumor microenvironment |
| T-regs | Regulatory T-cells |
| TAMs | Tumor-associated macrophages |
| TCA cycle | Tricarboxylic cycle |
| Trp | Tryptophan |
| Trp–Kyn–AhR | Tryptophan–Kynurenine–Aryl hydrocarbon receptor |
| TLR | Toll-like receptor |
References
- Granier, C.; De Guillebon, E.; Blanc, C.; Roussel, H.; Badoual, C.; Colin, E.; Saldmann, A.; Gey, A.; Oudard, S.; Tartour, E. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open 2017, 2, e000213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothlin, C.V.; Ghosh, S. Lifting the innate immune barriers to antitumor immunity. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Robert, C. A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 2020, 11, 1–3. [Google Scholar] [CrossRef]
- Einsele, H.; Briones, J.; Ciceri, F.; García-Cadenas, I.; Falkenburg, F.; Bolaños, N.; Heemskerk, H.M.; Houot, R.; Hudecek, M.; Locatelli, F. Immune-Based Therapies for Hematological Malignancies: An Update by the EHA SWG on Immunotherapy of Hematological Malignancies. HemaSphere 2020, 4, e423. [Google Scholar] [CrossRef]
- Carlo, M.I.; Voss, M.H.; Motzer, R.J. Checkpoint inhibitors and other novel immunotherapies for advanced renal cell carcinoma. Nat. Rev. Urol. 2016, 13, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queirolo, P.; Boutros, A.; Tanda, E.; Spagnolo, F.; Quaglino, P. Immune-Checkpoint inhibitors for the treatment of metastatic melanoma: A model of cancer immunotherapy. Semin. Cancer Biol. 2019, 59, 290–297. [Google Scholar] [CrossRef]
- Zimmermann, S.; Peters, S.; Owinokoko, T.; Gadgeel, S.M. Immune checkpoint inhibitors in the management of lung cancer. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 682–695. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Nair, V.S.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chew, V.; Toh, H.C.; Abastado, J.-P. Immune microenvironment in tumor progression: Characteristics and challenges for therapy. J. Oncol. 2012, 2012, 608406. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Liu, Y.; Guo, J.; Huang, L. Modulation of tumor microenvironment for immunotherapy: Focus on nanomaterial-based strategies. Theranostics 2020, 10, 3099. [Google Scholar] [CrossRef] [PubMed]
- Cassim, S.; Pouyssegur, J. Tumor microenvironment: A metabolic player that shapes the immune response. Int. J. Mol. Sci. 2020, 21, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Renner, K.; Singer, K.; Koehl, G.; Geissler, E.; Peter, K.; Siska, P.; Kreutz, M. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front. Immunol. 2017, 8, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouirand, V.; Guillaumond, F.; Vasseur, S. Influence of the tumor microenvironment on cancer cells metabolic reprogramming. Front. Oncol. 2018, 8, 117. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S. Molecular intricacies of aerobic glycolysis in cancer: Current insights into the classic metabolic phenotype. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 667–682. [Google Scholar] [CrossRef] [PubMed]
- Salmond, R.J. mTOR regulation of glycolytic metabolism in T-cells. Front. Cell Dev. Biol. 2018, 6, 122. [Google Scholar] [CrossRef]
- Scholz, G.; Jandus, C.; Zhang, L.; Grandclément, C.; Lopez-Mejia, I.C.; Soneson, C.; Delorenzi, M.; Fajas, L.; Held, W.; Dormond, O. Modulation of mTOR signalling triggers the formation of stem cell-like memory T-cells. EBioMedicine 2016, 4, 50–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, K.; Youngblood, B.; Ahmed, R. The role of mTOR in memory CD8+ T-cell differentiation. Immunol. Rev. 2010, 235, 234–243. [Google Scholar] [CrossRef]
- Coloff, J.L.; Macintyre, A.N.; Nichols, A.G.; Liu, T.; Gallo, C.A.; Plas, D.R.; Rathmell, J.C. Akt-Dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res. 2011, 71, 5204–5213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, L.; Xu, K.; Wei, Y.; Zhang, J.; Han, T.; Fry, C.; Zhang, Z.; Wang, Y.V.; Huang, L.; Yuan, M. Phosphorylation of EZH2 by AMPK suppresses PRC2 methyltransferase activity and oncogenic function. Mol. Cell 2018, 69, 279–291.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Sharma, S.; Rollins, R.A.; Paul, T.A. The complex role of EZH2 in the tumor microenvironment: Opportunities and challenges for immunotherapy combinations. Future Med. Chem. 2020, 12, 1415–1430. [Google Scholar] [CrossRef]
