
VTRNA2-1: Genetic Variation, Heritable Methylation and Disease Association


Abstract
1. Introduction
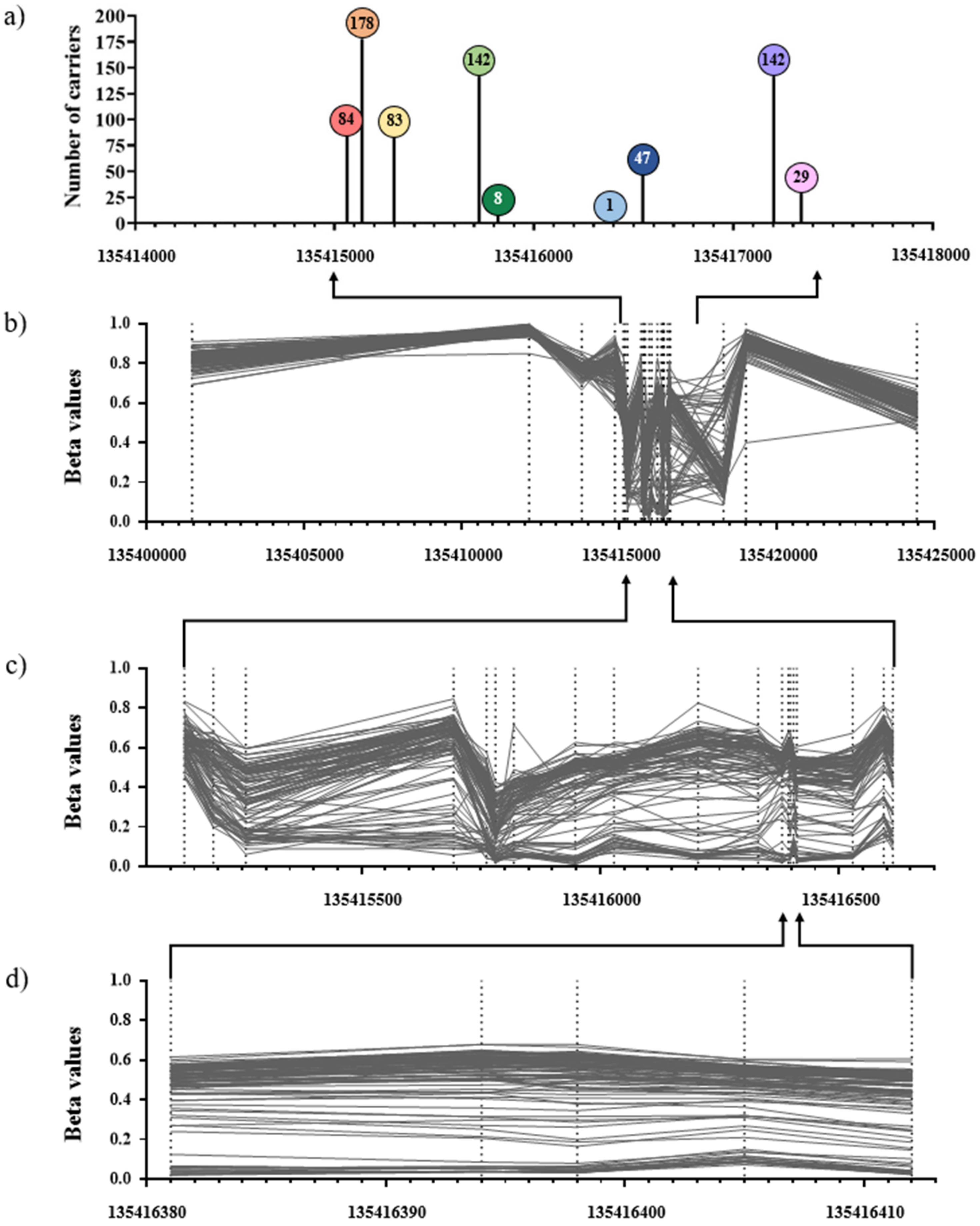
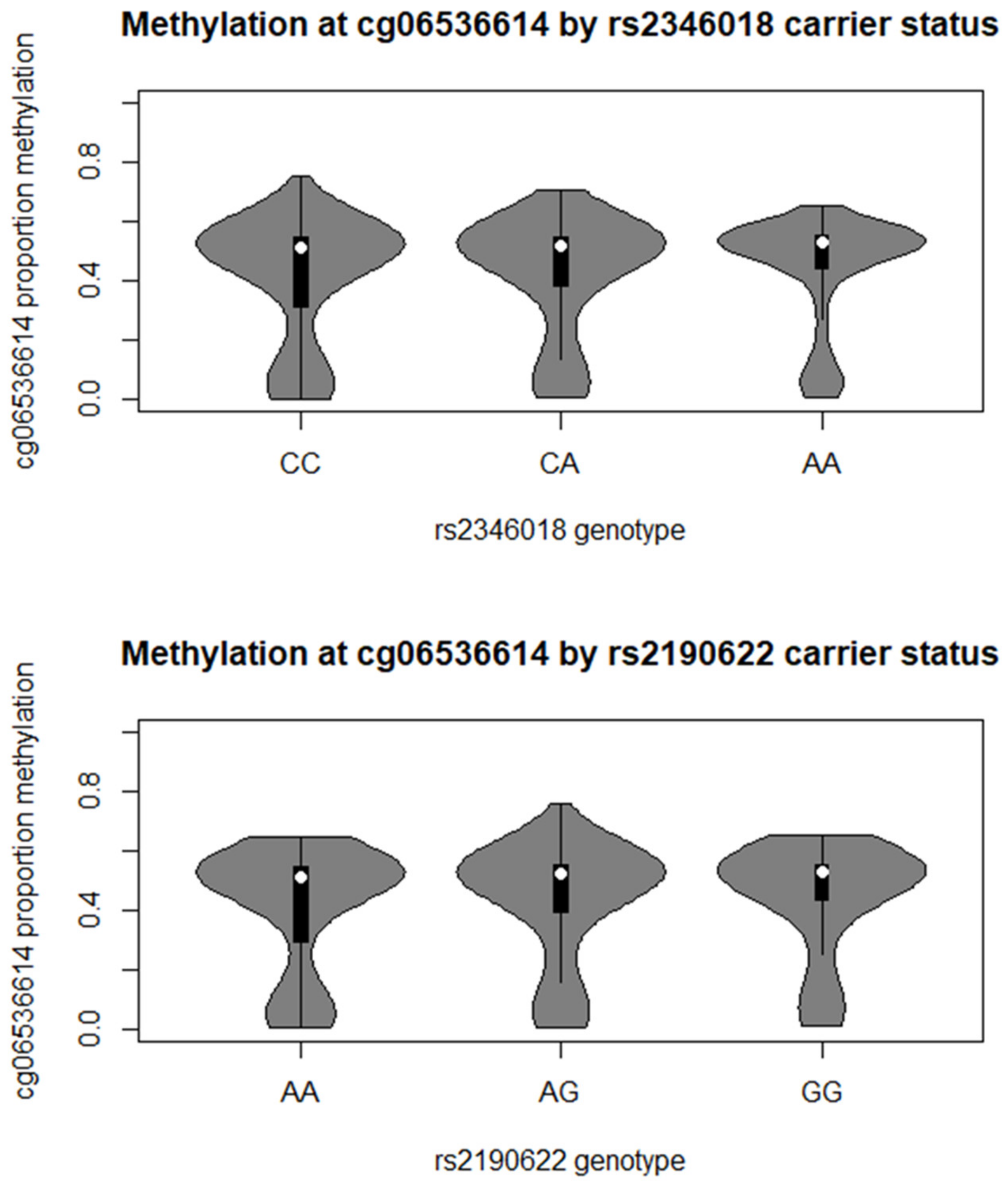
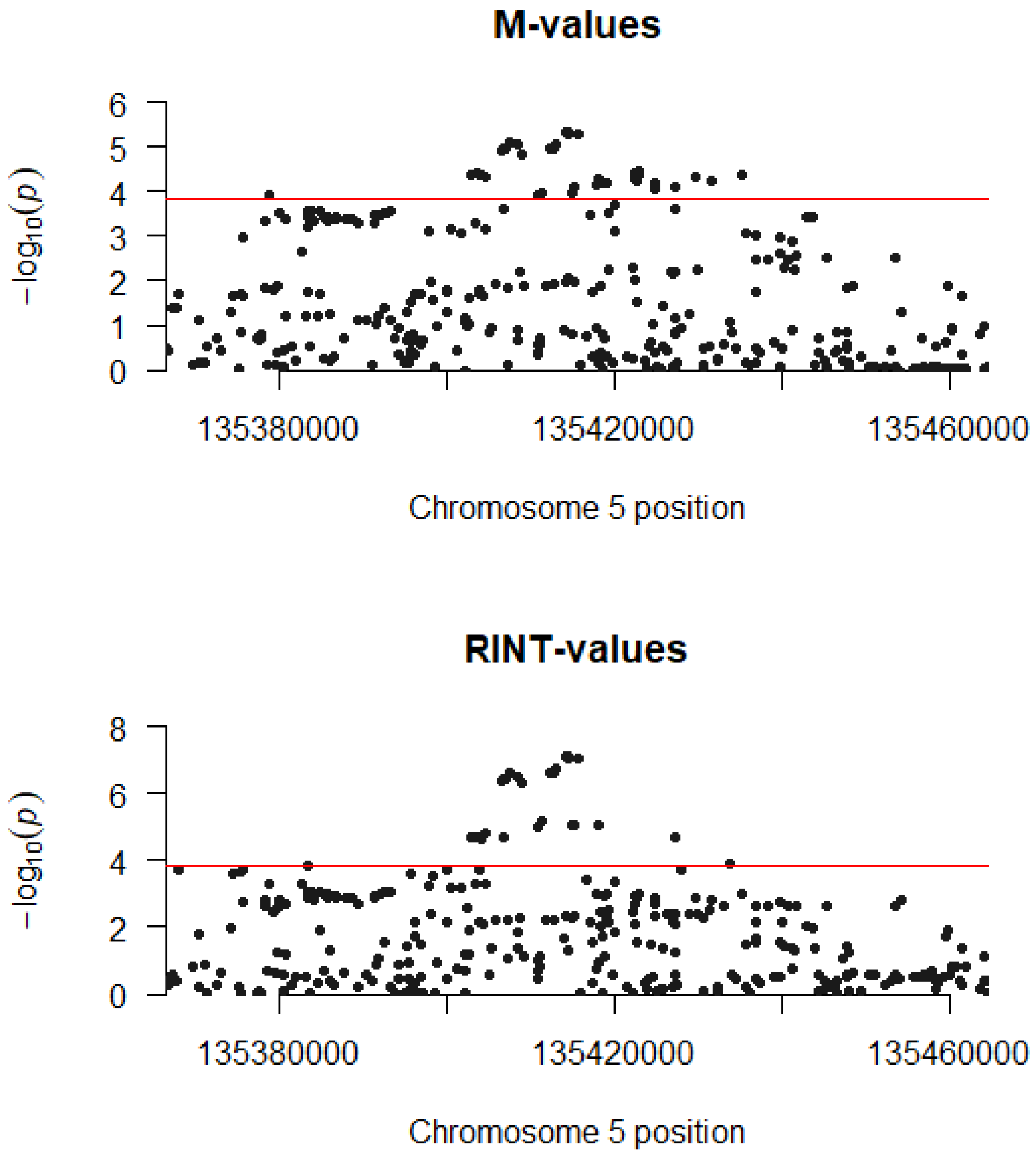
2. Results
3. Discussion
4. Materials and Methods
4.1. Data Sources
4.1.1. Prospective Cohort Study
4.1.2. Multiple-Case Breast Cancer Families
4.2. Genetic and DNA Methylation Data
4.3. Technical Validation of Methylation Measures Using Pyrosequencing
4.4. Statistical Analysis
4.4.1. Assessment of mQTLs
4.4.2. SNP-Based Heritability
4.4.3. Association of Non-Genetic Factors with VTRNA2-1 Methylation
4.4.4. Associations with Breast Cancer Risk
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Joo, J.E.; Dowty, J.G.; Milne, R.L.; Wong, E.M.; Dugue, P.A.; English, D.; Hopper, J.L.; Goldgar, D.E.; Giles, G.G.; Southey, M.C.; et al. Heritable DNA methylation marks associated with susceptibility to breast cancer. Nat. Commun. 2018, 9, 867. [Google Scholar] [CrossRef]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Jablonka, E.; Raz, G. Transgenerational epigenetic inheritance: Prevalence, mechanisms, and implications for the study of heredity and evolution. Q. Rev. Biol. 2009, 84, 131–176. [Google Scholar] [CrossRef]
- Horsthemke, B. A critical view on transgenerational epigenetic inheritance in humans. Nat. Commun. 2018, 9, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Calderon, B.M.; Conn, G.L. Human noncoding RNA 886 (nc886) adopts two structurally distinct conformers that are functionally opposing regulators of PKR. RNA 2017, 23, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Romanelli, V.; Nakabayashi, K.; Vizoso, M.; Moran, S.; Iglesias-Platas, I.; Sugahara, N.; Simón, C.; Hata, K.; Esteller, M.; Court, F.; et al. Variable maternal methylation overlapping the nc886/vtRNA2-1 locus is locked between hypermethylated repeats and is frequently altered in cancer. Epigenetics 2014, 9, 783–790. [Google Scholar] [CrossRef]
- Carpenter, B.L.; Zhou, W.; Madaj, Z.; DeWitt, A.K.; Ross, J.P.; Grønbæk, K.; Liang, G.; Clark, S.J.; Molloy, P.L.; Jones, P.A. Mother–child transmission of epigenetic information by tunable polymorphic imprinting. Proc. Natl. Acad. Sci. USA 2018, 115, E11970–E11977. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R.G. Structure and function of the protein Kinase R. In Interferon: The 50th Anniversary; Springer: Berlin/Heidelberg, Germany, 2007; Volume 316, pp. 253–292. [Google Scholar]
- Golec, E.; Lind, L.; Qayyum, M.; Blom, A.M.; King, B.C. The noncoding RNA nc886 regulates PKR signaling and cytokine production in human cells. J. Immunol. 2019, 202, 131–141. [Google Scholar] [CrossRef]
- Vlahopoulos, S.A. Aberrant control of NF-κB in cancer permits transcriptional and phenotypic plasticity, to curtail dependence on host tissue: Molecular mode. Cancer Biol. Med. 2017, 14, 254. [Google Scholar]
- Rakyan, V.K.; Blewitt, M.E.; Druker, R.; Preis, J.I.; Whitelaw, E. Metastable epialleles in mammals. Trends Genet. 2002, 18, 348–351. [Google Scholar] [CrossRef]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef]
