
Targeted Delivery of Soluble Guanylate Cyclase (sGC) Activator Cinaciguat to Renal Mesangial Cells via Virus-Mimetic Nanoparticles Potentiates Anti-Fibrotic Effects by cGMP-Mediated Suppression of the TGF-β Pathway

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
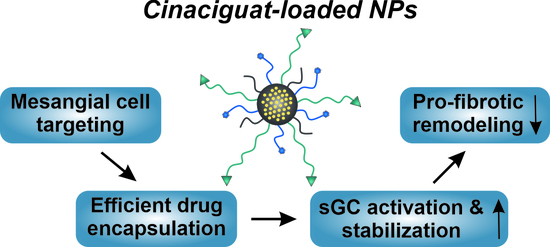
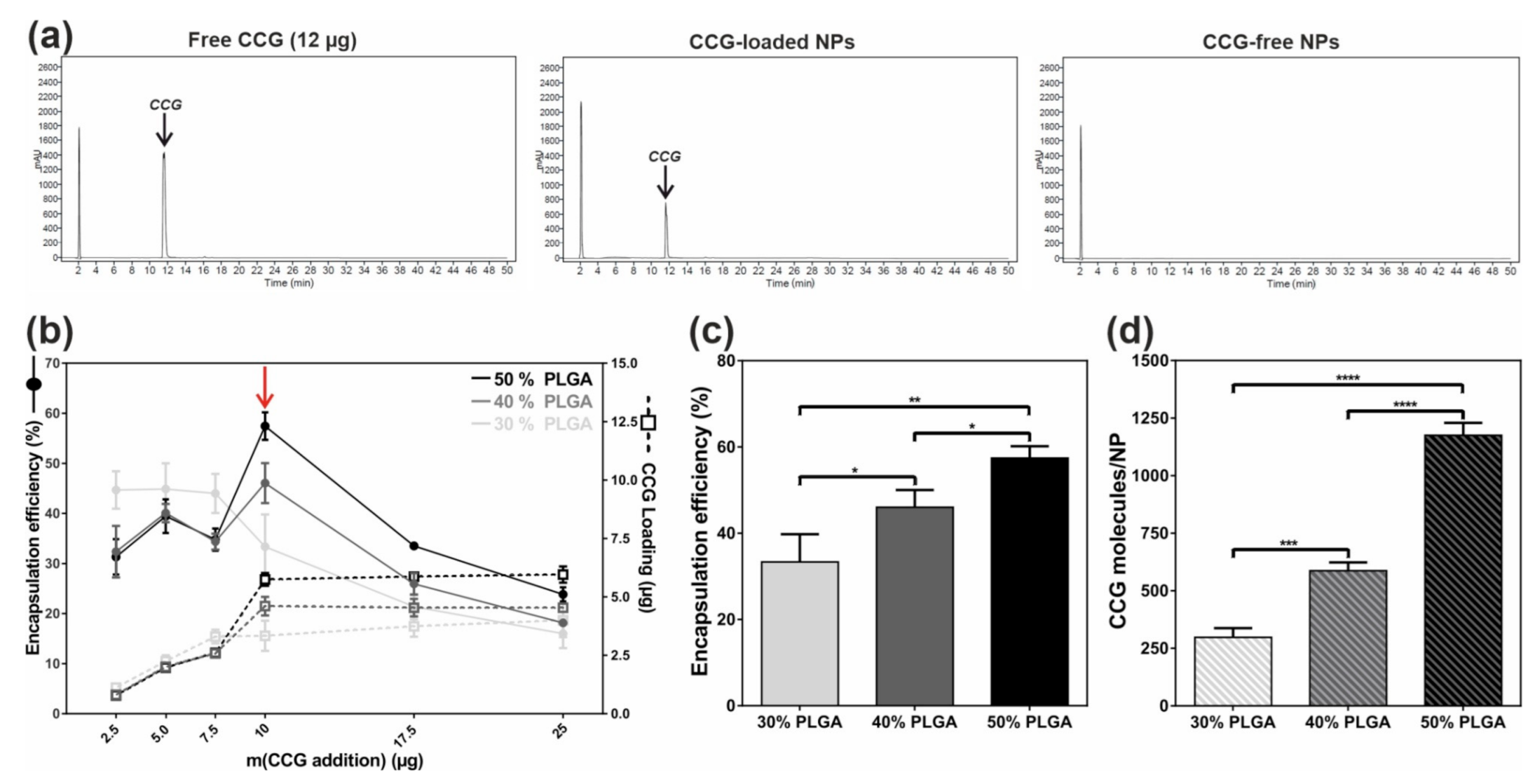
2.1. Efficient and Reproducible Encapsulation of CCG into Targeted NPs
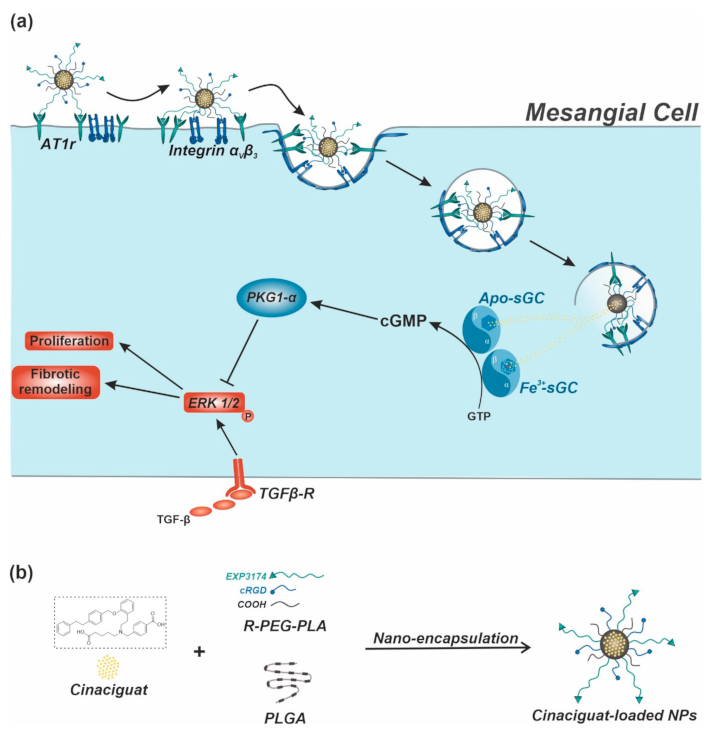
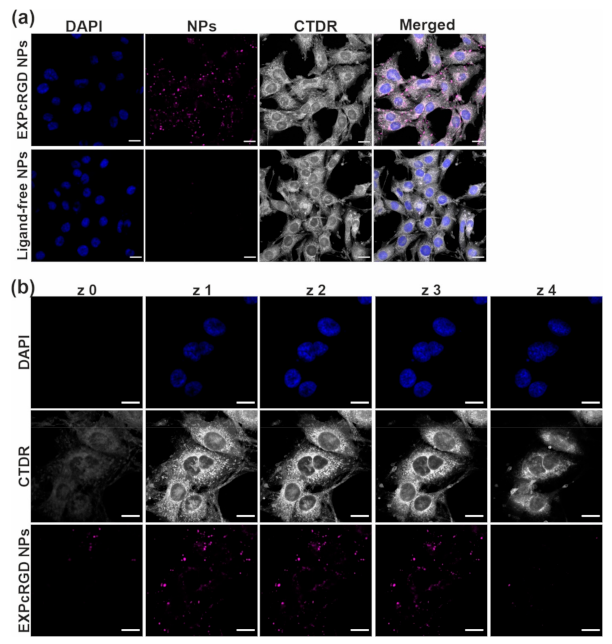
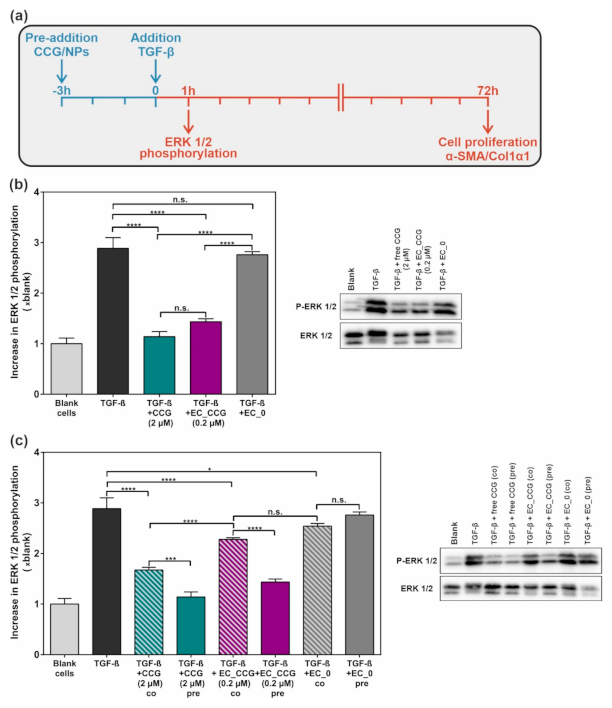
2.2. Targeted CCG Delivery Potentiates sGC Activation and Stabilization
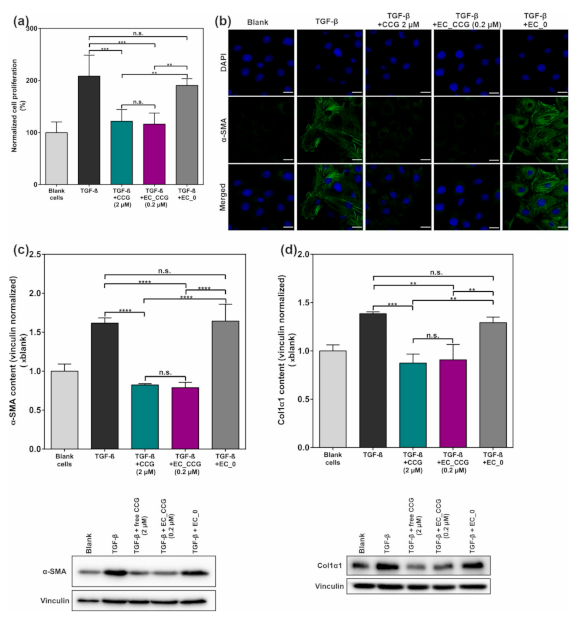
2.3. CCG-Loaded NPs Show A Substantial Anti-Fibrotic Effect via Suppression of the Non-Canonical TGF-β Pathway
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Synthesis and Functionalization of NP Polymer Components
3.2.2. NP Manufacture and CCG Encapsulation
3.2.3. NP Characterization
3.2.4. HPLC Analysis of Drug Encapsulation
3.2.5. Calculatory Determination of Encapsulation Parameters
3.2.6. CLSM Analysis of Uptake
3.2.7. Analysis of CCG Effect on Intracellular Targets
3.2.8. Analysis of Anti-Fibrotic CCG Effects
3.2.9. Cell Proliferation Assay
3.2.10. CLSM Analysis of α-SMA Expression
3.2.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cho, N.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global revalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.-J. Chronic kidney disease. Nat. Rev. Dis. Primers 2017, 3, 17088. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D.R. Global Prevalence of Chronic Kidney—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.M.; Zoungas, S.; Rossing, P.; Groop, P.-H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Primers 2015, 1, 15018. [Google Scholar] [CrossRef] [PubMed]
- Umanath, K.; Lewis, J.B. Update on Diabetic Nephropathy: Core Curriculum 2018. Am. J. Kidney Dis. 2018, 71, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Magee, C.; Grieve, D.J.; Watson, C.J.; Brazil, D.P. Diabetic Nephropathy: A Tangled Web to Unweave. Cardiovasc. Drugs Ther. 2017, 31, 579–592. [Google Scholar] [CrossRef] [Green Version]
- Rao, V.; Tan, S.H.; Candasamy, M.; Bhattamisra, S.K. Diabetic nephropathy: An update on pathogenesis and drug development. Diabetes Metab. Syndr. 2019, 13, 754–762. [Google Scholar] [CrossRef]
