
Secreted Signaling Molecules at the Neuromuscular Junction in Physiology and Pathology


{kind=link}
{kind=link}
Abstract
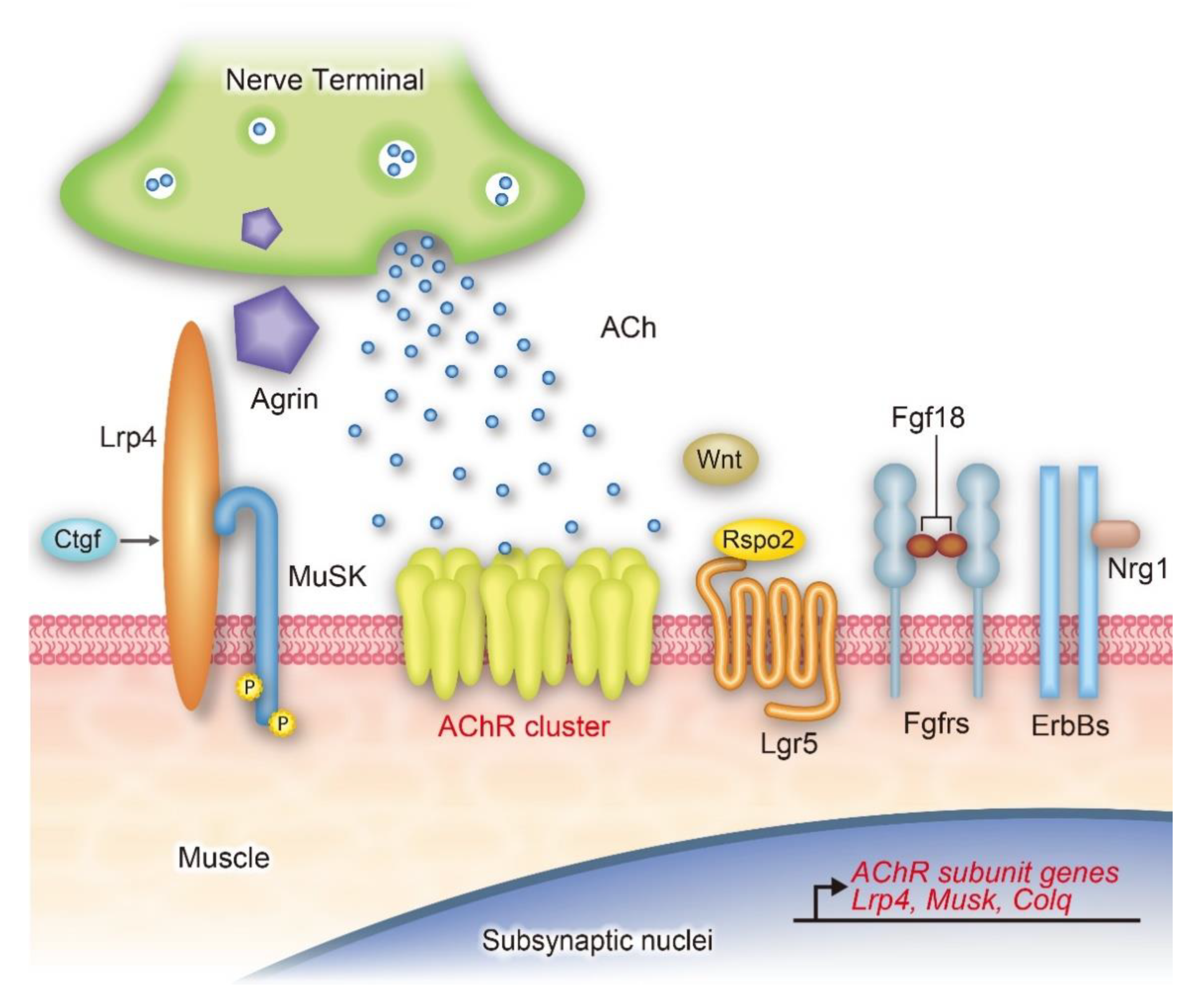
:1. Introduction
2. Environment of the Signaling Molecules: Extracellular Matrix (ECM)
3. Agrin-Lrp4-MuSK Signaling Pathway
3.1. Agrin
Agrin-Binding Partners: Laminins and α/β-dystroglycans
3.2. Low-Density Lipoprotein Receptor-Related Protein 4 (Lrp4)
Lrp4-Binding Partners: Amyloid β Precursor Protein (App), α-Sarcoglycan, and Connective Tissue Growth Factor (Ctgf)
3.3. Muscle-Specific Receptor Tyrosine Kinase (MuSK)
MuSK-Binding Partners: Collagen Q (ColQ), Biglycan, and Neuregulin-1/Erb2/4/Erbin
4. Acetylcholine (ACh)
5. Wnt Signaling Pathways
6. Fibroblast Growth Factors (FGFs) and Their Receptors
7. Neuregulins/ErbB Signaling Pathway
8. Other Signaling Pathways for AChR Clustering
9. Therapeutic Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Ohno, K.; Ohkawara, B.; Ito, M. Agrin-LRP4-MuSK signaling as a therapeutic target for myasthenia gravis and other neuromuscular disorders. Expert Opin. Ther. Targets 2017, 21, 949–958. [Google Scholar] [CrossRef]
- Gromova, A.; La Spada, A.R. Harmony Lost: Cell-Cell Communication at the Neuromuscular Junction in Motor Neuron Disease. Trends Neurosci. 2020, 43, 709–724. [Google Scholar] [CrossRef] [PubMed]
- York, A.L.; Zheng, J.Q. Super-Resolution Microscopy Reveals a Nanoscale Organization of Acetylcholine Receptors for Trans-Synaptic Alignment at Neuromuscular Synapses. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, G.; Xiong, W.C.; Mei, L. Rapsyn as a signaling and scaffolding molecule in neuromuscular junction formation and maintenance. Neurosci. Lett. 2020, 731, 135013. [Google Scholar] [CrossRef]
- Belotti, E.; Schaeffer, L. Regulation of Gene expression at the neuromuscular Junction. Neurosci. Lett. 2020, 735, 135163. [Google Scholar] [CrossRef]
- Engel, A.G. Genetic basis and phenotypic features of congenital myasthenic syndromes. Handb. Clin. Neurol. 2018, 148, 565–589. [Google Scholar] [CrossRef]
- Belhasan, D.C.; Akaaboune, M. The role of the dystrophin glycoprotein complex on the neuromuscular system. Neurosci. Lett. 2020, 722, 134833. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Shen, C.; Li, L.; Wu, H.; Xing, G.; Dong, Z.; Jing, H.; Chen, W.; Zhang, H.; Tan, Z.; et al. Sarcoglycan Alpha Mitigates Neuromuscular Junction Decline in Aged Mice by Stabilizing LRP4. J. Neurosci. 2018, 38, 8860–8873. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Ohno, K. Protein-anchoring therapy to target extracellular matrix proteins to their physiological destinations. Matrix Biol. 2018, 68–69, 628–636. [Google Scholar] [CrossRef]
- Hack, A.A.; Groh, M.E.; McNally, E.M. Sarcoglycans in muscular dystrophy. Microsc. Res. Tech. 2000, 48, 167–180. [Google Scholar] [CrossRef]
- Legay, C.; Dobbertin, A. Collagens at the vertebrate neuromuscular junction, from structure to pathologies. Neurosci. Lett. 2020, 735, 135155. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.S.; Nishimune, H. The role of laminins in the organization and function of neuromuscular junctions. Matrix Biol. 2017, 57–58, 86–105. [Google Scholar] [CrossRef] [Green Version]
- Fox, M.A.; Ho, M.S.; Smyth, N.; Sanes, J.R. A synaptic nidogen: Developmental regulation and role of nidogen-2 at the neuromuscular junction. Neural Dev. 2008, 3, 24. [Google Scholar] [CrossRef] [Green Version]
- Arikawa-Hirasawa, E.; Rossi, S.G.; Rotundo, R.L.; Yamada, Y. Absence of acetylcholinesterase at the neuromuscular junctions of perlecan-null mice. Nat. Neurosci. 2002, 5, 119–123. [Google Scholar] [CrossRef]
- Whitelock, J.M.; Melrose, J.; Iozzo, R.V. Diverse cell signaling events modulated by perlecan. Biochemistry 2008, 47, 11174–11183. [Google Scholar] [CrossRef] [Green Version]
