
Cellular Mechanisms of the Anti-Arrhythmic Effect of Cardiac PDE2 Overexpression

 , , , and
, , , and 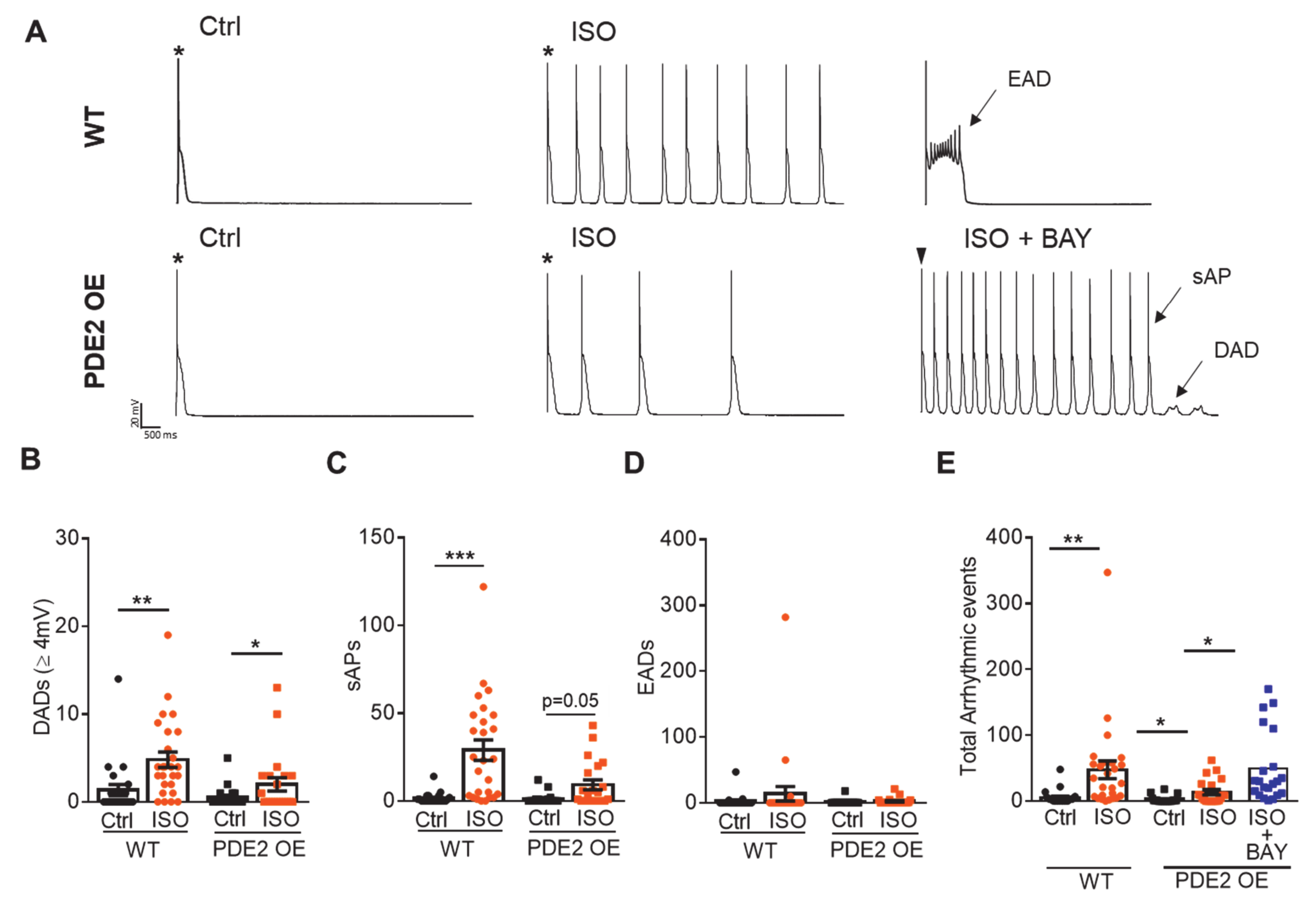{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
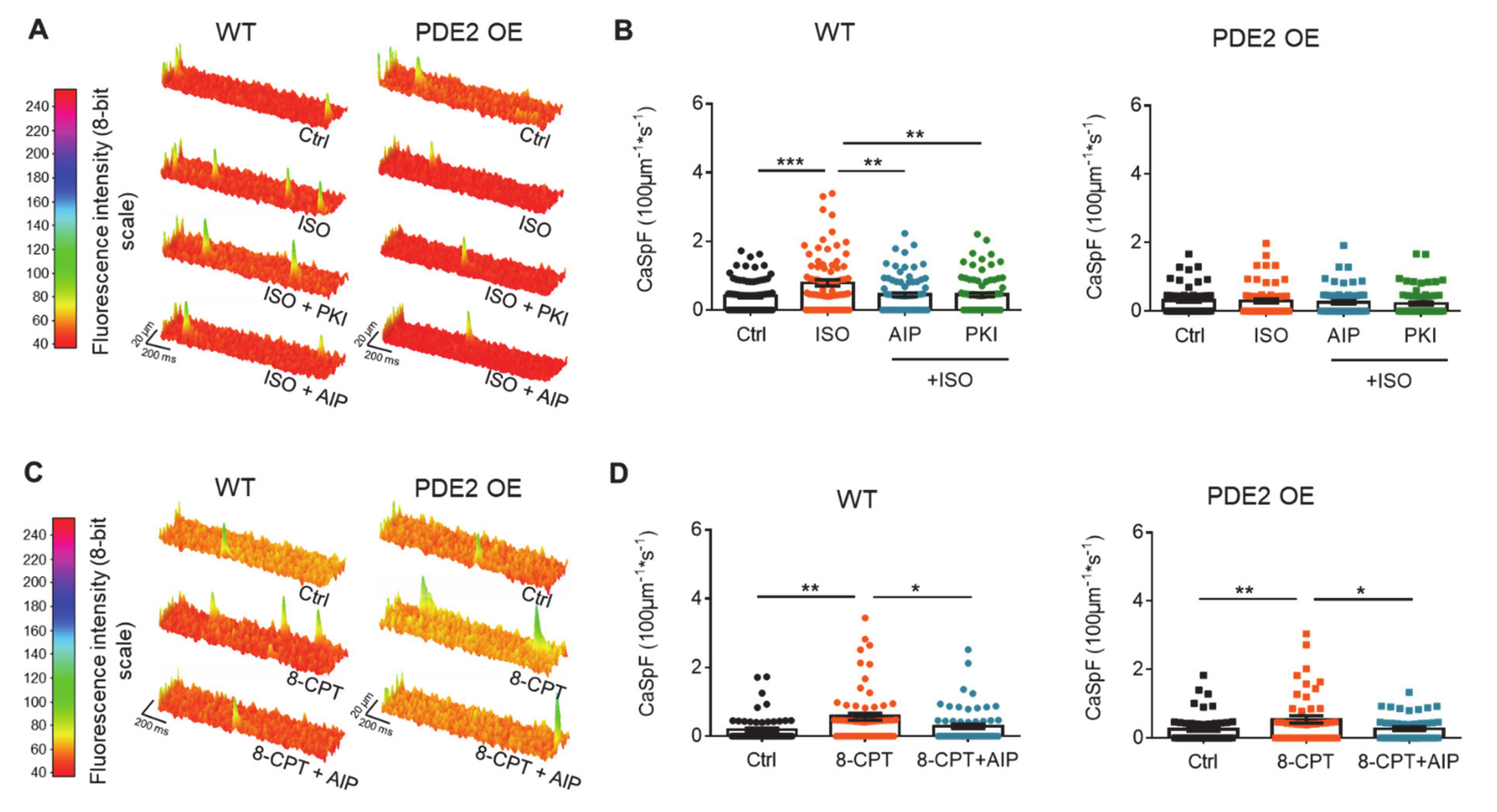
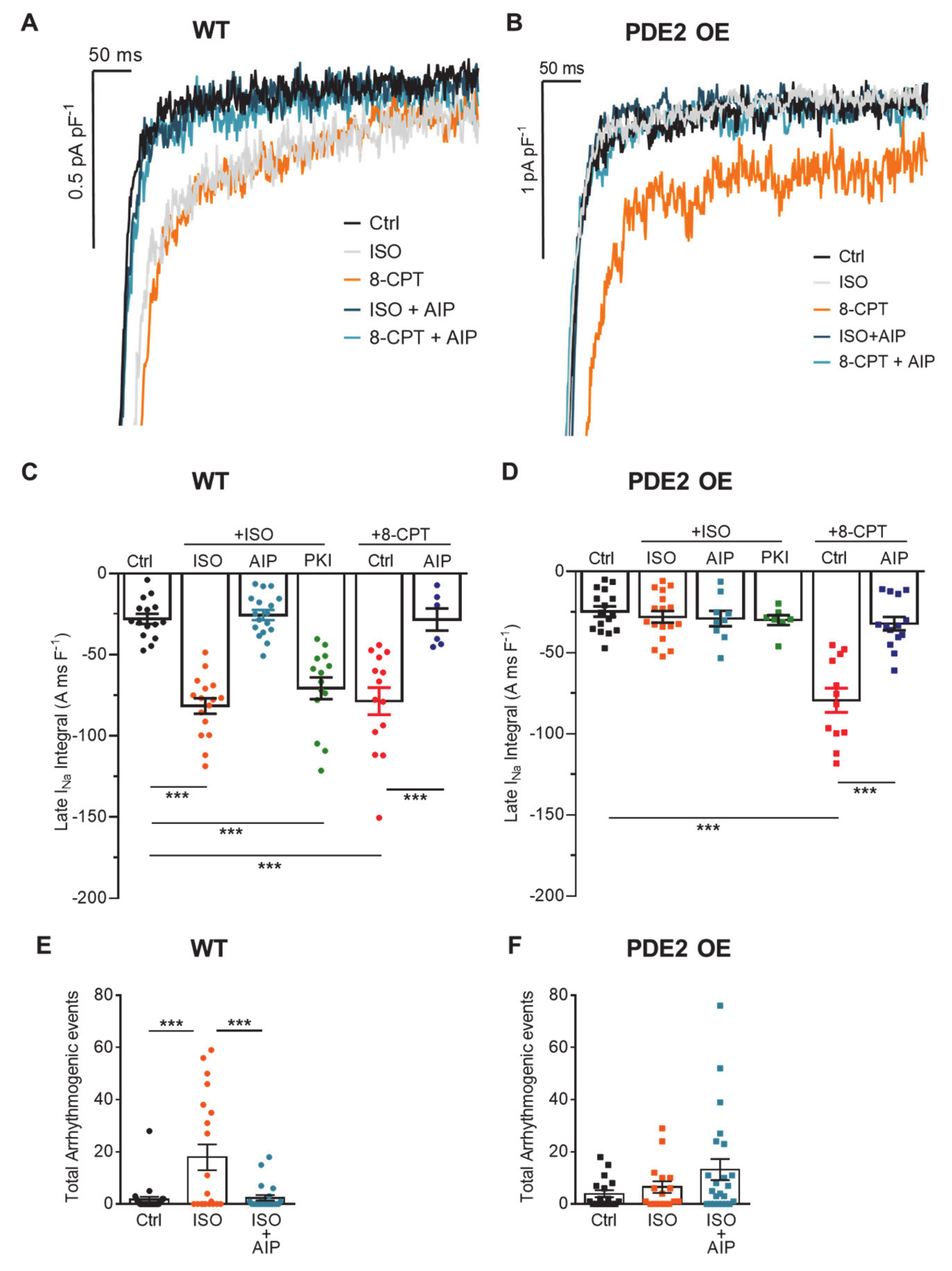
2. Results
3. Discussion
4. Materials and Methods
4.1. Animal Care and Adult Mouse Ventricular Myocyte Isolation
4.2. ECG Measurements of ex vivo Perfused Hearts
4.3. Electrophysiology
4.4. Ca2+ Spark Analysis
4.5. Protein Quantification
4.6. In Silico Modeling
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAR | Area at risk |
| AC | Adenylyl cyclase |
| AIP | Autocamptide-2 Related Inhibitor Peptide |
| AP | Action potential |
| APD | Action potential duration |
| ATP | Adenosine triphosphate |
| β-AR | β-adrenoceptor |
| CaMKII | Ca2+-calmodulin-dependent protein kinase II |
| CaSpF | Calcium spark frequency |
| cAMP | 3′,5′-cyclic adenosine monophosphate |
| cGMP | 3′,5′-cyclic guanosine monophosphate |
| 8-CPT | 8-(4-chlorophenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate |
| DAD | Delayed afterdepolarization |
| DAG | Diacylglycerol |
| DRP1 | Dynamin related protein 1 |
| EAD | Early afterdepolarization |
| ECC | Excitation-contraction coupling |
| ECG | Electrocardiogram |
| Epac | Exchange protein directly activated by cAMP |
| GAF | cGMP-specific phosphodiesterases, adenylyl cyclases and FhlA |
| HF | Heart failure |
| IP3 | Inositol triphosphate |
| ISO | isoprenaline |
| LAD | Left anterior descending coronary artery |
| LTCC | L-type Ca2+ channel |
| MI | Myocardial infarction |
| NCX | Na+/Ca2+ exchanger |
| NKA | Na+/K+ ATPase |
| NP | Natriuretic peptide |
| OE | Overexpression/ overexpressing |
| PDE | Phosphodiesterase |
| pGC | Particulate guanylyl cyclases |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PKI | Protein kinase inhibitor peptide |
| PLB | Phospholamban |
| PLM | Phospholemman |
| PVC | Premature ventricular complexes |
| RyR | Ryanodine receptors |
| sAC | Soluble adenylyl cyclases |
| sAP | Spontaneous action potential |
| SCD | Sudden cardiac death |
| SERCA2 | SR Ca2+-ATPase type-2a |