- Van Baak, T.E.; Coarfa, C.; Dugué, P.-A.; Fiorito, G.; Laritsky, E.; Baker, M.S.; Kessler, N.J.; Dong, J.; Duryea, J.D.; Silver, M.J.; et al. Epigenetic supersimilarity of monozygotic twin pairs. Genome Biol. 2018, 19, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Dugué, P.-A.; Dowty, J.G.; Joo, J.E.; Wong, E.M.; Makalic, E.; Schmidt, D.F.; English, D.R.; Hopper, J.L.; Pedersen, J.; Severi, G.; et al. Heritable methylation marks associated with breast and prostate cancer risk. Prostate 2018, 78, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Silver, M.J.; Kessler, N.J.; Hennig, B.J.; Dominguez-Salas, P.; Laritsky, E.; Baker, M.S.; Coarfa, C.; Hernandez-Vargas, H.; Castelino, J.M.; Routledge, M.N.; et al. Independent genomewide screens identify the tumor suppressor VTRNA2-1 as a human epiallele responsive to periconceptional environment. Genome Biol. 2015, 16, 118. [Google Scholar] [CrossRef]
- Richmond, R.C.; Sharp, G.C.; Herbert, G.; Atkinson, C.; Taylor, C.; Bhattacharya, S.; Campbell, D.; Hall, M.; Kazmi, N.; Gaunt, T.; et al. The long-term impact of folic acid in pregnancy on offspring DNA methylation: Follow-up of the Aberdeen Folic Acid Supplementation Trial (AFAST). Int. J. Epidemiol. 2018, 47, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, S.J.; Peters, T.J.; Buckley, M.; Zhou, J.; Jones, P.A.; Gibson, R.A.; Makrides, M.; Muhlhausler, B.S.; Molloy, P.L. DNA methylation in blood from neonatal screening cards and the association with BMI and insulin sensitivity in early childhood. Int. J. Obes. 2017, 42, 28–35. [Google Scholar] [CrossRef]
- Min, J.L.; Hemani, G.; Hannon, E.; Dekkers, K.F.; Castillo-Fernandez, J.; Luijk, R.; Carnero-Montoro, E.; Lawson, D.J.; Burrows, K.; Suderman, M. Genomic and phenomic insights from an atlas of genetic effects on DNA methylation. medRxiv 2020. [Google Scholar] [CrossRef]
- McRae, A.F.; Marioni, R.E.; Shah, S.; Yang, J.; Powell, J.E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Bowdler, L.; Painter, J.N.; et al. Identification of 55,000 replicated DNA methylation QTL. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Dugué, P.-A.; Jung, C.-H.; Joo, J.E.; Wang, X.; Wong, E.M.; Makalic, E.; Schmidt, D.F.; Baglietto, L.; Severi, G.; Southey, M.C.; et al. Smoking and blood DNA methylation: An epigenome-wide association study and assessment of reversibility. Epigenetics 2020, 15, 358–368. [Google Scholar] [CrossRef]
- Dugué, P.; Wilson, R.; Lehne, B.; Jayasekara, H.; Wang, X.; Jung, C.; Joo, J.E.; Makalic, E.; Schmidt, D.F.; Baglietto, L.; et al. Alcohol consumption is associated with widespread changes in blood DNA methylation: Analysis of cross-sectional and longitudinal data. Addict. Biol. 2019, e12855. [Google Scholar] [CrossRef]
- Geurts, Y.M.; Dugué, P.-A.; Joo, J.E.; Makalic, E.; Jung, C.-H.; Guan, W.; Nguyen, S.; Grove, M.L.; Wong, E.M.; Hodge, A.M.; et al. Novel associations between blood DNA methylation and body mass index in middle-aged and older adults. Int. J. Obes. 2018, 42, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Finer, S.; Iqbal, M.S.; Lowe, R.; Ogunkolade, B.W.; Pervin, S.; Mathews, C.; Smart, M.; Alam, D.S.; Hitman, G.A. Is famine exposure during developmental life in rural Bangladesh associated with a metabolic and epigenetic signature in young adulthood? A historical cohort study. BMJ Open 2016, 6, e011768. [Google Scholar] [CrossRef]
- Gaunt, T.R.; Shihab, H.A.; Hemani, G.; Min, J.L.; Woodward, G.; Lyttleton, O.; Zheng, J.; Duggirala, A.; McArdle, W.L.; Ho, K.; et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef]