- Kanwar, Y.S.; Sun, L.; Xie, P.; Liu, F.-Y.; Chen, S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu. Rev. Pathol. 2011, 6, 395–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolset, S.O.; Reinholt, F.P.; Jenssen, T. Diabetic Nephropathy and Extracellular Matrix. J. Histochem. Cytochem. 2012, 60, 976–986. [Google Scholar] [CrossRef] [Green Version]
- Scindia, Y.M.; Deshmukh, U.S.; Bagavant, H. Mesangial Pathology in Glomerular Disease: Targets for Therapeutic Intervention. Adv. Drug Delivery Rev. 2010, 62, 1337–1343. [Google Scholar] [CrossRef] [Green Version]
- Schlöndorff, D.; Banas, B. The Mesangial Cell Revisited: No Cell Is an Island. J. Am. Soc. Nephrol. 2009, 20, 1179–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arora, M.K.; Singh, U.K. Molecular mechanisms in the pathogenesis of diabetic nephropathy: An update. Vascul. Pharmacol. 2013, 58, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Huynh, P.; Chai, Z. Transforming growth factor β (TGF-β) and related molecules in chronic kidney disease (CKD). Clin. Sci. 2019, 133, 287–313. [Google Scholar] [CrossRef]
- Zeng, L.-F.; Xiao, Y.; Sun, L. A Glimpse of the Mechanisms Related to Renal Fibrosis in Diabetic Nephropathy. Adv. Exp. Med. Biol. 2019, 1165, 49–79. [Google Scholar] [CrossRef] [PubMed]
- Ruggenenti, P.; Cravedi, P.; Remuzzi, G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat. Rev. Nephrol. 2010, 6, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessari, P. Nitric oxide in the normal kidney and in patients with diabetic nephropathy. J. Nephrol. 2015, 28, 257–268. [Google Scholar] [CrossRef]
- Schinner, E.; Wetzl, V.; Schlossmann, J. Cyclic nucleotide signalling in kidney fibrosis. Int. J. Mol. Sci. 2015, 16, 2320–2351. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Johnson, D.W.; Gobe, G.C. The role of cGMP and its signaling pathways in kidney disease. Am. J. Physiol. Renal Physiol. 2016, 311, F671–F681. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Kraehling, J.; Eitner, F.; Bénardeau, A.; Sandner, P. The Impact of the Nitric Oxide (NO)/Soluble Guanylyl Cyclase (sGC) Signaling Cascade on Kidney Health and Disease: A Preclinical Perspective. Int. J. Mol. Sci. 2018, 19, 1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Vanhoutte, P.M. Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, A.J.; Stasch, J.-P. Soluble Guanylate Cyclase. In Nitric Oxide: Biology and Pathobiology; Academic Press: Cambridge, MA, USA, 2010. [Google Scholar]