- Cham, C.M.; Driessens, G.; O’Keefe, J.P.; Gajewski, T.F. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T-cells. Eur. J. Immunol. 2008, 38, 2438–2450. [Google Scholar] [CrossRef] [Green Version]
- Van der Windt, G.J.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Zin, A.A.M.; Ang, K.C.; Ch’ng, E.S. Evaluating the polarization of tumor-associated macrophages into M1 and M2 phenotypes in human cancer tissue: Technicalities and challenges in routine clinical practice. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Karimi Roshan, M.; Soltani, A.; Soleimani, A.; Rezaie Kahkhaie, K.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie 2019, 165, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Chen, M.; Yan, B.; He, X.; Chen, X.; Li, D. Identification of a role for the PI3K/AKT/mTOR signaling pathway in innate immune cells. PLoS ONE 2014, 9, e94496. [Google Scholar] [CrossRef]
- Sottnik, J.L.; Lori, J.C.; Rose, B.J.; Thamm, D.H. Glycolysis inhibition by 2-deoxy-D-glucose reverts the metastatic phenotype in vitro and in vivo. Clin. Exp. Metastasis 2011, 28, 865–875. [Google Scholar] [CrossRef]
- Terrén, I.; Orrantia, A.; Vitallé, J.; Zenarruzabeitia, O.; Borrego, F. NK cell metabolism and tumor microenvironment. Front. Immunol. 2019, 10, 2278. [Google Scholar] [CrossRef]
- Maratou, E.; Dimitriadis, G.; Kollias, A.; Boutati, E.; Lambadiari, V.; Mitrou, P.; Raptis, S. Glucose transporter expression on the plasma membrane of resting and activated white blood cells. Eur. J. Clin. Investig. 2007, 37, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Assmann, N.; O’Brien, K.L.; Donnelly, R.P.; Dyck, L.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Heinrich, P.; Oefner, P.J.; Lynch, L.; Gardiner, C.M. Srebp-Controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol. 2017, 18, 1197. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.L.; Finlay, D.K. Immunometabolism and natural killer cell responses. Nat. Rev. Immunol. 2019, 19, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Slattery, K.; Gardiner, C.M. NK cell metabolism and TGFβ-implications for immunotherapy. Front. Immunol. 2019, 10, 2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, J.P.; Amiel, E. Regulation of dendritic cell immune function and metabolism by cellular nutrient sensor mammalian target of rapamycin (mTOR). Front. Immunol. 2019, 9, 3145. [Google Scholar] [CrossRef] [Green Version]
- Guak, H.; Al Habyan, S.; Ma, E.H.; Aldossary, H.; Al-Masri, M.; Won, S.Y.; Ying, T.; Fixman, E.D.; Jones, R.G.; McCaffrey, L.M. Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christofi, M.; Le Sommer, S.; Mölzer, C.; Klaska, I.; Kuffova, L.; Forrester, J. Low-dose 2-deoxy glucose stabilises tolerogenic dendritic cells and generates potent in vivo immunosuppressive effects. Cell. Mol. Life Sci. 2020, 1–20. [Google Scholar] [CrossRef]
- Ananieva, E. Targeting amino acid metabolism in cancer growth and anti-tumor immune response. World J. Biol. Chem. 2015, 6, 281. [Google Scholar] [CrossRef]
- Brochez, L.; Chevolet, I.; Kruse, V. The rationale of indoleamine 2, 3-dioxygenase inhibition for cancer therapy. Eur. J. Cancer 2017, 76, 167–182. [Google Scholar] [CrossRef]
- Giannone, G.; Ghisoni, E.; Genta, S.; Scotto, G.; Tuninetti, V.; Turinetto, M.; Valabrega, G. Immuno-Metabolism and microenvironment in cancer: Key players for immunotherapy. Int. J. Mol. Sci. 2020, 21, 4414. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.; Puccetti, P. T-cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T-cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornyák, L.; Dobos, N.; Koncz, G.; Karányi, Z.; Páll, D.; Szabó, Z.; Halmos, G.; Székvölgyi, L. The role of indoleamine-2, 3-dioxygenase in cancer development, diagnostics, and therapy. Front. Immunol. 2018, 9, 151. [Google Scholar] [CrossRef]
- Harden, J.L.; Egilmez, N.K. Indoleamine 2, 3-dioxygenase and dendritic cell tolerogenicity. Immunol. Investig. 2012, 41, 738–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saravia, J.; Raynor, J.L.; Chapman, N.M.; Lim, S.A.; Chi, H. Signaling networks in immunometabolism. Cell Res. 2020, 30, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ganeshan, K.; Chawla, A. Metabolic regulation of immune responses. Annu. Rev. Immunol. 2014, 32, 609–634. [Google Scholar] [CrossRef] [Green Version]