- Amenta, A.R.; Creely, H.E.; Mercado, M.L.; Hagiwara, H.; McKechnie, B.A.; Lechner, B.E.; Rossi, S.G.; Wang, Q.; Owens, R.T.; Marrero, E.; et al. Biglycan is an extracellular MuSK binding protein important for synapse stability. J. Neurosci. 2012, 32, 2324–2334. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Luo, S.; Wang, Q.; Suzuki, T.; Xiong, W.C.; Mei, L. LRP4 serves as a coreceptor of agrin. Neuron 2008, 60, 285–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.; Stiegler, A.L.; Cameron, T.O.; Hallock, P.T.; Gomez, A.M.; Huang, J.H.; Hubbard, S.R.; Dustin, M.L.; Burden, S.J. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 2008, 135, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, Y.; Jin, R. Structural mechanisms of the agrin-LRP4-MuSK signaling pathway in neuromuscular junction differentiation. Cell. Mol. Life Sci. 2013, 70, 3077–3088. [Google Scholar] [CrossRef] [Green Version]
- Zong, Y.; Zhang, B.; Gu, S.; Lee, K.; Zhou, J.; Yao, G.; Figueiredo, D.; Perry, K.; Mei, L.; Jin, R. Structural basis of agrin-LRP4-MuSK signaling. Genes Dev. 2012, 26, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Coldefy, A.S.; Hubbard, S.R.; Burden, S.J. Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). J. Biol. Chem. 2011, 286, 40624–40630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, A.M.; Burden, S.J. The extracellular region of Lrp4 is sufficient to mediate neuromuscular synapse formation. Dev. Dyn. 2011, 240, 2626–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuka, K.; Ito, M.; Ohkawara, B.; Masuda, A.; Kawakami, Y.; Sahashi, K.; Nishida, H.; Mabuchi, N.; Takano, A.; Engel, A.G.; et al. Collagen Q and anti-MuSK autoantibody competitively suppress agrin/LRP4/MuSK signaling. Sci. Rep. 2015, 5, 13928. [Google Scholar] [CrossRef] [Green Version]
- Okada, K.; Inoue, A.; Okada, M.; Murata, Y.; Kakuta, S.; Jigami, T.; Kubo, S.; Shiraishi, H.; Eguchi, K.; Motomura, M.; et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science 2006, 312, 1802–1805. [Google Scholar] [CrossRef] [PubMed]
- Hallock, P.T.; Xu, C.F.; Park, T.J.; Neubert, T.A.; Curran, T.; Burden, S.J. Dok-7 regulates neuromuscular synapse formation by recruiting Crk and Crk-L. Genes Dev. 2010, 24, 2451–2461. [Google Scholar] [CrossRef] [Green Version]
- Hallock, P.T.; Chin, S.; Blais, S.; Neubert, T.A.; Glass, D.J. Sorbs1 and -2 Interact with CrkL and Are Required for Acetylcholine Receptor Cluster Formation. Mol. Cell. Biol. 2016, 36, 262–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burden, S.J. SnapShot: Neuromuscular Junction. Cell 2011, 144, 826. [Google Scholar] [CrossRef] [Green Version]
- Gautam, M.; Noakes, P.G.; Moscoso, L.; Rupp, F.; Scheller, R.H.; Merlie, J.P.; Sanes, J.R. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 1996, 85, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Burgess, R.W.; Dominguez, B.; Pfaff, S.L.; Sanes, J.R.; Lee, K.F. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 2001, 410, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Lu, Y.; Shen, C.; Patel, N.; Gan, L.; Xiong, W.C.; Mei, L. Distinct roles of muscle and motoneuron LRP4 in neuromuscular junction formation. Neuron 2012, 75, 94–107. [Google Scholar] [CrossRef] [Green Version]
- DeChiara, T.M.; Bowen, D.C.; Valenzuela, D.M.; Simmons, M.V.; Poueymirou, W.T.; Thomas, S.; Kinetz, E.; Compton, D.L.; Rojas, E.; Park, J.S.; et al. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 1996, 85, 501–512. [Google Scholar] [CrossRef] [Green Version]
- Fish, L.A.; Fallon, J.R. Multiple MuSK signaling pathways and the aging neuromuscular junction. Neurosci. Lett. 2020, 731, 135014. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.B.; Ruegg, M.A.; Winzen, U.; Halfter, W.; Engel, J.; Stetefeld, J. Mapping of the laminin-binding site of the N-terminal agrin domain (NtA). EMBO J. 2003, 22, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.W.; Skarnes, W.C.; Sanes, J.R. Agrin isoforms with distinct amino termini: Differential expression, localization, and function. J. Cell Biol. 2000, 151, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Ninni, A.; Sarappa, A.; D’Antonio, G. [Mechanism of action of antibiotics and microbial resistance]. Arch. Monaldi 1978, 33, 157–163. [Google Scholar] [PubMed]
