
Inhibition of Autophagy Does Not Re-Sensitize Acute Myeloid Leukemia Cells Resistant to Cytarabine




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Generation of Cytarabine Resistant Cell Lines
2.2. The Expression of General Autophagy Genes and Proteins Is Not Uniformly Elevated in AraC Resistant AML Cells
2.3. The Basal Level of Autophagic Flux Is Not Elevated in AraC Resistant Cells Compared to Parental Cells
2.4. Autophagy Is Activated Upon AraC Treatment in Parental but Not in AraC Resistant Cells
2.5. Autophagy Inhibitors Increase the Efficacy of AraC in Parental Cells, but Do Not Re-Sensitize AraC Resistant Cells for AraC
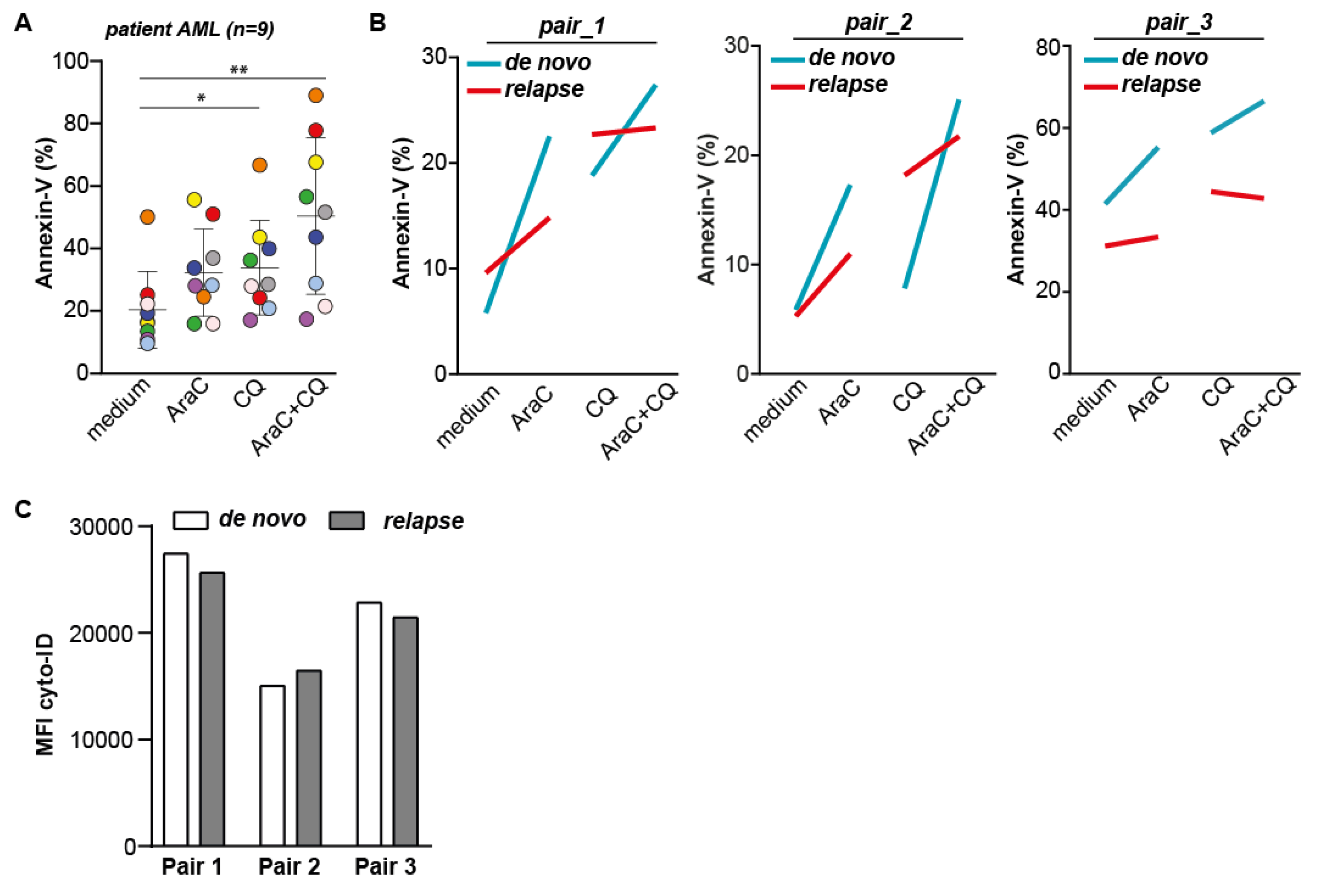
2.6. The Combination of CQ and AraC Is Most Effective in Inducing Cytotoxic Effects in AML Patient Samples, but Does Not Re-Sensitize Relapse AML Samples for AraC
3. Discussion
4. Material and Methods
4.1. Cell Lines and Generation of Cytarabine Resistant Cell Lines
4.2. Cytotoxicity Assays, Cytarabine, and Treatment with Autophagy Inhibitors
4.3. Western Blot-Based Detection of Autophagy Levels
4.4. RTqPCR Assays
4.5. Fluorescent Microscopy and Flow Cytometry
4.6. AML Patient Samples
4.7. Ex Vivo Culturing of Patient-Derived AML Samples
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, J.; Gu, Y.; Chen, B. Mechanisms of drug resistance in acute myeloid leukemia. OncoTargets Ther. 2019, 12, 1937–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zehra, S.; Najam, R.; Farzana, T.; Shamsi, T. Outcomes of 1st Remission Induction Chemotherapy in Acute Myeloid Leukemia Cytogenetic Risk Groups. Asian Pac. J. Cancer Prev. 2016, 17, 5251–5256. [Google Scholar]
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The multifaceted role of autophagy in cancer and the microenvironment. Med. Res. Rev. 2019, 39, 517–560. [Google Scholar] [CrossRef] [PubMed]
- Bosnjak, M.; Ristic, B.; Arsikin, K.; Mircic, A.; Suzin-Zivkovic, V.; Perovic, V.; Bogdanovic, A.; Paunovic, V.; Markovic, I.; Bumbasirevic, V.; et al. Inhibition of mTOR-dependent autophagy sensitizes leukemic cells to cytarabine-induced apoptotic death. PLoS ONE 2014, 9, e94374. [Google Scholar] [CrossRef]
- Kim, Y.; Eom, J.I.; Jeung, H.K.; Jang, J.E.; Kim, J.S.; Cheong, J.W.; Kim, Y.S.; Min, Y.H. Induction of cytosine arabinoside-resistant human myeloid leukemia cell death through autophagy regulation by hydroxychloroquine. Biomed. Pharmacother. 2015, 73, 87–96. [Google Scholar] [CrossRef]
- Cheong, J.W.; Kim, Y.; Eom, J.I.; Jeung, H.K.; Min, Y.H. Enhanced autophagy in cytarabine arabinoside-resistant U937 leukemia cells and its potential as a target for overcoming resistance. Mol. Med. Rep. 2016, 13, 3433–3440. [Google Scholar] [CrossRef] [Green Version]