- Rivadeneira, D.B.; Delgoffe, G.M. Antitumor T-cell reconditioning: Improving metabolic fitness for optimal cancer immunotherapy. Clin. Cancer Res. 2018, 24, 2473–2481. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W. Pharmacologic or genetic targeting of glutamine synthetase skews macrophages toward an M1-like phenotype and inhibits tumor metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M. L-Arginine modulates T-cell metabolism and enhances survival and anti-tumor activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [Green Version]
- Huber, J.P.; David Farrar, J. Regulation of effector and memory T-cell functions by type I interferon. Immunology 2011, 132, 466–474. [Google Scholar] [CrossRef]
- Kim, S.-H.; Roszik, J.; Grimm, E.A.; Ekmekcioglu, S. Impact of l-arginine metabolism on immune response and anticancer immunotherapy. Front. Oncol. 2018, 8, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terrén, I.; Orrantia, A.; Vitallé, J.; Astarloa-Pando, G.; Zenarruzabeitia, O.; Borrego, F. Modulating NK cell metabolism for cancer immunotherapy. Semin. Hematol. 2020, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Van Dalen, F.J.; Van Stevendaal, M.H.; Fennemann, F.L.; Verdoes, M.; Ilina, O. Molecular repolarisation of tumour-associated macrophages. Molecules 2019, 24, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkholder, B.; Huang, R.-Y.; Burgess, R.; Luo, S.; Jones, V.S.; Zhang, W.; Lv, Z.-Q.; Gao, C.-Y.; Wang, B.-L.; Zhang, Y.-M. Tumor-Induced perturbations of cytokines and immune cell networks. Biochim. Biophys. Acta (BBA) Rev. Cancer 2014, 1845, 182–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleve, A.; Durante, B.; Sica, A.; Consonni, F.M. Lipid Metabolism and Cancer Immunotherapy: Immunosuppressive Myeloid Cells at the Crossroad. Int. J. Mol. Sci. 2020, 21, 5845. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The metabolic signature of macrophage responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Puthenveetil, A.; Dubey, S. Metabolic reprograming of tumor-associated macrophages. Ann. Transl. Med. 2020, 8, 1030. [Google Scholar] [CrossRef]
- Remmerie, A.; Scott, C.L. Macrophages and lipid metabolism. Cell. Immunol. 2018, 330, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, J.E.; Bagdasarian, A.; Colman, R.W. Functional alpha1 protease inhibitor produced by a human hepatoma cell line. J. Lab. Clin. Med. 1982, 99, 108–117. [Google Scholar]
- Zhang, Y.; Sun, Y.; Rao, E.; Yan, F.; Li, Q.; Zhang, Y.; Silverstein, K.A.; Liu, S.; Sauter, E.; Cleary, M.P. Fatty acid-binding protein E-FABP restricts tumor growth by promoting IFN-β responses in tumor-associated macrophages. Cancer Res. 2014, 74, 2986–2998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusmartsev, S. Enhanced 15-lipoxygenase activity and elevated eicosanoid production in kidney tumor microenvironment contribute to the inflammation and immune suppression. Oncoimmunology 2012, 1, 249–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daurkin, I.; Eruslanov, E.; Stoffs, T.; Perrin, G.Q.; Algood, C.; Gilbert, S.M.; Rosser, C.J.; Su, L.-M.; Vieweg, J.; Kusmartsev, S. Tumor-associated macrophages mediate immunosuppression in the renal cancer microenvironment by activating the 15-lipoxygenase-2 pathway. Cancer Res. 2011, 71, 6400–6409. [Google Scholar] [CrossRef] [Green Version]
- Miao, H.; Ou, J.; Peng, Y.; Zhang, X.; Chen, Y.; Hao, L.; Xie, G.; Wang, Z.; Pang, X.; Ruan, Z. Macrophage ABHD5 promotes colorectal cancer growth by suppressing spermidine production by SRM. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Tang, Y.Q.; Miao, H. Metabolism in tumor microenvironment: Implications for cancer immunotherapy. MedComm 2020, 47–68. [Google Scholar] [CrossRef]
- Yin, Z.; Bai, L.; Li, W.; Zeng, T.; Tian, H.; Cui, J. Targeting T-cell metabolism in the tumor microenvironment: An anti-cancer therapeutic strategy. J. Exp. Clin. Cancer Res. 2019, 38, 403. [Google Scholar] [CrossRef] [PubMed]
- Bietz, A.; Zhu, H.; Xue, M.; Xu, C. Cholesterol metabolism in T-cells. Front. Immunol. 2017, 8, 1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Reynolds, J.M.; Stout, R.D.; Bernlohr, D.A.; Suttles, J. Regulation of Th17 Differentiation by Epidermal Fatty Acid-Binding Protein. J. Immunol. 2009, 182, 7625–7633. [Google Scholar] [CrossRef] [Green Version]