- Nicole, S.; Chaouch, A.; Torbergsen, T.; Bauche, S.; de Bruyckere, E.; Fontenille, M.J.; Horn, M.A.; van Ghelue, M.; Loseth, S.; Issop, Y.; et al. Agrin mutations lead to a congenital myasthenic syndrome with distal muscle weakness and atrophy. Brain 2014, 137, 2429–2443. [Google Scholar] [CrossRef] [Green Version]
- Huze, C.; Bauche, S.; Richard, P.; Chevessier, F.; Goillot, E.; Gaudon, K.; Ben Ammar, A.; Chaboud, A.; Grosjean, I.; Lecuyer, H.A.; et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am. J. Hum. Genet. 2009, 85, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Maselli, R.A.; Fernandez, J.M.; Arredondo, J.; Navarro, C.; Ngo, M.; Beeson, D.; Cagney, O.; Williams, D.C.; Wollmann, R.L.; Yarov-Yarovoy, V.; et al. LG2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Hum. Genet. 2012, 131, 1123–1135. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Dai, Y.; Han, J.N.; Chen, Z.H.; Ling, L.; Pu, C.Q.; Cui, L.Y.; Huang, X.S. A Novel AGRN Mutation Leads to Congenital Myasthenic Syndrome Only Affecting Limb-girdle Muscle. Chin. Med. J. (Engl.) 2017, 130, 2279–2282. [Google Scholar] [CrossRef]
- Wang, P.; Jing, Z.; Liu, C.; Xu, M.; Wang, P.; Wang, X.; Yin, Y.; Cui, Y.; Ren, D.; Rao, X. Hepatitis C virus infection and risk of thyroid cancer: A systematic review and meta-analysis. Arab J. Gastroenterol. 2017, 18, 1–5. [Google Scholar] [CrossRef]
- Xi, J.; Yan, C.; Liu, W.W.; Qiao, K.; Lin, J.; Tian, X.; Wu, H.; Lu, J.; Wong, L.J.; Beeson, D.; et al. Novel SEA and LG2 Agrin mutations causing congenital Myasthenic syndrome. Orphanet J. Rare Dis. 2017, 12, 182. [Google Scholar] [CrossRef] [Green Version]
- Rudell, J.B.; Maselli, R.A.; Yarov-Yarovoy, V.; Ferns, M.J. Pathogenic effects of agrin V1727F mutation are isoform specific and decrease its expression and affinity for HSPGs and LRP4. Hum. Mol. Genet. 2019, 28, 2648–2658. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Govindarajan, R. Presentation and management of congenital myasthenic syndrome with a homozygous Agrin variant (Pro1448Leu). Clin. Neurol. Neurosurg. 2020, 199, 106277. [Google Scholar] [CrossRef]
- Ohkawara, B.; Shen, X.; Selcen, D.; Nazim, M.; Bril, V.; Tarnopolsky, M.A.; Brady, L.; Fukami, S.; Amato, A.A.; Yis, U.; et al. Congenital myasthenic syndrome-associated agrin variants affect clustering of acetylcholine receptors in a domain-specific manner. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsen, G.; Halfter, W.; Kroger, S.; Cole, G.J. Agrin is a heparan sulfate proteoglycan. J. Biol. Chem. 1995, 270, 3392–3399. [Google Scholar] [CrossRef] [Green Version]
- Storms, S.D.; Kim, A.C.; Tran, B.H.; Cole, G.J.; Murray, B.A. NCAM-mediated adhesion of transfected cells to agrin. Cell Adhes. Commun. 1996, 3, 497–509. [Google Scholar] [CrossRef]
- Gesemann, M.; Brancaccio, A.; Schumacher, B.; Ruegg, M.A. Agrin is a high-affinity binding protein of dystroglycan in non-muscle tissue. J. Biol. Chem. 1998, 273, 600–605. [Google Scholar] [CrossRef] [Green Version]
- Ohno, K.; Rahman, M.A.; Nazim, M.; Nasrin, F.; Lin, Y.; Takeda, J.I.; Masuda, A. Splicing regulation and dysregulation of cholinergic genes expressed at the neuromuscular junction. J. Neurochem. 2017, 142 (Suppl. S2), 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Toole, J.J.; Deyst, K.A.; Bowe, M.A.; Nastuk, M.A.; McKechnie, B.A.; Fallon, J.R. Alternative splicing of agrin regulates its binding to heparin alpha-dystroglycan, and the cell surface. Proc. Natl. Acad. Sci. USA 1996, 93, 7369–7374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakaya, M.; Ceyhan-Birsoy, O.; Beggs, A.H.; Topaloglu, H. A Novel Missense Variant in the AGRN Gene; Congenital Myasthenic Syndrome Presenting with Head Drop. J. Clin. Neuromuscul. Dis. 2017, 18, 147–151. [Google Scholar] [CrossRef]