- McRae, A.F.; Powell, J.E.; Henders, A.K.; Bowdler, L.; Hemani, G.; Shah, S.; Painter, J.N.; Martin, N.G.; Visscher, P.M.; Montgomery, G.W. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol. 2014, 15, R73. [Google Scholar] [CrossRef]
- Van Dongen, J.; Nivard, M.; Willemsen, G.; Hottenga, J.J.; Helmer, Q.; Dolan, C.; Ehli, E.; Davies, G.; Van Iterson, M.; Breeze, C.E.; et al. Genetic and environmental influences interact with age and sex in shaping the human methylome. Nat. Commun. 2016, 7, 11115. [Google Scholar] [CrossRef]
- Zhang, H.; kConFab Investigators; Ahearn, T.U.; LeCarpentier, J.; Barnes, D.; Beesley, J.; Qi, G.; Jiang, O.; O’Mara, T.A.; Zhao, N.; et al. Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat. Genet. 2020, 52, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92. [Google Scholar] [CrossRef] [PubMed]
- Fort, R.S.; Mathó, C.; Geraldo, M.V.; Ottati, M.C.; Yamashita, A.S.; Saito, K.C.; Leite, K.R.M.; Méndez, M.; Maedo, N.; Méndez, L.; et al. Nc886 is epigenetically repressed in prostate cancer and acts as a tumor suppressor through the inhibition of cell growth. BMC Cancer 2018, 18, 127. [Google Scholar] [CrossRef]
- Kunkeaw, N.; Jeon, S.H.; Lee, K.; Johnson, B.H.; Tanasanvimon, S.; Javle, M.; Pairojkul, C.; Chamgramol, Y.; Wongfieng, W.; Gong, B.; et al. Cell death/proliferation roles for nc886, a non-coding RNA, in the protein kinase R pathway in cholangiocarcinoma. Oncogene 2013, 32, 3722–3731. [Google Scholar] [CrossRef]
- Lee, H.-S.; Lee, K.; Jang, H.-J.; Lee, G.K.; Park, J.-L.; Kim, S.-Y.; Kim, S.-B.; Johnson, B.H.; Zo, J.I.; Lee, J.-S.; et al. Epigenetic silencing of the non-coding RNA nc886 provokes oncogenes during human esophageal tumorigenesis. Oncotarget 2014, 5, 3472–3481. [Google Scholar] [CrossRef]
- Cao, J.; Song, Y.; Bi, N.; Shen, J.; Liu, W.; Fan, J.; Sun, G.; Tong, T.; He, J.; Shi, Y. DNA methylation-mediated repression of miR-886-3p predicts poor outcome of human small cell lung cancer. Cancer Res. 2013, 73, 3326–3335. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-S.; Park, J.-L.; Lee, K.; Richardson, L.E.; Johnson, B.H.; Lee, H.-S.; Lee, J.-S.; Kim, S.-B.; Kwon, O.-H.; Song, K.S.; et al. nc886, a non-coding RNA of anti-proliferative role, is suppressed by CpG DNA methylation in human gastric cancer. Oncotarget 2014, 5, 3944–3955. [Google Scholar] [CrossRef] [PubMed]
- Treppendahl, M.B.; Qiu, X.; Søgaard, A.; Yang, X.; Nandrup-Bus, C.; Hother, C.; Andersen, M.K.; Kjeldsen, L.; Möllgaard, L.; Hellström-Lindberg, E.; et al. Allelic methylation levels of the noncoding VTRNA2-1 located on chromosome 5q31.1 predict outcome in AML. Blood 2012, 119, 206–216. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, H.; Tang, L.; Lou, M.; Geng, Y. Silencing nc886, a non-coding RNA, induces apoptosis of human endometrial cancer cells-1A in vitro. Med. Sci. Monit. 2017, 23, 1317–1324. [Google Scholar] [CrossRef][Green Version]
- Lee, E.K.; Hong, S.-H.; Shin, S.; Lee, H.-S.; Lee, J.-S.; Park, E.J.; Choi, S.S.; Min, J.W.; Park, D.; Hwang, J.-A.; et al. nc886, a non-coding RNA and suppressor of PKR, exerts an oncogenic function in thyroid cancer. Oncotarget 2016, 7, 75000–75012. [Google Scholar] [CrossRef][Green Version]