- Fernández, L.P.; Gómez de Cedrón, M.; Ramírez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420. [Google Scholar] [CrossRef]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid-and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef]
- Kim, K.-Y.; Kim, J.K.; Han, S.H.; Lim, J.-S.; Kim, K.I.; Cho, D.H.; Lee, M.-S.; Lee, J.-H.; Yoon, D.-Y.; Yoon, S.R. Adiponectin is a negative regulator of NK cell cytotoxicity. J. Immunol. 2006, 176, 5958–5964. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Chapman, N.M.; Chi, H. Emerging roles of cellular metabolism in regulating dendritic cell subsets and function. Front. Cell Dev. Biol. 2018, 6, 152. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S. Paracrine Wnt5a-β-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity 2018, 48, 147–160.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.-I.; Celis, E.; Lennox, B. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880. [Google Scholar] [CrossRef] [Green Version]
- Shannon, A.M.; Bouchier-Hayes, D.J.; Condron, C.M.; Toomey, D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat. Rev. 2003, 29, 297–307. [Google Scholar] [CrossRef]
- Lequeux, A.; Noman, M.Z.; Xiao, M.; Sauvage, D.; Van Moer, K.; Viry, E.; Bocci, I.; Hasmim, M.; Bosseler, M.; Berchem, G. Impact of hypoxic tumor microenvironment and tumor cell plasticity on the expression of immune checkpoints. Cancer Lett. 2019, 458, 13–20. [Google Scholar] [CrossRef]
- Semenza, G.L. A compendium of proteins that interact with HIF-1α. Exp. Cell Res. 2017, 356, 128–135. [Google Scholar] [CrossRef]
- Kadomoto, S.; Izumi, K.; Mizokami, A. The CCL20-CCR6 Axis in Cancer Progression. Int. J. Mol. Sci. 2020, 21, 5186. [Google Scholar] [CrossRef]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarosz, E.L.; Chang, C.-H. The role of reactive oxygen species in regulating T-cell-mediated immunity and disease. Immune Netw. 2018, 18, e14. [Google Scholar] [CrossRef] [PubMed]
- Rashida Gnanaprakasam, J.N.; Wu, R.; Wang, R. Metabolic reprogramming in modulating T-cell reactive oxygen species generation and antioxidant capacity. Front. Immunol. 2018, 9, 1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frossi, B.; De Carli, M.; Piemonte, M.; Pucillo, C. Oxidative microenvironment exerts an opposite regulatory effect on cytokine production by Th1 and Th2 cells. Mol. Immunol. 2008, 45, 58–64. [Google Scholar] [CrossRef]
- Fu, G.; Xu, Q.; Qiu, Y.; Jin, X.; Xu, T.; Dong, S.; Wang, J.; Ke, Y.; Hu, H.; Cao, X. Suppression of Th17 cell differentiation by misshapen/NIK-related kinase MINK1. J. Exp. Med. 2017, 214, 1453–1469. [Google Scholar] [CrossRef]
- Kesarwani, P.; Murali, A.K.; Al-Khami, A.A.; Mehrotra, S. Redox regulation of T-cell function: From molecular mechanisms to significance in human health and disease. Antioxid. Redox Signal. 2013, 18, 1497–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsafti, A.; Scarpa, M.; Castagliuolo, I.; Scarpa, M. Reactive Oxygen Species and Antitumor Immunity—From Surveillance to Evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Noguchi, T.; Inoue, H.; Tanaka, T. The M1-and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J. Biol. Chem. 1986, 261, 13807–13812. [Google Scholar] [CrossRef]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef]
- Dayton, T.L.; Jacks, T.; Vander Heiden, M.G. PKM 2, cancer metabolism, and the road ahead. EMBO Rep. 2016, 17, 1721–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, J.; Manley, J.L. Turning on a fuel switch of cancer: hnRNP proteins regulate alternative splicing of pyruvate kinase mRNA. Cancer Res. 2010, 70, 8977–8980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riera-Domingo, C.; Audigé, A.; Granja, S.; Cheng, W.-C.; Ho, P.-C.; Baltazar, F.; Stockmann, C.; Mazzone, M. Immunity, Hypoxia, and Metabolism–the Ménage à Trois of Cancer: Implications for Immunotherapy. Physiol. Rev. 2020, 100, 1–102. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Sun, I.-M.; Oh, M.-H.; Sun, I.-H.; Wen, J.; Englert, J.; Powell, J.D. Inhibition of the adenosine A2a receptor modulates expression of T-cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol. Immunother. 