| SNP | Sodium nitroprusside |
| SR | Sarcoplasmic reticulum |
| TTC | 2,3,5-triphenyltetrazolium chloride |
| VES | Ventricular extrasystoles |
| WT | Wildtype |
References
- Huizar, J.F.; Ellenbogen, K.A.; Tan, A.Y.; Kaszala, K. Arrhythmia-Induced Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2328–2344. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.M.; Antoons, G. Arrhythmogenic Mechanisms in Heart Failure: Linking β-Adrenergic Stimulation, Stretch, and Calcium. Front. Physiol. 2018, 9, 1453. [Google Scholar] [CrossRef] [PubMed]
- Grandi, E.; Ripplinger, C.M. Antiarrhythmic mechanisms of beta blocker therapy. Pharmacol. Res. 2019, 146, 104274. [Google Scholar] [CrossRef] [PubMed]
- Mosterd, A.; Hoes, A.W. Clinical epidemiology of heart failure. Heart 2007, 93, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Van Heerebeek, L.; Hamdani, N.; Falcão-Pires, I.; Leite-Moreira, A.F.; Begieneman, M.P.; Bronzwaer, J.G.; van der Velden, J.; Stienen, G.J.; Laarman, G.J.; Somsen, A.; et al. Low Myocardial Protein Kinase G Activity in Heart Failure with Preserved Ejection Fraction. Circulation 2012, 126, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Jhund, P.; Rouleau, J.; Swedberg, K.; Zile, M.; Lefkowitz, M.; Prescott, M.; Shi, V.; Solomon, S.; Packer, M.; McMurray, J. Low Urinary Cgmp/Bnp Ratio Is Associated with Worse Outcomes in Heart Failure but Is Increased by Treatment with Sacubitril/Valsartan: An Analysis of Paradigm-HF. J. Am. Coll. Cardiol. 2017, 69, 679. [Google Scholar] [CrossRef]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin–Neprilysin Inhibition versus Enalapril in Heart Failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef]
- Blanton, R.M. cGMP Signaling and Modulation in Heart Failure. J. Cardiovasc. Pharmacol. 2020, 75, 385–398. [Google Scholar] [CrossRef]
- Emdin, M.; Aimo, A.; Castiglione, V.; Vergaro, G.; Georgiopoulos, G.; Saccaro, L.F.; Lombardi, C.M.; Passino, C.; Cerbai, E.; Metra, M.; et al. Targeting Cyclic Guanosine Monophosphate to Treat Heart Failure. J. Am. Coll. Cardiol. 2020, 76, 1795–1807. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef]