- Choi, H.Y.; Liu, Y.; Tennert, C.; Sugiura, Y.; Karakatsani, A.; Kroger, S.; Johnson, E.B.; Hammer, R.E.; Lin, W.; Herz, J. APP interacts with LRP4 and agrin to coordinate the development of the neuromuscular junction in mice. Elife 2013, 2, e00220. [Google Scholar] [CrossRef] [PubMed]
- Stanga, S.; Zanou, N.; Audouard, E.; Tasiaux, B.; Contino, S.; Vandermeulen, G.; Rene, F.; Loeffler, J.P.; Clotman, F.; Gailly, P.; et al. APP-dependent glial cell line-derived neurotrophic factor gene expression drives neuromuscular junction formation. FASEB J. 2016, 30, 1696–1711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkawara, B.; Kobayakawa, A.; Kanbara, S.; Hattori, T.; Kubota, S.; Ito, M.; Masuda, A.; Takigawa, M.; Lyons, K.M.; Ishiguro, N.; et al. CTGF/CCN2 facilitates LRP4-mediated formation of the embryonic neuromuscular junction. EMBO Rep. 2020, 21, e48462. [Google Scholar] [CrossRef] [PubMed]
- Rafii, M.S.; Hagiwara, H.; Mercado, M.L.; Seo, N.S.; Xu, T.; Dugan, T.; Owens, R.T.; Hook, M.; McQuillan, D.J.; Young, M.F.; et al. Biglycan binds to alpha- and gamma-sarcoglycan and regulates their expression during development. J. Cell. Physiol. 2006, 209, 439–447. [Google Scholar] [CrossRef] [Green Version]
- Takigawa, M. The CCN Proteins: An Overview. Methods Mol. Biol. 2017, 1489, 1–8. [Google Scholar] [CrossRef]
- Bao, J.; Zheng, J.J.; Wu, D. The structural basis of DKK-mediated inhibition of Wnt/LRP signaling. Sci. Signal 2012, 5, pe22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karner, C.M.; Dietrich, M.F.; Johnson, E.B.; Kappesser, N.; Tennert, C.; Percin, F.; Wollnik, B.; Carroll, T.J.; Herz, J. Lrp4 regulates initiation of ureteric budding and is crucial for kidney formation—A mouse model for Cenani-Lenz syndrome. PLoS ONE 2010, 5, e10418. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Tian, Q.B.; Endo, S.; Suzuki, T. A role for LRP4 in neuronal cell viability is related to apoE-binding. Brain Res. 2007, 1177, 19–28. [Google Scholar] [CrossRef]
- Ohkawara, B.; Cabrera-Serrano, M.; Nakata, T.; Milone, M.; Asai, N.; Ito, K.; Ito, M.; Masuda, A.; Ito, Y.; Engel, A.G.; et al. LRP4 third beta-propeller domain mutations cause novel congenital myasthenia by compromising agrin-mediated MuSK signaling in a position-specific manner. Hum. Mol. Genet. 2014, 23, 1856–1868. [Google Scholar] [CrossRef] [Green Version]
- Al-Qattan, M.M.; Alkuraya, F.S. Cenani-Lenz syndrome and other related syndactyly disorders due to variants in LRP4, GREM1/FMN1, and APC: Insight into the pathogenesis and the relationship to polyposis through the WNT and BMP antagonistic pathways. Am J Med Genet A 2019, 179, 266–279. [Google Scholar] [CrossRef]
- Li, Y.; Pawlik, B.; Elcioglu, N.; Aglan, M.; Kayserili, H.; Yigit, G.; Percin, F.; Goodman, F.; Nurnberg, G.; Cenani, A.; et al. LRP4 mutations alter Wnt/beta-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am. J. Hum. Genet. 2010, 86, 696–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leupin, O.; Piters, E.; Halleux, C.; Hu, S.; Kramer, I.; Morvan, F.; Bouwmeester, T.; Schirle, M.; Bueno-Lozano, M.; Fuentes, F.J.; et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J. Biol. Chem. 2011, 286, 19489–19500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selcen, D.; Ohkawara, B.; Shen, X.M.; McEvoy, K.; Ohno, K.; Engel, A.G. Impaired Synaptic Development, Maintenance, and Neuromuscular Transmission in LRP4-Related Myasthenia. JAMA Neurol. 2015, 72, 889–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasi, S.; Spina, V.; Bruscaggin, A.; Vaisitti, T.; Tripodo, C.; Forconi, F.; De Paoli, L.; Fangazio, M.; Sozzi, E.; Cencini, E.; et al. A variant of the LRP4 gene affects the risk of chronic lymphocytic leukaemia transformation to Richter syndrome. Br. J. Haematol. 2011, 152, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Swanberg, M.; McGuigan, F.; Callreus, M.; Gerdhem, P.; Akesson, K. LRP4 association to bone properties and fracture and interaction with genes in the Wnt- and BMP signaling pathways. Bone 2011, 49, 343–348. [Google Scholar] [CrossRef]