2018, 67, 1271–1284. [Google Scholar] [CrossRef]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol. 2020, 17, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Csóka, B.; Selmeczy, Z.; Koscsó, B.; Németh, Z.H.; Pacher, P.; Murray, P.J.; Kepka-Lenhart, D.; Morris, S.M., Jr.; Gause, W.C.; Leibovich, S.J. Adenosine promotes alternative macrophage activation via A2A and A2B receptors. FASEB J. 2012, 26, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Guo, G.; Huang, L.; Deng, L.; Chang, C.-S.; Achyut, B.R.; Canning, M.; Xu, N.; Arbab, A.S.; Bollag, R.J. CD73 on cancer-associated fibroblasts enhanced by the A 2B-mediated feedforward circuit enforces an immune checkpoint. Nat. Commun. 2020, 11, 1–17. [Google Scholar]
- Allard, B.; Cousineau, I.; Allard, D.; Buisseret, L.; Pommey, S.; Chrobak, P.; Stagg, J. Adenosine A2a receptor promotes lymphangiogenesis and lymph node metastasis. Oncoimmunology 2019, 8, 1601481. [Google Scholar] [CrossRef] [Green Version]
- Hammami, A.; Allard, D.; Allard, B.; Stagg, J. Targeting the adenosine pathway for cancer immunotherapy. Semin. Immunol. 2019, 101304. [Google Scholar] [CrossRef]
- Linnemann, C.; Schildberg, F.A.; Schurich, A.; Diehl, L.; Hegenbarth, S.I.; Endl, E.; Lacher, S.; Müller, C.E.; Frey, J.; Simeoni, L. Adenosine regulates CD8 T-cell priming by inhibition of membrane-proximal T-cell receptor signalling. Immunology 2009, 128, e728–e737. [Google Scholar] [CrossRef]
- Mastelic-Gavillet, B.; Rodrigo, B.N.; Décombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derré, L.; Valerio, M. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8+ T-cells. J. Immunother. Cancer 2019, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.B.; Prado-Garcia, H.; Sánchez-García, F.J. Lactate contribution to the tumor microenvironment: Mechanisms, effects on immune cells and therapeutic relevance. Front. Immunol. 2016, 7, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, S.K.; Mueller-Klieser, W.; Pouysségur, J. Lactate and acidity in the cancer microenvironment. Annu. Rev. Cancer Biol. 2020, 4, 141–158. [Google Scholar] [CrossRef]
- Marchiq, I.; Le Floch, R.; Roux, D.; Simon, M.-P.; Pouyssegur, J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. 2015, 75, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. The monocarboxylate transporter family—Structure and functional characterization. IUBMB Life 2012, 64, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Dietl, K.; Renner, K.; Dettmer, K.; Timischl, B.; Eberhart, K.; Dorn, C.; Hellerbrand, C.; Kastenberger, M.; Kunz-Schughart, L.A.; Oefner, P.J. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J. Immunol. 2010, 184, 1200–1209. [Google Scholar] [CrossRef]
- Fischer, B.; Müller, B.; Fisch, P.; Kreutz, W. An Acidic Microenvironment Inhibits Antitumoral Non-Major Histocompatibility Complex-Restricted Cytotoxicity: Implications for Cancer Immunotherapy. J. Immunother. 2000, 23, 196–207. [Google Scholar] [CrossRef]
- Müller, B.; Fischer, B.; Kreutz, W. An acidic microenvironment impairs the generation of non-major histocompatibility complex-restricted killer cells. Immunology 2000, 99, 375–384. [Google Scholar] [CrossRef]
- Bosticardo, M.; Ariotti, S.; Losana, G.; Bernabei, P.; Forni, G.; Novelli, F. Biased activation of human T lymphocytes due to low extracellular pH is antagonized by B7/CD28 costimulation. Eur. J. Immunol. 2001, 31, 2829–2838. [Google Scholar] [CrossRef]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Cova, A.; Canese, R.; Jachetti, E.; Rossetti, M. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72, 2746–2756. [Google Scholar] [CrossRef] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mulé, J.J.; Ibrahim-Hashim, A. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef] [Green Version]
- Renner, K.; Bruss, C.; Schnell, A.; Koehl, G.; Becker, H.M.; Fante, M.; Menevse, A.-N.; Kauer, N.; Blazquez, R.; Hacker, L. Restricting glycolysis preserves T-cell effector functions and augments checkpoint therapy. Cell Rep. 2019, 29, 135–150.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkach, M.; Kowal, J.; Théry, C. Why the need and how to approach the functional diversity of extracellular vesicles. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20160479. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Gupta, P.; Chaluvally-Raghavan, P.; Pradeep, S. Emerging Role of Extracellular Vesicles in Immune Regulation and Cancer Progression. Cancers 2020, 12, 3563. [Google Scholar] [CrossRef] [PubMed]