- Palmeira dos Santos, C.; Pereira, G.J.; Barbosa, C.M.; Jurkiewicz, A.; Smaili, S.S.; Bincoletto, C. Comparative study of autophagy inhibition by 3MA and CQ on Cytarabineinduced death of leukaemia cells. J. Cancer Res. Clin. Oncol. 2014, 140, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Takagaki, K.; Katsuma, S.; Kaminishi, Y.; Horio, T.; Nakagawa, S.; Tanaka, T.; Ohgi, T.; Yano, J. Gene-expression profiling reveals down-regulation of equilibrative nucleoside transporter 1 (ENT1) in Ara-C-resistant CCRF-CEM-derived cells. J. Biochem. 2004, 136, 733–740. [Google Scholar] [CrossRef]
- Norgaard, J.M.; Bukh, A.; Langkjer, S.T.; Clausen, N.; Palshof, T.; Hokland, P. MDR1 gene expression and drug resistance of AML cells. Br. J. Haematol. 1998, 100, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel-Eibrink, M.M.; Wiemer, E.A.; Kuijpers, M.; Pieters, R.; Sonneveld, P. Absence of mutations in the deoxycytidine kinase (dCK) gene in patients with relapsed and/or refractory acute myeloid leukemia (AML). Leukemia 2001, 15, 855–856. [Google Scholar] [CrossRef] [Green Version]
- Nishi, R.; Shigemi, H.; Negoro, E.; Okura, M.; Hosono, N.; Yamauchi, T. Venetoclax and alvocidib are both cytotoxic to acute myeloid leukemia cells resistant to cytarabine and clofarabine. BMC Cancer 2020, 20, 984. [Google Scholar] [CrossRef]
- Nishi, R.; Yamauchi, T.; Negoro, E.; Takemura, H.; Ueda, T. Combination of guanine arabinoside and Bcl-2 inhibitor YC137 overcomes the cytarabine resistance in HL-60 leukemia cell line. Cancer Sci. 2013, 104, 502–507. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Clark, M.; Wunderlich, M.; Fan, C.; Davis, A.; Chen, S.; Guan, J.L.; Mulloy, J.C.; Kumar, A.; Zheng, Y. Autophagy is dispensable for Kmt2a/Mll-Mllt3/Af9 AML maintenance and anti-leukemic effect of chloroquine. Autophagy 2017, 13, 955–966. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Guo, P.; Zhang, Y.; Li, X.; Jia, P.; Tong, J.; Li, J. Autophagy is an important event for low-dose cytarabine treatment in acute myeloid leukemia cells. Leuk. Res. 2017, 60, 44–52. [Google Scholar] [CrossRef]
- Panina, S.B.; Baran, N.; Brasil da Costa, F.H.; Konopleva, M.; Kirienko, N.V. A mechanism for increased sensitivity of acute myeloid leukemia to mitotoxic drugs. Cell Death Dis. 2019, 10, 617. [Google Scholar] [CrossRef] [Green Version]
- Piya, S.; Kornblau, S.M.; Ruvolo, V.R.; Mu, H.; Ruvolo, P.P.; McQueen, T.; Davis, R.E.; Hail, N.; Kantarjian, H.; Andreeff, M.; et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood 2016, 128, 1260–1269. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.; Zhou, G.; Cao, S. Targeted inhibition of ULK1 enhances daunorubicin sensitivity in acute myeloid leukemia. Life Sci. 2020, 243, 117234. [Google Scholar] [CrossRef] [PubMed]
- Azadniv, M.; Myers, J.R.; McMurray, H.R.; Guo, N.; Rock, P.; Coppage, M.L.; Ashton, J.; Becker, M.W.; Calvi, L.M.; Liesveld, J.L. Bone marrow mesenchymal stromal cells from acute myelogenous leukemia patients demonstrate adipogenic differentiation propensity with implications for leukemia cell support. Leukemia 2020, 34, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, O.; Gjertsen, B.T.; Foss, B.; Huang, T.S. New strategies in the treatment of acute myelogenous leukemia (AML): In vitro culture of aml cells--the present use in experimental studies and the possible importance for future therapeutic approaches. Stem Cells 2001, 19, 1–11. [Google Scholar] [CrossRef]