- Murad, F. Nitric Oxide and Cyclic GMP in Cell Signaling and Drug Development. N. Engl. J. Med. 2006, 355, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Gladwin, M.T.; Weitzberg, E. Strategies to increase nitric oxide signalling in cardiovascular disease. Nat. Rev. Drug Discov. 2015, 14, 623–641. [Google Scholar] [CrossRef] [PubMed]
- Mátyás, C.; Németh, B.T.; Oláh, A.; Hidi, L.; Birtalan, E.; Kellermayer, D.; Ruppert, M.; Korkmaz-Icöz, S.; Kökény, G.; Horváth, E.M.; et al. The soluble guanylate cyclase activator cinaciguat prevents cardiac dysfunction in a rat model of type-1 diabetes mellitus. Cardiovasc. Diabetol. 2015, 14, 145. [Google Scholar] [CrossRef] [Green Version]
- Kalk, P.; Godes, M.; Relle, K.; Rothkegel, C.; Hucke, A.; Stasch, J.-P.; Hocher, B. NO-independent activation of soluble guanylate cyclase prevents disease progression in rats with 5/6 nephrectomy. Br. J. Pharmacol. 2006, 148, 853–859. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, L.S.; Kretschmer, A.; Lawrenz, B.; Hocher, B.; Stasch, J.-P. Chronic Activation of Heme Free Guanylate Cyclase Leads to Renal Protection in Dahl Salt-Sensitive Rats. PLoS ONE 2015, 10, e0145048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohenstein, B.; Daniel, C.; Wagner, A.; Stasch, J.-P.; Hugo, C. Stimulation of soluble guanylyl cyclase inhibits mesangial cell proliferation and matrix accumulation in experimental glomerulonephritis. Am. J. Physiol. Renal Physiol. 2005, 288, F685–F693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czirok, S.; Fang, L.; Radovits, T.; Szabó, G.; Szénási, G.; Rosivall, L.; Merkely, B.; Kökény, G. Cinaciguat ameliorates glomerular damage by reducing ERK1/2 activity and TGF-ß expression in type-1 diabetic rats. Sci. Rep. 2017, 7, 11218. [Google Scholar] [CrossRef] [PubMed]
- Beyer, C.; Zenzmaier, C.; Palumbo-Zerr, K.; Mancuso, R.; Distler, A.; Dees, C.; Zerr, P.; Huang, J.; Maier, C.; Pachowsky, M.L.; et al. Stimulation of the soluble guanylate cyclase (sGC) inhibits fibrosis by blocking non-canonical TGFβ signalling. Ann. Rheum. Dis. 2015, 74, 1408–1416. [Google Scholar] [CrossRef] [Green Version]