- Kumari, N.; Gaur, H.; Bhargava, A. Cardiac voltage gated calcium channels and their regulation by β-adrenergic signaling. Life Sci. 2018, 194, 139–149. [Google Scholar] [CrossRef]
- Van der Heyden, M.A.; Wijnhoven, T.J.; Opthof, T. Molecular aspects of adrenergic modulation of the transient outward current. Cardiovasc. Res. 2006, 71, 430–442. [Google Scholar] [CrossRef]
- Iqbal, S.M.; Lemmens-Gruber, R. Phosphorylation of cardiac voltage-gated sodium channel: Potential players with multiple dimensions. Acta Physiol. 2019, 225, e13210. [Google Scholar] [CrossRef]
- Grimm, M.; Brown, J.H. Beta-adrenergic receptor signaling in the heart: Role of CaMKII. J. Mol. Cell. Cardiol. 2010, 48, 322–330. [Google Scholar] [CrossRef]
- Saad, N.S.; Elnakish, M.T.; Ahmed, A.A.; Janssen, P.M. Protein Kinase A as a Promising Target for Heart Failure Drug Development. Arch. Med Res. 2018, 49, 530–537. [Google Scholar] [CrossRef]
- Enriquez, A.; Frankel, D.S.; Baranchuk, A. Pathophysiology of ventricular tachyarrhythmias: From automaticity to reentry. Herzschrittmacherther. Elektrophysiol. 2017, 28, 149–156. [Google Scholar] [CrossRef]
- Weber, S.; Zeller, M.; Guan, K.; Wunder, F.; Wagner, M.; El-Armouche, A. PDE2 at the crossway between cAMP and cGMP signalling in the heart. Cell. Signal. 2017, 38, 76–84. [Google Scholar] [CrossRef]
- Sadek, M.S.; Cachorro, E.; El-Armouche, A.; Kämmerer, S. Therapeutic Implications for PDE2 and cGMP/cAMP Mediated Crosstalk in Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 7462. [Google Scholar] [CrossRef]
- Rosman, G.J.; Martins, T.J.; Sonnenburg, W.K.; Beavo, J.A.; Ferguson, K.; Loughney, K. Isolation and characterization of human cDNAs encoding a cGMP-stimulated 3’,5’-cyclic nucleotide phosphodiesterase. Gene 1997, 191, 89–95. [Google Scholar] [CrossRef]
- Mongillo, M.; Tocchetti, C.G.; Terrin, A.; Lissandron, V.; Cheung, Y.F.; Dostmann, W.R.; Pozzan, T.; Kass, D.A.; Paolocci, N.; Houslay, M.D.; et al. Compartmentalized phosphodiesterase-2 activity blunts beta-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ. Res. 2006, 98, 226–234. [Google Scholar] [CrossRef]