- Milne, R.L.; Fletcher, A.S.; MacInnis, R.J.; Hodge, A.M.; Hopkins, A.H.; Bassett, J.K.; Bruinsma, F.J.; Lynch, B.M.; Dugue, P.A.; Jayasekara, H.; et al. Cohort profile: The Melbourne collaborative cohort study (Health 2020). Int. J. Epidemiol. 2017, 46, 1757–1757i. [Google Scholar] [CrossRef]
- Dugué, P.-A.; English, D.R.; MacInnis, R.J.; Jung, C.-H.; Bassett, J.K.; Fitzgerald, L.M.; Wong, E.M.; Joo, J.E.; Hopper, J.L.; Southey, M.C.; et al. Reliability of DNA methylation measures from dried blood spots and mononuclear cells using the human methylation 450k bead array. Sci. Rep. 2016, 6, 30317. [Google Scholar] [CrossRef]
- Dugué, P.-A.; Hodge, A.M.; Brinkman, M.T.; Bassett, J.K.; Shivappa, N.; Hebert, J.R.; Hopper, J.L.; English, D.R.; Milne, R.L.; Giles, G.G. Association between selected dietary scores and the risk of urothelial cell carcinoma: A prospective cohort study. Int. J. Cancer 2016, 139, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Chiuve, S.E.; Fung, T.T.; Rimm, E.B.; Hu, F.B.; McCullough, M.L.; Wang, M.; Stampfer, M.J.; Willett, W.C. Alternative dietary indices both strongly predict risk of chronic disease. J. Nutr. 2012, 142, 1009–1018. [Google Scholar] [CrossRef]
- Osborne, R.H.; Hopper, J.L.; Kirk, J.A.; Chenevix-Trench, G.; Thorne, H.J.; Sambrook, J.F.; Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer. kConFab: A research resource of Australasian breast cancer families. Med. J. Aust. 2000, 172, 463–464. [Google Scholar] [CrossRef] [PubMed]
- John, E.M.; Hopper, J.L.; Beck, J.C.; Knight, J.A.; Neuhausen, S.L.; Senie, R.T.; Ziogas, A.; Andrulis, I.L.; Anton-Culver, H.; Boyd, N.; et al. The Breast Cancer Family Registry: An infrastructure for cooperative multinational, interdisciplinary and translational studies of the genetic epidemiology of breast cancer. Breast Cancer Res. 2004, 6, R375–R389. [Google Scholar] [CrossRef]
- Terry, M.B.; Phillips, K.A.; Daly, M.B.; John, E.M.; Andrulis, I.L.; Buys, S.S.; Goldgar, D.E.; Knight, J.A.; Whittemore, A.S.; Chung, W.K.; et al. Cohort profile: The breast cancer Prospective Family Study Cohort (ProF-SC). Int. J. Epidemiol. 2016, 45, 683–692. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef]
- Nguyen-Dumont, T.; Steen, J.A.; Winship, I.; Park, D.J.; Pope, B.J.; Hammet, F.; Mahmoodi, M.; Tsimiklis, H.; Theys, D.; Clendenning, M.; et al. Mismatch repair gene pathogenic germline variants in a population-based cohort of breast cancer. Fam. Cancer 2020, 19, 197–202. [Google Scholar] [CrossRef]
- Amos, C.I.; Dennis, J.; Wang, Z.; Byun, J.; Schumacher, F.R.; Gayther, S.A.; Casey, G.; Hunter, D.J.; Sellers, T.A.; Gruber, S.B.; et al. The OncoArray Consortium: A network for understanding the genetic architecture of common cancers. Cancer Epidemiol. Biomark. Prev. 2017, 26, 126–135. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Howie, B.N.; Donnelly, P.; Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009, 5, e1000529. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.-K.; Adams, M.J.; Davies, G.; Howard, D.M.; Hall, L.S.; Padmanabhan, S.; Murray, A.D.; Smith, B.H.; Campbell, A.; Hayward, C.; et al. Genome-wide association study of alcohol consumption and genetic overlap with other health-related traits in UK Biobank (N = 112 117). Mol. Psychiatry 2017, 22, 1376–1384. [Google Scholar] [CrossRef]