- Erdmann, E.; Semigran, M.J.; Nieminen, M.S.; Gheorghiade, M.; Agrawal, R.; Mitrovic, V.; Mebazaa, A. Cinaciguat, a soluble guanylate cyclase activator, unloads the heart but also causes hypotension in acute decompensated heart failure. Eur. Heart J. 2013, 34, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Glassman, P.M.; Muzykantov, V.R. Pharmacokinetic and Pharmacodynamic Properties of Drug Delivery Systems. J. Pharmacol. Exp. Ther. 2019, 370, 570–580. [Google Scholar] [CrossRef]
- Yoo, J.; Park, C.; Lee, D.; Koo, H. Active Targeting Strategies Using Biological Ligands for Nanoparticle Drug Delivery Systems. Cancers 2019, 11, 640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nag, O.K.; Delehanty, J.B. Active Cellular and Subcellular Targeting of Nanoparticles for Drug Delivery. Pharmaceutics 2019, 11, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Wen, H.F.; Yu, D.G.; Yang, Y.; Zhang, D.F. Electrosprayed hydrophilic nanocomposites coated with shellac for colon-specific delayed drug delivery. Mater. Des. 2018, 143, 248–255. [Google Scholar] [CrossRef]
- Colino, C.I.; Lanao, J.M.; Gutierrez-Milan, C. Targeting of Hepatic Macrophages by Therapeutic Nanoparticles. Front. Immunol. 2020, 11, 218. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zeng, Y.; Peng, K.; Liu, C.; Zhang, Z.; Zhang, L. Design and evaluation of glomerulus mesangium-targeted PEG-PLGA nanoparticles loaded with dexamethasone acetate. Acta Pharm. Sin. 2019, 40, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, D.; Maslanka Figueroa, S.; Beck, S.; Abstiens, K.; Witzgall, R.; Schweda, F.; Tauber, P.; Goepferich, A. Adenovirus-Mimetic Nanoparticles: Sequential Ligand-Receptor Interplay as a Universal Tool for Enhanced In Vitro/In Vivo Cell Identification. ACS Appl. Mater. Interfaces 2020. [Google Scholar] [CrossRef] [PubMed]
- Maslanka Figueroa, S.; Fleischmann, D.; Goepferich, A. Biomedical nanoparticle design: What we can learn from viruses. J. Control. Release 2020. [Google Scholar] [CrossRef]