- Boudin, E.; Steenackers, E.; de Freitas, F.; Nielsen, T.L.; Andersen, M.; Brixen, K.; Van Hul, W.; Piters, E. A common LRP4 haplotype is associated with bone mineral density and hip geometry in men-data from the Odense Androgen Study (OAS). Bone 2013, 53, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Yumoto, N.; Kim, N.; Burden, S.J. Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature 2012, 489, 438–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, A.; Kattamuri, C.; Ozdeslik, R.N.; Schmiedel, C.; Mentzer, S.; Schorl, C.; Oancea, E.; Thompson, T.B.; Fallon, J.R. MuSK is a BMP co-receptor that shapes BMP responses and calcium signaling in muscle cells. Sci. Signal. 2016, 9, ra87. [Google Scholar] [CrossRef] [Green Version]
- Stiegler, A.L.; Burden, S.J.; Hubbard, S.R. Crystal structure of the agrin-responsive immunoglobulin-like domains 1 and 2 of the receptor tyrosine kinase MuSK. J. Mol. Biol. 2006, 364, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopf, C.; Hoch, W. Dimerization of the muscle-specific kinase induces tyrosine phosphorylation of acetylcholine receptors and their aggregation on the surface of myotubes. J. Biol. Chem. 1998, 273, 6467–6473. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.; Topf, A.; Preethish-Kumar, V.; Lorenzoni, P.J.; Vroling, B.; Scola, R.H.; Dias-Tosta, E.; Geraldo, A.; Polavarapu, K.; Nashi, S.; et al. Recessive variants of MuSK are associated with late onset CMS and predominant limb girdle weakness. Am. J. Med. Genet. A 2018, 176, 1594–1601. [Google Scholar] [CrossRef] [PubMed]
- Wadwekar, V.; Pillai, R.R.; Sesh, S.; Nair, S.S.; Nair, M. Pregnancy-associated respiratory failure in muscle specific kinase congenital myasthenic syndrome. Muscle Nerve 2019, 59, E24–E26. [Google Scholar] [CrossRef] [PubMed]
- Gallenmuller, C.; Muller-Felber, W.; Dusl, M.; Stucka, R.; Guergueltcheva, V.; Blaschek, A.; von der Hagen, M.; Huebner, A.; Muller, J.S.; Lochmuller, H.; et al. Salbutamol-responsive limb-girdle congenital myasthenic syndrome due to a novel missense mutation and heteroallelic deletion in MUSK. Neuromuscul. Disord. 2014, 24, 31–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, V.; Salih, M.A.; Mukhtar, M.M.; Abuzeid, H.A.; El-Sadig, S.M.; von der Hagen, M.; Huebner, A.; Nurnberg, G.; Abicht, A.; Muller, J.S.; et al. Refinement of the clinical phenotype in musk-related congenital myasthenic syndromes. Neurology 2009, 73, 1926–1928. [Google Scholar] [CrossRef]
- Liu, Y.; Qiao, K.; Yan, C.; Song, J.; Huan, X.; Luo, S.; Lu, J.; Zhao, C.; Xi, J. Congenital myasthenia syndrome in a Chinese family with mutations in MUSK: A hotspot mutation and literature review. J. Clin. Neurosci. 2020, 76, 161–165. [Google Scholar] [CrossRef]
- Wu, H.; Xiong, W.C.; Mei, L. To build a synapse: Signaling pathways in neuromuscular junction assembly. Development 2010, 137, 1017–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, M.M.; Sultana, N.; Benedetti, A.; Obermair, G.J.; Linde, N.F.; Papadopoulos, S.; Dayal, A.; Grabner, M.; Flucher, B.E. Calcium Influx and Release Cooperatively Regulate AChR Patterning and Motor Axon Outgrowth during Neuromuscular Junction Formation. Cell Rep. 2018, 23, 3891–3904. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Liu, Y.; Sugiura, Y.; Allen, P.D.; Gregg, R.G.; Lin, W. Neuromuscular synaptic patterning requires the function of skeletal muscle dihydropyridine receptors. Nat. Neurosci. 2011, 14, 570–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejat, A.; Ramond, F.; Bassel-Duby, R.; Khochbin, S.; Olson, E.N.; Schaeffer, L. Histone deacetylase 9 couples neuronal activity to muscle chromatin acetylation and gene expression. Nat. Neurosci. 2005, 8, 313–321. [Google Scholar] [CrossRef]