- Marees, A.T.; De Kluiver, H.; Stringer, S.; Vorspan, F.; Curis, E.; Marie-Claire, C.; Derks, E.M. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int. J. Methods Psychiatr. Res. 2018, 27, e1608. [Google Scholar] [CrossRef]
- Joo, J.E.; Wong, E.M.; Baglietto, L.; Jung, C.-H.; Tsimiklis, H.; Park, D.J.; Wong, N.C.; English, D.R.; Hopper, J.L.; Severi, G.; et al. The use of DNA from archival dried blood spots with the Infinium Human Methylation 450 array. BMC Biotechnol. 2013, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Zhang, X.; Huang, C.C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value Methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef]
- Van Rooij, J.; Mandaviya, P.R.; Claringbould, A.; Felix, J.F.; Van Dongen, J.; Jansen, R.; Franke, L.; ’t Hoen, P.A.C.; Heijmans, B.; Van Meurs, J.B.; et al. Evaluation of commonly used analysis strategies for epigenome- and transcriptome-wide association studies through replication of large-scale population studies. Genome Biol. 2019, 20, 235-14. [Google Scholar] [CrossRef] [PubMed]
- Luijk, R.; Wu, H.; Ward-Caviness, C.K.; Hannon, E.; Carnero-Montoro, E.; Min, J.L.; Mandaviya, P.; Müller-Nurasyid, M.; Mei, H.; van der Maarel, S.M.; et al. Autosomal genetic variation is associated with DNA methylation in regions variably escaping X-chromosome inactivation. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.E.; Irizarry, R.A. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014, 15, R31. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.R.; Platt, R.W.; Schisterman, E.F.; Chu, H.; Westreich, D.; Richardson, D.; Poole, C. Illustrating bias due to conditioning on a collider. Int. J. Epidemiol. 2010, 39, 417–420. [Google Scholar] [CrossRef]
- Greenland, S. quantifying biases in causal models: Classical confounding vs collider-stratification bias. Epidemiology 2003, 14, 300–306. [Google Scholar] [CrossRef]
- Pearce, N.; Richiardi, L. Commentary: Three worlds collide: Berkson’s bias, selection bias and collider bias. Int. J. Epidemiol. 2014, 43, 521–524. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Benyamin, B.; McEvoy, B.P.; Gordon, S.D.; Henders, A.K.; Nyholt, D.R.; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010, 42, 565–569. [Google Scholar] [CrossRef]
- Consortium, I.H. Integrating common and rare genetic variation in diverse human populations. Nature 2010, 467, 52. [Google Scholar] [CrossRef]
- Ripke, S.; O’Dushlaine, C.; Chambert, K.; Moran, J.L.; Kähler, A.K.; Akterin, S.; Bergen, S.E.; Collins, A.L.; Crowley, J.J.; Fromer, M.; et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet. 2013, 45, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Bulik-Sullivan, B.; Finucane, H.K.; Anttila, V.; Gusev, A.; Day, F.R.; Loh, P.R.; ReproGen Consortium; Psychiatric Genomics Consortium; Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium; Duncan, L.; et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015, 47, 1236–1241. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Controls (n = 2272) | Cases (n = 2228) | |
|---|---|---|
| Population-based study (MCCS) | ||
| Age at blood draw (median [IQR]) | 59.8 [52.7–65.0] | 60.3 [53.3–65.6] |
| Sex (female) | 919 (40%) | 901 (40%) |
| Country of birth | ||
| Australia/NZ | 1602 (70%) | 1571 (69%) |