- Ricklefs, F.L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.L.; Lee, K.; Balaj, L.; Passaro, C. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci. Adv. 2018, 4. [Google Scholar] [CrossRef] [Green Version]
- Cianciaruso, C.; Beltraminelli, T.; Duval, F.; Nassiri, S.; Hamelin, R.; Mozes, A.; Gallart-Ayala, H.; Torres, G.C.; Torchia, B.; Ries, C.H. Molecular profiling and functional analysis of macrophage-derived tumor extracellular vesicles. Cell Rep. 2019, 27, 3062–3080.e11. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Luo, G.; Zhang, K.; Cao, J.; Huang, C.; Jiang, T.; Liu, B.; Su, L.; Qiu, Z. Hypoxic tumor-derived exosomal miR-301a mediates M2 macrophage polarization via PTEN/PI3Kγ to promote pancreatic cancer metastasis. Cancer Res. 2018, 78, 4586–4598. [Google Scholar] [CrossRef] [Green Version]
- Theodoraki, M.-N.; Yerneni, S.S.; Hoffmann, T.K.; Gooding, W.E.; Whiteside, T.L. Clinical significance of PD-L1+ exosomes in plasma of head and neck cancer patients. Clin. Cancer Res. 2018, 24, 896–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Poggio, M.; Hu, T.; Pai, C.C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell 2019, 177, 414–427.e13. [Google Scholar] [CrossRef] [Green Version]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; de Peredo, A.G. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Garris, C.S.; Blaho, V.A.; Hla, T.; Han, M.H. Sphingosine-1-phosphate receptor 1 signalling in T-cells: Trafficking and beyond. Immunology 2014, 142, 347–353. [Google Scholar] [CrossRef]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef]
- Schwab, S.R.; Pereira, J.P.; Matloubian, M.; Xu, Y.; Huang, Y.; Cyster, J.G. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science 2005, 309, 1735–1739. [Google Scholar] [CrossRef]
- Taylor-Papadimitriou, J.; Burchell, J.M.; Graham, R.; Beatson, R. Latest developments in MUC1 immunotherapy. Biochem. Soc. Trans. 2018, 46, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Bandyopadhyay, D. MUC1: A target molecule for cancer therapy. Cancer Biol. Ther. 2007, 6, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Aksoylar, H.-I.; Tijaro-Ovalle, N.M.; Boussiotis, V.A.; Patsoukis, N. T-cell Metabolism in Cancer Immunotherapy. Immunometabolism 2020, 2. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T-cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naik, G.S.; Waikar, S.S.; Johnson, A.E.; Buchbinder, E.I.; Haq, R.; Hodi, F.S.; Schoenfeld, J.D.; Ott, P.A. Complex inter-relationship of body mass index, gender and serum creatinine on survival: Exploring the obesity paradox in melanoma patients treated with checkpoint inhibition. J. Immunother. Cancer 2019, 7, 89. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T-cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-L.; Roh, W.; Reuben, A.; Cooper, Z.A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Bassett, R.L.; Gopalakrishnan, V.; Wani, K. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016, 6, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325. [Google Scholar] [CrossRef]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [Green Version]
- Rassy, E.E.; Ghosn, M.; Rassy, N.A.; Assi, T.; Robert, C. Do immune checkpoint inhibitors perform identically in patients with weight extremes? Future Med. 2018, 10. [Google Scholar] [CrossRef]
- Clements, V.K.; Long, T.; Long, R.; Figley, C.; Smith, D.M.; Ostrand-Rosenberg, S. Frontline Science: High fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J. Leukoc. Biol. 2018, 103, 395–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesinger, S.; Siegert, S.; Koch, M.; Walter, J.; Heits, N.; Hinz, S.; Jacobs, G.; Hampe, J.; Schafmayer, C.; Nöthlings, U. Postdiagnosis body mass index and risk of mortality in colorectal cancer survivors: A prospective study and meta-analysis. Cancer Causes Control 2014, 25, 1407–1418. [Google Scholar] [CrossRef]
- Lennon, H.; Sperrin, M.; Badrick, E.; Renehan, A.G. The obesity paradox in cancer: A review. Curr. Oncol. Rep. 2016, 18, 56. [Google Scholar] [CrossRef]