- Tang, H.; Goldman, D. Activity-dependent gene regulation in skeletal muscle is mediated by a histone deacetylase (HDAC)-Dach2-myogenin signal transduction cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 16977–16982. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Macpherson, P.; Marvin, M.; Meadows, E.; Klein, W.H.; Yang, X.J.; Goldman, D. A histone deacetylase 4/myogenin positive feedback loop coordinates denervation-dependent gene induction and suppression. Mol. Biol. Cell 2009, 20, 1120–1131. [Google Scholar] [CrossRef] [Green Version]
- Castets, P.; Rion, N.; Theodore, M.; Falcetta, D.; Lin, S.; Reischl, M.; Wild, F.; Guerard, L.; Eickhorst, C.; Brockhoff, M.; et al. mTORC1 and PKB/Akt control the muscle response to denervation by regulating autophagy and HDAC4. Nat. Commun. 2019, 10, 3187. [Google Scholar] [CrossRef] [Green Version]
- Cohen, T.J.; Waddell, D.S.; Barrientos, T.; Lu, Z.; Feng, G.; Cox, G.A.; Bodine, S.C.; Yao, T.P. The histone deacetylase HDAC4 connects neural activity to muscle transcriptional reprogramming. J. Biol. Chem. 2007, 282, 33752–33759. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Luo, Z.G.; Wang, Q.; Zhou, J.Z.; Wang, J.; Luo, Z.; Liu, M.; He, X.; Wynshaw-Boris, A.; Xiong, W.C.; Lu, B.; et al. Regulation of AChR clustering by Dishevelled interacting with MuSK and PAK1. Neuron 2002, 35, 489–505. [Google Scholar] [CrossRef] [Green Version]
- Henriquez, J.P.; Webb, A.; Bence, M.; Bildsoe, H.; Sahores, M.; Hughes, S.M.; Salinas, P.C. Wnt signaling promotes AChR aggregation at the neuromuscular synapse in collaboration with agrin. Proc. Natl. Acad. Sci. USA 2008, 105, 18812–18817. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Luo, S.; Dong, X.P.; Zhang, X.; Liu, C.; Luo, Z.; Xiong, W.C.; Mei, L. Beta-catenin regulates acetylcholine receptor clustering in muscle cells through interaction with rapsyn. J. Neurosci. 2007, 27, 3968–3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Jing, Z.; Zhang, L.; Zhou, G.; Braun, J.; Yao, Y.; Wang, Z.Z. Regulation of acetylcholine receptor clustering by the tumor suppressor APC. Nat. Neurosci. 2003, 6, 1017–1018. [Google Scholar] [CrossRef]
- Serebryanov, O.V.; Stemberg, A.S.; Zabludovskii, A.L. Optimization of reinforcing current parameters in motor-defensive conditioning in rats. Neurosci. Behav. Physiol. 1989, 19, 480–482. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Barik, A.; Lu, Y.; Shen, C.; Bowman, A.; Li, L.; Sathyamurthy, A.; Lin, T.W.; Xiong, W.C.; Mei, L. Slit2 as a beta-catenin/Ctnnb1-dependent retrograde signal for presynaptic differentiation. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Messeant, J.; Dobbertin, A.; Girard, E.; Delers, P.; Manuel, M.; Mangione, F.; Schmitt, A.; Le Denmat, D.; Molgo, J.; Zytnicki, D.; et al. MuSK frizzled-like domain is critical for mammalian neuromuscular junction formation and maintenance. J. Neurosci. 2015, 35, 4926–4941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barik, A.; Zhang, B.; Sohal, G.S.; Xiong, W.C.; Mei, L. Crosstalk between Agrin and Wnt signaling pathways in development of vertebrate neuromuscular junction. Dev. Neurobiol. 2014, 74, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Remedio, L.; Gribble, K.D.; Lee, J.K.; Kim, N.; Hallock, P.T.; Delestree, N.; Mentis, G.Z.; Froemke, R.C.; Granato, M.; Burden, S.J. Diverging roles for Lrp4 and Wnt signaling in neuromuscular synapse development during evolution. Genes Dev. 2016, 30, 1058–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Liang, C.; Bates, R.; Yin, Y.; Xiong, W.C.; Mei, L. Wnt proteins regulate acetylcholine receptor clustering in muscle cells. Mol. Brain 2012, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, H.; Ohkawara, B.; Ishigaki, S.; Fukudome, T.; Ito, K.; Tsushima, M.; Konishi, H.; Okuno, T.; Yoshimura, T.; Ito, M.; et al. R-spondin 2 promotes acetylcholine receptor clustering at the neuromuscular junction via Lgr5. Sci. Rep. 2016, 6, 28512. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ito, M.; Ohkawara, B.; Masuda, A.; Ohno, K. Differential effects of spinal motor neuron-derived and skeletal muscle-derived Rspo2 on acetylcholine receptor clustering at the neuromuscular junction. Sci. Rep. 2018, 8, 13577. [Google Scholar] [CrossRef]