| UK/Northern Europe | 161 (7%) | 156 (7%) |
| Italy | 311 (14%) | 297 (13%) |
| Greece | 198 (9%) | 204 (9%) |
| Smoking status | ||
| Never | 1154 (51%) | 1106 (49%) |
| Former | 854 (38%) | 888 (39%) |
| Current | 263 (11%) | 233 (10%) |
| BMI (kg/m2) | 26.4 [24.1–29.1] | 26.8 [24.4–29.7] |
| Alcohol consumption (g/day) | 4.3 [0.0–17.1] | 4.3 [0.0–17.1] |
| Alternate Healthy Eating Index 2010 | 63.5 [56.0–71.0] | 64.0 [56.5–71.5] |
| Family-based studies (ABCFR/kConFab) a | ||
| Sex (female) | 123 (100%) | 87 (100%) |
| Chromosome | Position | Variant | REF a | ALT a | BETA a | SE a | Pa | R2 a | MAF a |
|---|---|---|---|---|---|---|---|---|---|
| 5 | 135378781 | rs3805700 | A | G | 0.093 | 0.024 | 1.2 × 10-4 | 0.003 | 0.26 |
| 5 | 135402852 | rs11956252 | G | C | 0.092 | 0.023 | 4.1 × 10-4 | 0.004 | 0.32 |
| 5 | 135403529 | rs6899012 | G | A | 0.093 | 0.022 | 3.9 × 10-4 | 0.004 | 0.32 |
| 5 | 135403745 | rs74634331 | AGTG | AG | 0.092 | 0.023 | 4.2 × 10-4 | 0.004 | 0.32 |
| 5 | 135404173 | rs9986124 | G | T | 0.093 | 0.023 | 4.1 × 10-4 | 0.004 | 0.32 |
| 5 | 135404613 | rs9986287 | T | C | 0.093 | 0.023 | 4.7 × 10-4 | 0.004 | 0.32 |
| 5 | 135406459 | rs7725702 | C | G | 0.097 | 0.022 | 1.2 × 10-4 | 0.004 | 0.36 |
| 5 | 135406534 | rs7725447 | G | A | 0.097 | 0.022 | 1.2 × 10-4 | 0.004 | 0.35 |
| 5 | 135406658 | rs2058043 | A | G | 0.098 | 0.022 | 1.0 × 10-4 | 0.004 | 0.36 |
| 5 | 135406894 | rs2058042 | G | A | 0.098 | 0.022 | 1.1 × 10-4 | 0.004 | 0.35 |
| 5 | 135407572 | rs4976470 | A | G | 0.099 | 0.022 | 8.0 × 10-4 | 0.004 | 0.36 |
| 5 | 135408325 | rs4976471 | T | A | 0.099 | 0.022 | 8.6 × 10-4 | 0.004 | 0.36 |
| 5 | 135409014 | rs6861956 | T | C | 0.096 | 0.022 | 1.5 × 10-4 | 0.004 | 0.36 |
| 5 | 135410863 | rs11742191 | A | G | 0.087 | 0.022 | 1.2 × 10-4 | 0.003 | 0.33 |
| 5 | 135411281 | rs11749522 | C | T | 0.087 | 0.022 | 1.1 × 10-4 | 0.003 | 0.33 |
| 5 | 135412195 | rs10079215 | A | G | 0.097 | 0.022 | 1.1 × 10-4 | 0.004 | 0.36 |
| 5 | 135412675 | rs35137944 | A | G | 0.097 | 0.022 | 1.0 × 10-4 | 0.004 | 0.36 |
| 5 | 135413026 | rs7724672 | A | G | 0.098 | 0.022 | 8.7 × 10-4 | 0.004 | 0.36 |
| 5 | 135414280 | rs2190622 | A | G | 0.100 | 0.022 | 4.6 × 10-4 | 0.005 | 0.36 |
| 5 | 135414455 | rs4246798 | A | G | 0.100 | 0.022 | 4.6 × 10-4 | 0.005 | 0.36 |
| 5 | 135414510 | rs4246799 | G | A | 0.100 | 0.022 | 5.3 × 10-4 | 0.005 | 0.36 |
| 5 | 135414866 | rs17169806 | C | T | 0.087 | 0.022 | 1.0 × 10-4 | 0.003 | 0.33 |
| 5 | 135415064 | rs62365993 | A | G | 0.087 | 0.022 | 1.1 × 10-4 | 0.003 | 0.33 |
| 5 | 135415300 | rs2346018 | C | A | 0.089 | 0.023 | 8.2 × 10-4 | 0.004 | 0.33 |
| 5 | 135415726 | rs2346019 | A | G | 0.101 | 0.022 | 5.4 × 10-4 | 0.005 | 0.36 |
| 5 | 135417898 | rs12653557 | G | T | 0.083 | 0.021 | 7.4 × 10-4 | 0.004 | 0.49 |
| 5 | 135418032 | rs917303 | G | A | 0.090 | 0.022 | 5.4 × 10-4 | 0.004 | 0.34 |
| 5 | 135418717 | rs4976472 | G | C | 0.083 | 0.021 | 6.6 × 10-4 | 0.004 | 0.49 |
| 5 | 135419159 | rs4976473 | C | A | 0.084 | 0.021 | 6.2 × 10-4 | 0.004 | 0.49 |
| 5 | 135422443 | rs11242311 | T | C | 0.086 | 0.021 | 3.9 × 10-4 | 0.004 | 0.49 |
| 5 | 135422507 | rs34835264 | G | GA | −0.087 | 0.022 | 5.3 × 10-4 | 0.004 | 0.50 |
| 5 | 135422598 | rs11242312 | G | A | 0.086 | 0.021 | 4.2 × 10-4 | 0.004 | 0.49 |