- Fleischmann, D.; Maslanka Figueroa, S.; Goepferich, A. Steric Shielding of cRGD-Functionalized Nanoparticles from Premature Exposition to Off-Target Endothelial Cells under a Physiological Flow. ACS Appl. Bio Mater. 2020. [Google Scholar] [CrossRef]
- Maslanka Figueroa, S.; Fleischmann, D.; Beck, S.; Goepferich, A. Thermodynamic, Spatial and Methodological Considerations for the Manufacturing of Therapeutic Polymer Nanoparticles. Pharm. Res. 2020, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakral, S.; Thakral, N.K. Prediction of drug-polymer miscibility through the use of solubility parameter based Flory-Huggins interaction parameter and the experimental validation: PEG as model polymer. J. Pharm. Sci. 2013, 102, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Satchell, S.C.; Braet, F. Glomerular Endothelial Cell Fenestrations: An Integral Component of the Glomerular Filtration Barrier. Am. J. Physiol. Renal Physiol. 2009, 296, 947–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, H.Y.; Kwon, S.M.; Chung, H.; Lee, S.-Y.; Kwon, S.-H.; Jeon, H.; Kim, Y.; Park, J.H.; Kim, J.; Her, S.; et al. Cellular uptake mechanism and intracellular fate of hydrophobically modified glycol chitosan nanoparticles. J. Control. Release 2009, 135, 259–267. [Google Scholar] [CrossRef]
- Seo, S.-J.; Chen, M.; Wang, H.; Kang, M.S.; Leong, K.W.; Kim, H.-W. Extra- and intra-cellular fate of nanocarriers under dynamic interactions with biology. Nano Today 2017, 14, 84–99. [Google Scholar] [CrossRef]
- Meurer, S.; Pioch, S.; Pabst, T.; Opitz, N.; Schmidt, P.M.; Beckhaus, T.; Wagner, K.; Matt, S.; Gegenbauer, K.; Geschka, S.; et al. Nitric oxide-independent vasodilator rescues heme-oxidized soluble guanylate cyclase from proteasomal degradation. Circ. Res. 2009, 105, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evgenov, O.V.; Pacher, P.; Schmidt, P.M.; Hasko, G.; Schmidt, H.H.H.W.; Stasch, J.-P. NO-independent stimulators and activators of soluble guanylate cyclase: Discovery and therapeutic potential. Nat. Rev. Drug Discov. 2006, 5, 755–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, F.; Baskaran, P.; Ma, X.; Dunten, P.W.; Schaefer, M.; Stasch, J.-P.; Beuve, A.; van den Akker, F. Structure of cinaciguat (BAY 58–2667) bound to Nostoc H-NOX domain reveals insights into heme-mimetic activation of the soluble guanylyl cyclase. J. Biol. Chem. 2010, 285, 22651–22657. [Google Scholar] [CrossRef] [Green Version]