- Garrido, S.M.; Appelbaum, F.R.; Willman, C.L.; Banker, D.E. Acute myeloid leukemia cells are protected from spontaneous and drug-induced apoptosis by direct contact with a human bone marrow stromal cell line (HS-5). Exp. Hematol. 2001, 29, 448–457. [Google Scholar] [CrossRef]
- Pabst, C.; Krosl, J.; Fares, I.; Boucher, G.; Ruel, R.; Marinier, A.; Lemieux, S.; Hebert, J.; Sauvageau, G. Identification of small molecules that support human leukemia stem cell activity ex vivo. Nat. Methods 2014, 11, 436–442. [Google Scholar] [CrossRef]
- Cucchi, D.G.J.; Groen, R.W.J.; Janssen, J.; Cloos, J. Ex vivo cultures and drug testing of primary acute myeloid leukemia samples: Current techniques and implications for experimental design and outcome. Drug Resist. Updat. 2020, 53, 100730. [Google Scholar] [CrossRef]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Vogl, D.T.; Stadtmauer, E.A.; Tan, K.S.; Heitjan, D.F.; Davis, L.E.; Pontiggia, L.; Rangwala, R.; Piao, S.; Chang, Y.C.; Scott, E.C.; et al. Combined autophagy and proteasome inhibition: A phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014, 10, 1380–1390. [Google Scholar] [CrossRef] [Green Version]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorburn, A.; Thamm, D.H.; Gustafson, D.L. Autophagy and cancer therapy. Mol. Pharmacol. 2014, 85, 830–838. [Google Scholar] [CrossRef] [Green Version]
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017, 8, e2927. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.; Dykatra, K.; Cronin, T.; Lutgen-Dunckley, L.; Martens, B.L.; Guzman, M.L.; Wang, E.S. Chloroquine derivate Lys05 overcomes hypoxia-induced chemoresistance in acute myeloid leukemia through metabolic disruption. Blood 2018, 132, 3948. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Han, Y.; Visconte, V.; Przychodzen, B.; Espitia, C.M.; Phillips, J.; Anwer, F.; Advani, A.; Carraway, H.E.; Kelly, K.R.; et al. The novel autophagy inhibitor ROC-325 augments the antileukemic activity of azacitidine. Leukemia 2019, 33, 2971–2974. [Google Scholar] [CrossRef]
- Follo, C.; Cheng, Y.; Richards, W.G.; Bueno, R.; Broaddus, V.C. Inhibition of autophagy initiation potentiates chemosensitivity in mesothelioma. Mol. Carcinog. 2018, 57, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Zahedi, S.; Fitzwalter, B.E.; Morin, A.; Grob, S.; Desmarais, M.; Nellan, A.; Green, A.L.; Vibhakar, R.; Hankinson, T.C.; Foreman, N.K.; et al. Effect of early-stage autophagy inhibition in BRAF(V600E) autophagy-dependent brain tumor cells. Cell Death Dis. 2019, 10, 679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, Y.; Shen, C.; Li, C.; Liu, Y.; Gao, D.; Shi, C.; Peng, F.; Liu, Z.; Zhao, B.; Zheng, Z.; et al. Inhibition of autophagy induced by quercetin at a late stage enhances cytotoxic effects on glioma cells. Tumour. Biol. 2016, 37, 3549–3560. [Google Scholar] [CrossRef] [PubMed]