- Lugnier, C.; Keravis, T.; le Bec, A.; Pauvert, O.; Proteau, S.; Rousseau, E. Characterization of cyclic nucleotide phos-phodiesterase isoforms associated to isolated cardiac nuclei. Biochim. Biophys. Acta Gen. Subj. 1999, 1472, 431–446. [Google Scholar] [CrossRef]
- Liu, D.; Wang, Z.; Nicolas, V.; Lindner, M.; Mika, D.; Vandecasteele, G.; Fischmeister, R.; Brenner, C. PDE2 regulates membrane potential, respiration and permeability transition of rodent subsarcolemmal cardiac mitochondria. Mitochondrion 2019, 47, 64–75. [Google Scholar] [CrossRef]
- Monterisi, S.; Lobo, M.J.; Livie, C.; Castle, J.C.; Weinberger, M.; Baillie, G.S.; Surdo, N.C.; Musheshe, N.; Stangherlin, A.; Gottlieb, E.; et al. PDE2A2 regulates mitochondria morphology and apoptotic cell death via local modulation of cAMP/PKA signalling. eLife 2017, 6, e21374. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Russwurm, M.; Günnewig, K.; Gertz, M.; Zoidl, G.; Ramos, L.; Buck, J.; Levin, L.R.; Rassow, J.; Manfredi, G.; et al. A Phosphodiesterase 2A Isoform Localized to Mitochondria Regulates Respiration. J. Biol. Chem. 2011, 286, 30423–30432. [Google Scholar] [CrossRef]
- Mongillo, M.; McSorley, T.; Evellin, S.; Sood, A.; Lissandron, V.; Terrin, A.; Huston, E.; Hannawacker, A.; Lohse, M.J.; Pozzan, T.; et al. Fluorescence Resonance Energy Transfer–Based Analysis of cAMP Dynamics in Live Neonatal Rat Cardiac Myocytes Reveals Distinct Functions of Compartmentalized Phosphodiesterases. Circ. Res. 2004, 95, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, J.U.; Perera, R.K.; Steinbrecher, J.H.; Lehnart, S.E.; Maier, L.S.; Hasenfuss, G.; Nikolaev, V.O. In vivo model with targeted cAMP biosensor reveals changes in receptor–microdomain communication in cardiac disease. Nat. Commun. 2015, 6, 6965. [Google Scholar] [CrossRef] [PubMed]
- Mehel, H.; Emons, J.; Vettel, C.; Wittkopper, K.; Seppelt, D.; Dewenter, M.; Lutz, S.; Sossalla, S.; Maier, L.S.; Lechene, P.; et al. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts beta-adrenergic responses in cardiomyocytes. J. Am. Coll. Cardiol. 2013, 62, 1596–1606. [Google Scholar] [CrossRef] [PubMed]