- Smolenski, A.; Burckhardt, M.; Eigenthaler, M.; Butt, E.; Gambaryan, S.; Lohmann, S.M.; Walter, U. Functional analysis of cGMP-dependent protein kinases I and II as mediators of NO/cGMP effects. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1998, 358, 134–139. [Google Scholar] [CrossRef]
- Geiselhöringer, A.; Gaisa, M.; Hofmann, F.; Schlossmann, J. Distribution of IRAG and cGKI-isoforms in murine tissues. FEBS Lett. 2004, 575, 19–22. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, A.; Jelkmann, W.; Bauer, C. Mesangial Cells Derived from Glomeruli Produce an Erythropoiesis Stimulating Factor in Cell Culture. FEBS Lett. 1982, 137, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Pfeilschifter, J.; Schalkwijk, C.; Briner, V.A.; van den Bosch, H. Cytokine-stimulated secretion of group II phospholipase A2 by rat mesangial cells. Its contribution to arachidonic acid release and prostaglandin synthesis by cultured rat glomerular cells. J. Clin. Investig. 1993, 92, 2516–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslanka Figueroa, S.; Fleischmann, D.; Beck, S.; Tauber, P.; Witzgall, R.; Schweda, F.; Goepferich, A. Nanoparticles Mimicking Viral Cell Recognition Strategies Are Superior Transporters into Mesangial Cells. Adv. Sci. 2020, 7, 1903204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abstiens, K.; Gregoritza, M.; Goepferich, A.M. Ligand Density and Linker Length are Critical Factors for Multivalent Nanoparticle-Receptor Interactions. ACS Appl. Mater. Interfaces 2019, 11, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Maslanka Figueroa, S.; Veser, A.; Abstiens, K.; Fleischmann, D.; Beck, S.; Goepferich, A. Influenza A Virus Mimetic Nanoparticles Trigger Selective Cell Uptake. Proc. Natl. Acad. Sci. USA 2019, 116, 9831–9836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muenster, U.; Becker-Pelster, E.-M.; Mao, S.; Ni, R.; Liang, Z. Process for the Preparation of Porous Microparticles. U.S. Patent 15,735,421, 7 June 2016. [Google Scholar]
- Kansal, A.R.; Torquato, S.; Stillingser, F.H. Computer generation of dense polydisperse sphere packings. J. Chem. Phys. 2002, 117, 8212–8218. [Google Scholar] [CrossRef] [Green Version]