- Li, P.P.; Zhou, J.J.; Meng, M.; Madhavan, R.; Peng, H.B. Reciprocal regulation of axonal Filopodia and outgrowth during neuromuscular junction development. PLoS ONE 2012, 7, e44759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, M.A.; Sanes, J.R.; Borza, D.B.; Eswarakumar, V.P.; Fassler, R.; Hudson, B.G.; John, S.W.; Ninomiya, Y.; Pedchenko, V.; Pfaff, S.L.; et al. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell 2007, 129, 179–193. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Ohkawara, B.; Yagi, H.; Nakashima, H.; Tsushima, M.; Ota, K.; Konishi, H.; Masuda, A.; Imagama, S.; Kiyama, H.; et al. Lack of Fgf18 causes abnormal clustering of motor nerve terminals at the neuromuscular junction with reduced acetylcholine receptor clusters. Sci. Rep. 2018, 8, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandrock, A.W., Jr.; Goodearl, A.D.; Yin, Q.W.; Chang, D.; Fischbach, G.D. ARIA is concentrated in nerve terminals at neuromuscular junctions and at other synapses. J. Neurosci. 1995, 15, 6124–6136. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Chu, G.C.; Merlie, J.P. ARIA/HRG regulates AChR epsilon subunit gene expression at the neuromuscular synapse via activation of phosphatidylinositol 3-kinase and Ras/MAPK pathway. J. Cell Biol. 1996, 134, 465–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, D.; Birchmeier, C. Multiple essential functions of neuregulin in development. Nature 1995, 378, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Negro, A.; Brar, B.K.; Lee, K.F. Essential roles of Her2/erbB2 in cardiac development and function. Recent Prog. Horm. Res. 2004, 59, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gassmann, M.; Casagranda, F.; Orioli, D.; Simon, H.; Lai, C.; Klein, R.; Lemke, G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 1995, 378, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Simeone, L.; Straubinger, M.; Khan, M.A.; Nalleweg, N.; Cheusova, T.; Hashemolhosseini, S. Identification of Erbin interlinking MuSK and ErbB2 and its impact on acetylcholine receptor aggregation at the neuromuscular junction. J. Neurosci. 2010, 30, 6620–6634. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, N.; Akaaboune, M.; Gajendran, N.; Martinez-Pena y Valenzuela, I.; Wakefield, S.; Thurnheer, R.; Brenner, H.R. Neuregulin/ErbB regulate neuromuscular junction development by phosphorylation of alpha-dystrobrevin. J. Cell Biol. 2011, 195, 1171–1184. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Shen, C.; Lu, Y.; Huang, Z.; Li, L.; Rand, C.D.; Pan, J.; Sun, X.D.; Tan, Z.; Wang, H.; et al. Muscle Yap Is a Regulator of Neuromuscular Junction Formation and Regeneration. J. Neurosci. 2017, 37, 3465–3477. [Google Scholar] [CrossRef] [Green Version]
- Ham, D.J.; Borsch, A.; Lin, S.; Thurkauf, M.; Weihrauch, M.; Reinhard, J.R.; Delezie, J.; Battilana, F.; Wang, X.; Kaiser, M.S.; et al. The neuromuscular junction is a focal point of mTORC1 signaling in sarcopenia. Nat. Commun. 2020, 11, 4510. [Google Scholar] [CrossRef]
- Castets, P.; Ham, D.J.; Ruegg, M.A. The TOR Pathway at the Neuromuscular Junction: More Than a Metabolic Player? Front. Mol. Neurosci. 2020, 13, 162. [Google Scholar] [CrossRef]