- Vettel, C.; Lindner, M.; Dewenter, M.; Lorenz, K.; Schanbacher, C.; Riedel, M.; Lammle, S.; Meinecke, S.; Mason, F.E.; Sossalla, S.; et al. Phosphodiesterase 2 Protects Against Catechola-mine-Induced Arrhythmia and Preserves Contractile Function After Myocardial Infarction. Circ. Res. 2017, 120, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M.; Morotti, S. Ca (2+) current facilitation is CaMKII-dependent and has arrhythmogenic consequences. Front. Pharmacol. 2014, 5, 144. [Google Scholar] [CrossRef]
- Sag, C.M.; Mallwitz, A.; Wagner, S.; Hartmann, N.; Schotola, H.; Fischer, T.H.; Ungeheuer, N.; Herting, J.; Shah, A.M.; Maier, L.S.; et al. Enhanced late INa induces proarrhythmogenic SR Ca leak in a CaMKII-dependent manner. J. Mol. Cell. Cardiol. 2014, 76, 94–105. [Google Scholar] [CrossRef]
- Métrich, M.; Berthouze, M.; Morel, E.; Crozatier, B.; Gomez, A.M.; Lezoualc’h, F. Role of the cAMP-binding protein Epac in cardiovascular physiology and pathophysiology. Pflug. Arch. Eur. J. of Physiol. 2010, 459, 535–546. [Google Scholar] [CrossRef]
- Neef, S.; Heijman, J.; Otte, K.; Dewenter, M.; Saadatmand, A.R.; Meyer-Roxlau, S.; Antos, C.L.; Backs, J.; Dobrev, D.; Wagner, M.; et al. Chronic loss of inhibitor-1 diminishes cardiac RyR2 phosphorylation despite exaggerated CaMKII activity. Naunyn Schmiedeberg Arch. Pharmacol. 2017, 390, 857–862. [Google Scholar] [CrossRef]
- Wagner, S.; Hacker, E.; Grandi, E.; Weber, S.L.; Dybkova, N.; Sossalla, S.; Sowa, T.; Fabritz, L.; Kirchhof, P.; Bers, N.M.; et al. Ca/Calmodulin Kinase II Differentially Modulates Potassium Currents. Circ. Arrhythmia Electrophysiol. 2009, 2, 285–294. [Google Scholar] [CrossRef]
- Ding, B.; Abe, J.I.; Wei, H.; Huang, Q.; Walsh, R.A.; Molina, C.A.; Zhao, A.; Sadoshima, J.; Blaxall, B.C.; Berk, B.C.; et al. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: Implication in heart failure. Circulation 2005, 111, 2469–2476. [Google Scholar] [CrossRef]
- Bristow, M.R. Treatment of chronic heart failure with β-adrenergic receptor antagonists: A convergence of receptor pharmacology and clinical cardiology. Circ. Res. 2011, 109, 1176–1194. [Google Scholar] [CrossRef]