| 5 | 135422698 | rs10900843 | G | A | 0.084 | 0.021 | 5.7 × 10-4 | 0.004 | 0.49 |
| 5 | 135422738 | rs10900844 | A | G | 0.086 | 0.021 | 4.2 × 10-4 | 0.004 | 0.49 |
| 5 | 135422864 | rs11242313 | G | A | 0.087 | 0.021 | 3.4 × 10-4 | 0.004 | 0.49 |
| 5 | 135423029 | rs11242314 | T | C | 0.086 | 0.021 | 4.4 × 10-4 | 0.004 | 0.49 |
| 5 | 135424756 | rs13186426 | C | A | 0.083 | 0.021 | 8.5 × 10-4 | 0.004 | 0.48 |
| 5 | 135424847 | 5:135424847 | A | AAT | 0.083 | 0.021 | 7.0 × 10-4 | 0.004 | 0.49 |
| 5 | 135424922 | rs1465239 | A | G | 0.084 | 0.021 | 6.3 × 10-4 | 0.004 | 0.49 |
| 5 | 135427371 | rs1974552 | T | A | 0.089 | 0.022 | 8.1 × 10-4 | 0.004 | 0.34 |
| 5 | 135429640 | rs1558095 | C | T | 0.086 | 0.021 | 4.4 × 10-4 | 0.004 | 0.49 |
| 5 | 135431590 | rs1203219753 | A | G | 0.085 | 0.021 | 5.8 × 10-4 | 0.004 | 0.49 |
| 5 | 135435140 | rs1544486 | C | T | 0.087 | 0.021 | 4.1 × 10-45 | 0.004 | 0.49 |
| CpG | Chromosome | Position | Name | Location | Relation to Island | Enhancer | h2 (M-Values) | 95% CI (M-Values) | h2 (RINT-Values | 95% CI (RINT-Values) |
|---|---|---|---|---|---|---|---|---|---|---|
| cg08836729 | 5 | 135401437 | Yes | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |||
| cg16402693 | 5 | 135412139 | N_Shelf | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |||
| cg17974054 | 5 | 135413810 | N_Shore | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |||
| cg11852404 | 5 | 135414858 | N_Shore | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |||
| cg16684184 | 5 | 135415129 | Island | 0 | −0.14; 0.14 | 0.03 | −0.11; 0.17 | |||
| cg00308130 | 5 | 135415190 | Island | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |||
| cg15837280 | 5 | 135415258 | Island | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |||
| cg07158503 | 5 | 135415693 | N_Shore | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |||
| cg04515200 | 5 | 135415762 | N_Shore | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |||
| cg13581155 | 5 | 135415781 | N_Shore | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |||
| cg11978884 | 5 | 135415819 | N_Shore | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |||
| cg11608150 | 5 | 135415948 | N_Shore | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |||
| cg06478886 | 5 | 135416029 | N_Shore | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |||
| cg04481923 | 5 | 135416205 | MIR886 | Body | Island | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |
| cg18678645 | 5 | 135416331 | MIR886 | TSS200 | Island | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |
| cg06536614 | 5 | 135416381 | MIR886 | TSS200 | Island | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |
| cg26328633 | 5 | 135416394 | MIR886 | TSS200 | Island | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |
| cg25340688 | 5 | 135416398 | MIR886 | TSS200 | Island | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |
| cg26896946 | 5 | 135416405 | MIR886 | TSS200 | Island | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |
| cg00124993 | 5 | 135416412 | MIR886 | TSS200 | Island | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |
| cg08745965 | 5 | 135416529 | MIR886 | TSS1500 | S_Shore | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |
| cg16615357 | 5 | 135416594 | MIR886 | TSS1500 | S_Shore | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |
| cg18797653 | 5 | 135416613 | MIR886 | TSS1500 | S_Shore | 0 | −0.14; 0.14 | 0 | −0.14; 0.14 | |
| cg12897067 | 5 | 135418308 | S_Shore | 0.95 | 0.81; 1.09 | 0.76 | 0.62; 0.90 | |||
| cg05631625 | 5 | 135419019 | S_Shelf | 0.14 | 0.00; 0.28 | 0.15 | 0.01; 0.29 | |||
| cg01930756 | 5 | 135424444 | Yes | 0.09 | −0.05; 0.23 | 0.1 | −0.04; 0.24 |
| Estimate a | 95% CI | p-Value | |
|---|---|---|---|
| Age (years) | −0.005 | −0.012; 0.002 | 0.18 |
| Sex (female) | 0.047 | −0.114; 0.207 | 0.57 |
| Greece vs. Aus/NZ | −0.043 | −0.250; 0.164 | 0.68 |
| Italy vs. Aus/NZ | −0.068 | −0.239; 0.102 | 0.43 |
| Northern Europe vs. Aus/NZ | 0.171 | −0.049; 0.391 | 0.13 |
| Current vs. never smoker | −0.031 | −0.223; 0.162 | 0.76 |
| Former vs. never smoker | −0.050 | −0.176; 0.076 | 0.43 |
| BMI (in kg/m2) | 0.002 | −0.012; 0.016 | 0.79 |
| Alcohol consumption (g/day) | 0.001 | −0.003; 0.004 | 0.70 |
| Healthy eating index | 0.003 | −0.003; 0.008 | 0.36 |
| CD4 + T cells | −1.760 | −6.108; 2.589 | 0.43 |
| CD8+ T cells | −0.380 | −4.054; 3.294 | 0.84 |
| NK cells | −0.858 | −5.242; 3.525 | 0.70 |
| B cells | −2.616 | −6.391; 1.159 | 0.17 |
| Granulocytes | −1.852 | −5.969; 2.264 | 0.38 |
| Monocytes | −0.355 | −4.699; 3.990 | 0.87 |
| CpG | Chromosome | Position | ∆l a | Not Adjusted for SNPs b | Adjusted for rs2346018 | ||
|---|---|---|---|---|---|---|---|
| Biased HR (95% CI) c | p-Value | Biased HR (95% CI) c | p-Value | ||||
| cg06536614 | 5 | 135416381 | 143.6 | 3.1 (2.1–4.6) | 7 × 10−9 | 3.0 (2.0–4.3) | 3 × 10−8 |
| cg00124993 | 5 | 135416412 | 108.0 | 3.2 (2.2–4.7) | 2 × 10−8 | 3.0 (2.0–4.4) | 9 × 10−8 |
| cg26328633 | 5 | 135416394 | 107.5 | 3.2 (2.2–4.8) | 2 × 10−8 | 3.0 (2.0–4.5) | 4 × 10−8 |
| cg25340688 | 5 | 135416398 | 105.9 | 3.2 (2.1–4.7) | 3 × 10−8 | 2.9 (2.0–4.3) | 1 × 10−7 |
| cg26896946 | 5 | 135416405 | 92.1 | 3.6 (2.4–5.4) | 2 × 10−9 | 3.3 (2.2–5.0) | 8 × 10−9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dugué, P.-A.; Yu, C.; McKay, T.; Wong, E.M.; Joo, J.E.; Tsimiklis, H.; Hammet, F.; Mahmoodi, M.; Theys, D.; kConFab; et al. VTRNA2-1: Genetic Variation, Heritable Methylation and Disease Association. Int. J. Mol. Sci. 2021, 22, 2535. https://doi.org/10.3390/ijms22052535
Dugué P-A, Yu C, McKay T, Wong EM, Joo JE, Tsimiklis H, Hammet F, Mahmoodi M, Theys D, kConFab, et al. VTRNA2-1: Genetic Variation, Heritable Methylation and Disease Association. International Journal of Molecular Sciences. 2021; 22(5):2535. https://doi.org/10.3390/ijms22052535
Chicago/Turabian StyleDugué, Pierre-Antoine, Chenglong Yu, Timothy McKay, Ee Ming Wong, Jihoon Eric Joo, Helen Tsimiklis, Fleur Hammet, Maryam Mahmoodi, Derrick Theys, kConFab, and et al. 2021. "VTRNA2-1: Genetic Variation, Heritable Methylation and Disease Association" International Journal of Molecular Sciences 22, no. 5: 2535. https://doi.org/10.3390/ijms22052535
APA StyleDugué, P.-A., Yu, C., McKay, T., Wong, E. M., Joo, J. E., Tsimiklis, H., Hammet, F., Mahmoodi, M., Theys, D., kConFab, Hopper, J. L., Giles, G. G., Milne, R. L., Steen, J. A., Dowty, J. G., Nguyen-Dumont, T., & Southey, M. C. (2021). VTRNA2-1: Genetic Variation, Heritable Methylation and Disease Association. International Journal of Molecular Sciences, 22(5), 2535. https://doi.org/10.3390/ijms22052535