- Mantilla, C.B.; Stowe, J.M.; Sieck, D.C.; Ermilov, L.G.; Greising, S.M.; Zhang, C.; Shokat, K.M.; Sieck, G.C. TrkB kinase activity maintains synaptic function and structural integrity at adult neuromuscular junctions. J. Appl. Physiol. (1985) 2014, 117, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Belluardo, N.; Westerblad, H.; Mudo, G.; Casabona, A.; Bruton, J.; Caniglia, G.; Pastoris, O.; Grassi, F.; Ibanez, C.F. Neuromuscular junction disassembly and muscle fatigue in mice lacking neurotrophin-4. Mol. Cell. Neurosci. 2001, 18, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsim, K.W.; Barnard, E.A. The signaling pathways mediated by P2Y nucleotide receptors in the formation and maintenance of the skeletal neuromuscular junction. Neurosignals 2002, 11, 58–64. [Google Scholar] [CrossRef]
- Ryten, M.; Koshi, R.; Knight, G.E.; Turmaine, M.; Dunn, P.; Cockayne, D.A.; Ford, A.P.; Burnstock, G. Abnormalities in neuromuscular junction structure and skeletal muscle function in mice lacking the P2X2 nucleotide receptor. Neuroscience 2007, 148, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Tung, E.K.; Choi, R.C.; Siow, N.L.; Jiang, J.X.; Ling, K.K.; Simon, J.; Barnard, E.A.; Tsim, K.W. P2Y2 receptor activation regulates the expression of acetylcholinesterase and acetylcholine receptor genes at vertebrate neuromuscular junctions. Mol. Pharmacol. 2004, 66, 794–806. [Google Scholar] [CrossRef] [Green Version]
- Feng, G.; Laskowski, M.B.; Feldheim, D.A.; Wang, H.; Lewis, R.; Frisen, J.; Flanagan, J.G.; Sanes, J.R. Roles for ephrins in positionally selective synaptogenesis between motor neurons and muscle fibers. Neuron 2000, 25, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Butt, B.; Ip, F.C.; Dai, Y.; Jiang, L.; Yung, W.H.; Greenberg, M.E.; Fu, A.K.; Ip, N.Y. Ephexin1 is required for structural maturation and neurotransmission at the neuromuscular junction. Neuron 2010, 65, 204–216. [Google Scholar] [CrossRef] [Green Version]
- Henriquez, J.P.; Krull, C.E.; Osses, N. The Wnt and BMP families of signaling morphogens at the vertebrate neuromuscular junction. Int. J. Mol. Sci. 2011, 12, 8924–8946. [Google Scholar] [CrossRef] [Green Version]
- Badawi, Y.; Nishimune, H. Impairment Mechanisms and Intervention Approaches for Aged Human Neuromuscular Junctions. Front. Mol. Neurosci. 2020, 13, 568426. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Brengman, J.; Tsujino, A.; Engel, A.G. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (ColQ) of the asymmetric enzyme. Proc. Natl. Acad. Sci. USA 1998, 95, 9654–9659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Suzuki, Y.; Okada, T.; Fukudome, T.; Yoshimura, T.; Masuda, A.; Takeda, S.; Krejci, E.; Ohno, K. Protein-anchoring strategy for delivering acetylcholinesterase to the neuromuscular junction. Mol. Ther. 2012, 20, 1384–1392. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Ehara, Y.; Li, J.; Inada, K.; Ohno, K. Protein-Anchoring Therapy of Biglycan for Mdx Mouse Model of Duchenne Muscular Dystrophy. Hum. Gene Ther. 2017, 28, 428–436. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohkawara, B.; Ito, M.; Ohno, K. Secreted Signaling Molecules at the Neuromuscular Junction in Physiology and Pathology. Int. J. Mol. Sci. 2021, 22, 2455. https://doi.org/10.3390/ijms22052455
Ohkawara B, Ito M, Ohno K. Secreted Signaling Molecules at the Neuromuscular Junction in Physiology and Pathology. International Journal of Molecular Sciences. 2021; 22(5):2455. https://doi.org/10.3390/ijms22052455
Chicago/Turabian StyleOhkawara, Bisei, Mikako Ito, and Kinji Ohno. 2021. "Secreted Signaling Molecules at the Neuromuscular Junction in Physiology and Pathology" International Journal of Molecular Sciences 22, no. 5: 2455. https://doi.org/10.3390/ijms22052455
APA StyleOhkawara, B., Ito, M., & Ohno, K. (2021). Secreted Signaling Molecules at the Neuromuscular Junction in Physiology and Pathology. International Journal of Molecular Sciences, 22(5), 2455. https://doi.org/10.3390/ijms22052455