- Holmes, J.R.; Kubo, S.H.; Cody, R.J.; Kligfield, P. Milrinone in congestive heart failure: Observations on ambulatory ventricular arrhythmias. Am. Hear. J. 1985, 110, 800–806. [Google Scholar] [CrossRef]
- Cohn, J.N.; Goldstein, S.O.; Greenberg, B.H.; Lorell, B.H.; Bourge, R.C.; Jaski, B.E.; Gottlieb, S.O.; McGrew, I.F.; Demets, D.L.; White, B.G. A dose-dependent increase in mortality with vesnarinone among patients with severe heart failure. N. Eng. J. Med. 1998, 339, 1810–1816. [Google Scholar] [CrossRef]
- Moreno, J.D.; Clancy, C.E. Pathophysiology of the cardiac late Na current and its potential as a drug target. J. Mol. Cell. Cardiol. 2012, 52, 608–619. [Google Scholar] [CrossRef]
- Wagner, S.; Maier, L.S.; Bers, D.M. Role of sodium and calcium dysregulation in tachyarrhythmias in sudden cardiac death. Circ. Res. 2015, 116, 1956–1970. [Google Scholar] [CrossRef]
- Shattock, M.J.; Ottolia, M.; Bers, D.M.; Blaustein, M.P.; Boguslavskyi, A.; Bossuyt, J.; Bridge, J.H.; Chen-Izu, Y.; Clancy, C.E.; Edwards, A.; et al. Na+/Ca2+ ex-change and Na+/K+-ATPase in the heart. J. Physiol. 2015, 593, 1361–1382. [Google Scholar] [CrossRef]
- Rivet-Bastide, M.; Vandecasteele, G.; Hatem, S.; Verde, I.; Benardeau, A.; Mercadier, J.J.; Fischmeister, R. cGMP-stimulated cyclic nucleotide phosphodiesterase regulates the basal calcium current in human atrial myocytes. J. Clin. Investig. 1997, 99, 2710–2718. [Google Scholar]
- Hua, R.; Adamczyk, A.; Robbins, C.; Ray, G.; Rose, R.A. Distinct Patterns of Constitutive Phosphodiesterase Activity in Mouse Sinoatrial Node and Atrial Myocardium. PLoS ONE 2012, 7, e47652. [Google Scholar] [CrossRef]
- Galindo-Tovar, A.; Vargas, M.L.; Kaumann, A.J. Phosphodiesterase PDE2 activity, increased by isoprenaline, does not reduce beta-adrenoceptor-mediated chronotropic and inotropic effects in rat heart. Naunyn Schmiedeberg Arch. Pharmacol. 2018, 391, 571–585. [Google Scholar] [CrossRef]
- Vincent, K.P.; McCulloch, A.D.; Edwards, A.G. Toward a hierarchy of mechanisms in CaMKII-mediated arrhythmia. Front. Pharmacol. 2014, 5, 110. [Google Scholar] [CrossRef]