- Dubin, S.A.; Silverstein, P.I.; Wakefield, M.L.; Jense, H.G. Comparison of the effects of oral famotidine and ranitidine on gastric volume and pH. Anesth. Analg. 1989, 69, 680–683. [Google Scholar] [CrossRef]
- Stegmann, A.P.; Mitchel, B.S. Transfection of wild-type deoxycytidine kinase (dck) cDNA into an AraC- and DAC-resistant rat leukemic cell line of clonal origin fully restores drug sensitivity. Blood 1995, 85, 1188–1194. [Google Scholar] [CrossRef] [Green Version]
- Hapke, D.M.; Stegmann, A.P.; Mitchell, B.S. Retroviral transfer of deoxycytidine kinase into tumor cell lines enhances nucleoside toxicity. Cancer Res. 1996, 56, 2343–2347. [Google Scholar] [PubMed]
- Liu, Q.; Garcia, M.; Wang, S.; Chen, C.W. Therapeutic Target Discovery Using High-Throughput Genetic Screens in Acute Myeloid Leukemia. Cells 2020, 9, 1888. [Google Scholar] [CrossRef]
- Jin, G.; Matsushita, H.; Asai, S.; Tsukamoto, H.; Ono, R.; Nosaka, T.; Yahata, T.; Takahashi, S.; Miyachi, H. FLT3-ITD induces ara-C resistance in myeloid leukemic cells through the repression of the ENT1 expression. Biochem. Biophys. Res. Commun. 2009, 390, 1001–1006. [Google Scholar] [CrossRef]
- Hu, Q.; Qin, Y.; Zhang, B.; Liang, C.; Ji, S.; Shi, S.; Xu, W.; Xiang, J.; Liang, D.; Ni, Q.; et al. FBW7 increases the chemosensitivity of pancreatic cancer cells to gemcitabine through upregulation of ENT1. Oncol. Rep. 2017, 38, 2069–2077. [Google Scholar] [CrossRef] [Green Version]
- Wiersma, V.R.; de Bruyn, M.; Wei, Y.; van Ginkel, R.J.; Hirashima, M.; Niki, T.; Nishi, N.; Zhou, J.; Pouwels, S.D.; Samplonius, D.F.; et al. The epithelial polarity regulator LGALS9/galectin-9 induces fatal frustrated autophagy in KRAS mutant colon carcinoma that depends on elevated basal autophagic flux. Autophagy 2015, 11, 1373–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Visser, N.; Lourens, H.J.; Huls, G.; Bremer, E.; Wiersma, V.R. Inhibition of Autophagy Does Not Re-Sensitize Acute Myeloid Leukemia Cells Resistant to Cytarabine. Int. J. Mol. Sci. 2021, 22, 2337. https://doi.org/10.3390/ijms22052337
Visser N, Lourens HJ, Huls G, Bremer E, Wiersma VR. Inhibition of Autophagy Does Not Re-Sensitize Acute Myeloid Leukemia Cells Resistant to Cytarabine. International Journal of Molecular Sciences. 2021; 22(5):2337. https://doi.org/10.3390/ijms22052337
Chicago/Turabian StyleVisser, Nienke, Harm Jan Lourens, Gerwin Huls, Edwin Bremer, and Valerie R. Wiersma. 2021. "Inhibition of Autophagy Does Not Re-Sensitize Acute Myeloid Leukemia Cells Resistant to Cytarabine" International Journal of Molecular Sciences 22, no. 5: 2337. https://doi.org/10.3390/ijms22052337
APA StyleVisser, N., Lourens, H. J., Huls, G., Bremer, E., & Wiersma, V. R. (2021). Inhibition of Autophagy Does Not Re-Sensitize Acute Myeloid Leukemia Cells Resistant to Cytarabine. International Journal of Molecular Sciences, 22(5), 2337. https://doi.org/10.3390/ijms22052337