- McQuade, J.L.; Daniel, C.R.; Hess, K.R.; Mak, C.; Wang, D.Y.; Rai, R.R.; Park, J.J.; Haydu, L.E.; Spencer, C.; Wongchenko, M. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: A retrospective, multicohort analysis. Lancet Oncol. 2018, 19, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.T.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R. Paradoxical effects of obesity on T-cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 2019, 25, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buono, R.; Cheng, C.-W.; Cacciottolo, M.; Martin-Montalvo, A.; de Cabo, R.; Wei, M. Fasting-Mimicking diet reduces HO-1 to promote T-cell-mediated tumor cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbiano, S.; Suárez-Zamorano, N.; Rigo, D.; Veyrat-Durebex, C.; Dokic, A.S.; Colin, D.J.; Trajkovski, M. Caloric restriction leads to browning of white adipose tissue through type 2 immune signaling. Cell Metab. 2016, 24, 434–446. [Google Scholar] [CrossRef]
- Messaoudi, I.; Warner, J.; Fischer, M.; Park, B.; Hill, B.; Mattison, J.; Lane, M.A.; Roth, G.S.; Ingram, D.K.; Picker, L.J. Delay of T-cell senescence by caloric restriction in aged long-lived nonhuman primates. Proc. Natl. Acad. Sci. USA 2006, 103, 19448–19453. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Tao, S.; Chen, Z.; Koliesnik, I.O.; Calmes, P.G.; Hoerr, V.; Han, B.; Gebert, N.; Zörnig, M.; Löffler, B. Dietary restriction improves repopulation but impairs lymphoid differentiation capacity of hematopoietic stem cells in early aging. J. Exp. Med. 2016, 213, 535–553. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-E.; Rayyan, M.; Liao, A.; Edery, I.; Pletcher, S.D. Acute dietary restriction acts via TOR, PP2A, and Myc signaling to boost innate immunity in Drosophila. Cell Rep. 2017, 20, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farazi, M.; Nguyen, J.; Goldufsky, J.; Linnane, S.; Lukaesko, L.; Weinberg, A.D.; Ruby, C.E. Caloric restriction maintains OX40 agonist-mediated tumor immunity and CD4 T-cell priming during aging. Cancer Immunol. Immunother. 2014, 63, 615–626. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Jing, Y.; Li, L.; Mills, G.B.; Diao, L.; Liu, H.; Han, L. Sex-Associated molecular differences for cancer immunotherapy. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Ju, Q.; Jia, K.; Yu, J.; Shi, H.; Wu, H.; Jiang, M. Correlation between sex and efficacy of immune checkpoint inhibitors (PD-1 and CTLA-4 inhibitors). Int. J. Cancer 2018, 143, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wykes, M.N.; Lewin, S.R. Immune checkpoint blockade in infectious diseases. Nat. Rev. Immunol. 2018, 18, 91. [Google Scholar] [CrossRef] [PubMed]
- Gatto, L.; Franceschi, E.; Nunno, V.D.; Brandes, A.A. Potential protective and therapeutic role of immune checkpoint inhibitors against viral infections and COVID-19. Future Med. 2020, 12. [Google Scholar] [CrossRef]
- Picchi, H.; Mateus, C.; Chouaid, C.; Besse, B.; Marabelle, A.; Michot, J.; Champiat, S.; Voisin, A.; Lambotte, O. Infectious complications associated with the use of immune checkpoint inhibitors in oncology: Reactivation of tuberculosis after anti PD-1 treatment. Clin. Microbiol. Infect. 2018, 24, 216–218. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Ayyoub, M.; Routy, B.; Kroemer, G. Microbiome and anticancer immunosurveillance. Cell 2016, 165, 276–287. [Google Scholar] [CrossRef] [Green Version]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia 2017, 19, 848–855. [Google Scholar] [CrossRef]
- Chalabi, M.; Cardona, A.; Nagarkar, D.; Scala, A.D.; Gandara, D.; Rittmeyer, A.; Albert, M.; Powles, T.; Kok, M.; Herrera, F. Efficacy of chemotherapy and atezolizumab in patients with non-small-cell lung cancer receiving antibiotics and proton pump inhibitors: Pooled post hoc analyses of the oak and poplar trials. Ann. Oncol. 2020, 31, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.A.; Goodrich, J.K.; Maxan, M.-E.; Freedberg, D.E.; Abrams, J.A.; Poole, A.C.; Sutter, J.L.; Welter, D.; Ley, R.E.; Bell, J.T. Proton pump inhibitors alter the composition of the gut microbiota. Gut 2016, 65, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.; Karpinets, T.; Prieto, P.; Vicente, D.; Hoffman, K.; Wei, S. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.K.; Man, J.; Lord, S.; Cooper, W.; Links, M.; Gebski, V.; Herbst, R.S.; Gralla, R.J.; Mok, T.; Yang, J.C.-H. Clinical and molecular characteristics associated with survival among patients treated with checkpoint inhibitors for advanced non–small cell lung carcinoma: A systematic review and meta-analysis. JAMA Oncol. 2018, 4, 210–216. [Google Scholar] [CrossRef]