- Glynn, P.; Musa, H.; Wu, X.; Unudurthi, S.D.; Little, S.; Qian, L.; Wright, P.J.; Radwanski, P.B.; Györke, S.; Mohler, P.J.; et al. Voltage-Gated Sodium Channel Phosphorylation at Ser571 Regulates Late Current, Arrhythmia, and Cardiac Function In Vivo. Circulation 2015, 132, 567–577. [Google Scholar] [CrossRef]
- Pereira, L.; Bare, D.J.; Galice, S.; Shannon, T.R.; Bers, D.M. β-Adrenergic induced SR Ca 2+ leak is mediated by an Epac-NOS pathway. J. Mol. Cell. Cardiol. 2017, 108, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Dybkova, N.; Wagner, S.; Backs, J.; Hund, T.J.; Mohler, P.J.; Sowa, T.; Nikolaev, V.O.; Maier, L.S. Tubulin polymerization disrupts cardiac β-adrenergic regulation of late INa. Cardiovasc. Res. 2014, 103, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Zoccarato, A.; Surdo, N.C.; Aronsen, J.M.; Fields, L.A.; Mancuso, L.; Dodoni, G.; Stangherlin, A.; Livie, C.; Jiang, H.; Sin, Y.Y.; et al. Cardiac Hypertrophy Is Inhibited by a Local Pool of cAMP Regulated by Phosphodiesterase 2. Circ. Res. 2015, 117, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Baliga, R.S.; Preedy, M.E.J.; Dukinfield, M.S.; Chu, S.M.; Aubdool, A.A.; Bubb, K.J.; Moyes, A.J.; Tones, M.A.; Hobbs, A.J. Phosphodiesterase 2 inhibition preferentially promotes NO/guanylyl cyclase/cGMP signaling to reverse the development of heart failure. Proc. Natl. Acad. Sci. USA 2018, 115, e7428–e7437. [Google Scholar] [PubMed]
- Yang, K.-C.; Bonini, M.G.; Dudley, S.C. Mitochondria and arrhythmias. Free. Radic. Biol. Med. 2014, 71, 351–361. [Google Scholar] [CrossRef]
- Subramanian, H.; Froese, A.; Jönsson, P.; Schmidt, H.; Gorelik, J.; Nikolaev, V.O. Distinct submembrane localisation compartmentalises cardiac NPR1 and NPR2 signalling to cGMP. Nat. Commun. 2018, 9, 2446. [Google Scholar] [CrossRef]
- Börner, S.; Schwede, F.; Schlipp, A.; Berisha, F.; Calebiro, D.; Lohse, M.J.; Nikolaev, V.O. FRET measurements of intra-cellular cAMP concentrations and cAMP analog permeability in intact cells. Nat. Protoc. 2011, 6, 427–438. [Google Scholar] [CrossRef]
- Stables, C.L.; Curtis, M.J. Development and characterization of a mouse in vitro model of ischaemia-induced ventricular fibrillation. Cardiovasc. Res. 2009, 83, 397–404. [Google Scholar] [CrossRef]