- Fedors, R.F. A method for estimating both the solubility parameters and molar volumes of liquids. Polym. Eng. Sci. 1974, 14, 147–154. [Google Scholar] [CrossRef]
- Schramm, A.; Mueller-Thuemen, P.; Littmann, T.; Harloff, M.; Ozawa, T.; Schlossmann, J. Establishing a Split Luciferase Assay for Proteinkinase G (PKG) Interaction Studies. Int. J. Mol. Sci. 2018, 19, 1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowry, O.H.; Rosebrough, N.J.; Lewis Farr, A.; Randall, R.J. Protein Measurement with the Folin Phenol Reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Image Lab 6.1 Software; Bio-Rad Laboratories GmbH: Munich, Germany, 2020.
- Danielpour, D.; Kim, K.Y.; Dart, L.L.; Watanabe, S.; Roberts, A.B.; Sporn, M. Sandwich Enzyme-Linked Immunosorbent Assays (Selisas) Quantitate and Distinguish Two Forms of Transforming Growth Factor-Beta (TGF-β1 and TGF-β2) in Complex Biological Fluids. Growth Factors 1989, 2, 61–71. [Google Scholar] [CrossRef]
- GraphPad Prism 6 Software; GraphPad Software: San Diego, CA, USA, 2020.


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fleischmann, D.; Harloff, M.; Maslanka Figueroa, S.; Schlossmann, J.; Goepferich, A. Targeted Delivery of Soluble Guanylate Cyclase (sGC) Activator Cinaciguat to Renal Mesangial Cells via Virus-Mimetic Nanoparticles Potentiates Anti-Fibrotic Effects by cGMP-Mediated Suppression of the TGF-β Pathway. Int. J. Mol. Sci. 2021, 22, 2557. https://doi.org/10.3390/ijms22052557
Fleischmann D, Harloff M, Maslanka Figueroa S, Schlossmann J, Goepferich A. Targeted Delivery of Soluble Guanylate Cyclase (sGC) Activator Cinaciguat to Renal Mesangial Cells via Virus-Mimetic Nanoparticles Potentiates Anti-Fibrotic Effects by cGMP-Mediated Suppression of the TGF-β Pathway. International Journal of Molecular Sciences. 2021; 22(5):2557. https://doi.org/10.3390/ijms22052557
Chicago/Turabian StyleFleischmann, Daniel, Manuela Harloff, Sara Maslanka Figueroa, Jens Schlossmann, and Achim Goepferich. 2021. "Targeted Delivery of Soluble Guanylate Cyclase (sGC) Activator Cinaciguat to Renal Mesangial Cells via Virus-Mimetic Nanoparticles Potentiates Anti-Fibrotic Effects by cGMP-Mediated Suppression of the TGF-β Pathway" International Journal of Molecular Sciences 22, no. 5: 2557. https://doi.org/10.3390/ijms22052557