- Abdel-Rahman, O. Smoking and EGFR status may predict outcomes of advanced NSCLC treated with PD-(L) 1 inhibitors beyond first line: A meta-analysis. Clin. Respir. J. 2018, 12, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Huang, X.; Fu, L. Impact of smoking on efficacy of PD-1/PD-L1 inhibitors in non-small cell lung cancer patients: A meta-analysis. Onco Targets Ther. 2018, 11, 3691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, J.; Hu, X.; Gu, L.; Chen, B.; Khadaroo, P.A.; Shen, Z.; Dong, L.; Lv, Y.; Chitumba, M.N.; Liu, J. Smokers or non-smokers: Who benefits more from immune checkpoint inhibitors in treatment of malignancies? An up-to-date meta-analysis. World J. Surg. Oncol. 2020, 18, 15. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.L.; Liu, Y.; Dimou, A.; Patil, T.; Aisner, D.L.; Dong, Z.; Jiang, T.; Su, C.; Wu, C.; Ren, S. Predictive value of oncogenic driver subtype, programmed death-1 ligand (PD-L1) score, and smoking status on the efficacy of PD-1/PD-L1 inhibitors in patients with oncogene-driven non–small cell lung cancer. Cancer 2019, 125, 1038–1049. [Google Scholar] [CrossRef]
- Singal, G.; Miller, P.G.; Agarwala, V.; Li, G.; Kaushik, G.; Backenroth, D.; Gossai, A.; Frampton, G.M.; Torres, A.Z.; Lehnert, E.M. Association of patient characteristics and tumor genomics with clinical outcomes among patients with non–small cell lung cancer using a clinicogenomic database. JAMA 2019, 321, 1391–1399. [Google Scholar] [CrossRef] [Green Version]
- Verdura, S.; Cuyàs, E.; Martin-Castillo, B.; Menendez, J.A. Metformin as an archetype immuno-metabolic adjuvant for cancer immunotherapy. Oncoimmunology 2019, 8, e1633235. [Google Scholar] [CrossRef]
- Li, K.; Fang, D.; Xiong, Z.; Luo, R. Inhibition of the hedgehog pathway for the treatment of cancer using Itraconazole. Onco Targets Ther. 2019, 12, 6875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Liu, W.; Wang, J.Q.; Tang, Z. “Hedgehog pathway”: A potential target of itraconazole in the treatment of cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 297–304. [Google Scholar] [CrossRef] [PubMed]
- El Halabi, I.; Bejjany, R.; Nasr, R.; Mukherji, D.; Temraz, S.; Nassar, F.J.; El Darsa, H.; Shamseddine, A. Ascorbic acid in colon cancer: From the basic to the clinical applications. Int. J. Mol. Sci. 2018, 19, 2752. [Google Scholar] [CrossRef] [Green Version]
- Roa, F.J.; Peña, E.; Gatica, M.; Escobar-Acuña, K.; Saavedra, P.; Maldonado, M.; Cuevas, M.E.; Moraga-Cid, G.; Rivas, C.I.; Muñoz-Montesino, C. Therapeutic Use of Vitamin C in Cancer: Physiological Considerations. Front. Pharmacol. 2020, 11, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, L.; Xu, W.; Lei, W.; Ma, X.; Gong, Z.; Zhang, S. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar]
- Ayyagari, V.N.; Wang, X.; Diaz-Sylvester, P.L.; Groesch, K.; Brard, L. Assessment of acyl-CoA cholesterol acyltransferase (ACAT-1) role in ovarian cancer progression—An in vitro study. PLoS ONE 2020, 15, e0228024. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Moderators of Immune Cells’ Activity and Metabolism | |
|---|---|
| Nutrients | Other Mechanisms and Pathways |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Sayed, R.; Haibe, Y.; Amhaz, G.; Bouferraa, Y.; Shamseddine, A. Metabolic Factors Affecting Tumor Immunogenicity: What Is Happening at the Cellular Level? Int. J. Mol. Sci. 2021, 22, 2142. https://doi.org/10.3390/ijms22042142
El Sayed R, Haibe Y, Amhaz G, Bouferraa Y, Shamseddine A. Metabolic Factors Affecting Tumor Immunogenicity: What Is Happening at the Cellular Level? International Journal of Molecular Sciences. 2021; 22(4):2142. https://doi.org/10.3390/ijms22042142
Chicago/Turabian StyleEl Sayed, Rola, Yolla Haibe, Ghid Amhaz, Youssef Bouferraa, and Ali Shamseddine. 2021. "Metabolic Factors Affecting Tumor Immunogenicity: What Is Happening at the Cellular Level?" International Journal of Molecular Sciences 22, no. 4: 2142. https://doi.org/10.3390/ijms22042142