- Foltz, W.U.; Wagner, M.; Rudakova, E.; Volk, T. N-acetylcysteine prevents electrical remodeling and attenuates cellular hypertrophy in epicardial myocytes of rats with ascending aortic stenosis. Basic Res. Cardiol. 2012, 107, 1–18. [Google Scholar] [CrossRef]
- Heijman, J.; Volders, P.G.; Westra, R.L.; Rudy, Y. Local control of β-adrenergic stimulation: Effects on ventricular myocyte electrophysiology and Ca(2+)-transient. J. Mol. Cell. Cardiol. 2011, 50, 863–871. [Google Scholar] [CrossRef]
- Clerx, M.; Collins, P.; de Lange, E.; Volders, P.G. Myokit: A simple interface to cardiac cellular electrophysiology. Prog. Biophys. Mol. Biol. 2016, 120, 100–114. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagner, M.; Sadek, M.S.; Dybkova, N.; Mason, F.E.; Klehr, J.; Firneburg, R.; Cachorro, E.; Richter, K.; Klapproth, E.; Kuenzel, S.R.; et al. Cellular Mechanisms of the Anti-Arrhythmic Effect of Cardiac PDE2 Overexpression. Int. J. Mol. Sci. 2021, 22, 4816. https://doi.org/10.3390/ijms22094816
Wagner M, Sadek MS, Dybkova N, Mason FE, Klehr J, Firneburg R, Cachorro E, Richter K, Klapproth E, Kuenzel SR, et al. Cellular Mechanisms of the Anti-Arrhythmic Effect of Cardiac PDE2 Overexpression. International Journal of Molecular Sciences. 2021; 22(9):4816. https://doi.org/10.3390/ijms22094816
Chicago/Turabian StyleWagner, Michael, Mirna S. Sadek, Nataliya Dybkova, Fleur E. Mason, Johann Klehr, Rebecca Firneburg, Eleder Cachorro, Kurt Richter, Erik Klapproth, Stephan R. Kuenzel, and et al. 2021. "Cellular Mechanisms of the Anti-Arrhythmic Effect of Cardiac PDE2 Overexpression" International Journal of Molecular Sciences 22, no. 9: 4816. https://doi.org/10.3390/ijms22094816
APA StyleWagner, M., Sadek, M. S., Dybkova, N., Mason, F. E., Klehr, J., Firneburg, R., Cachorro, E., Richter, K., Klapproth, E., Kuenzel, S. R., Lorenz, K., Heijman, J., Dobrev, D., El-Armouche, A., Sossalla, S., & Kämmerer, S. (2021). Cellular Mechanisms of the Anti-Arrhythmic Effect of Cardiac PDE2 Overexpression. International Journal of Molecular Sciences, 22(9), 4816. https://doi.org/10.3390/ijms22094816