
Interactions of SARS-CoV-2 with the Blood–Brain Barrier

 , ,
, ,  and
and 
Abstract
:1. Introduction
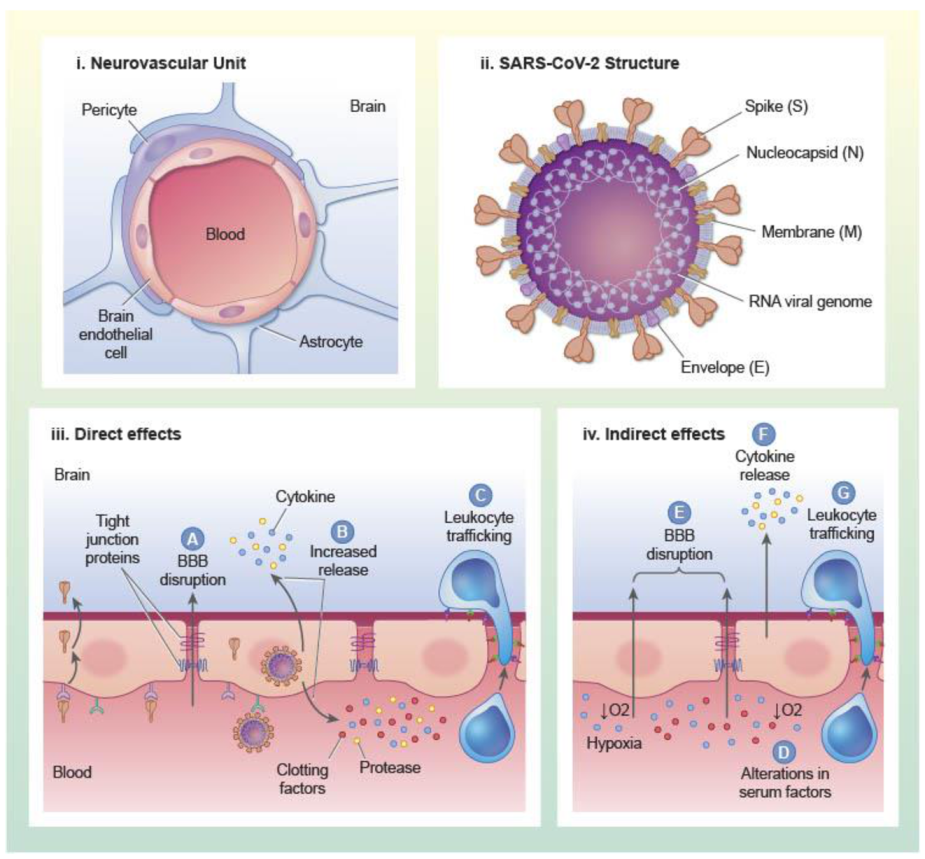
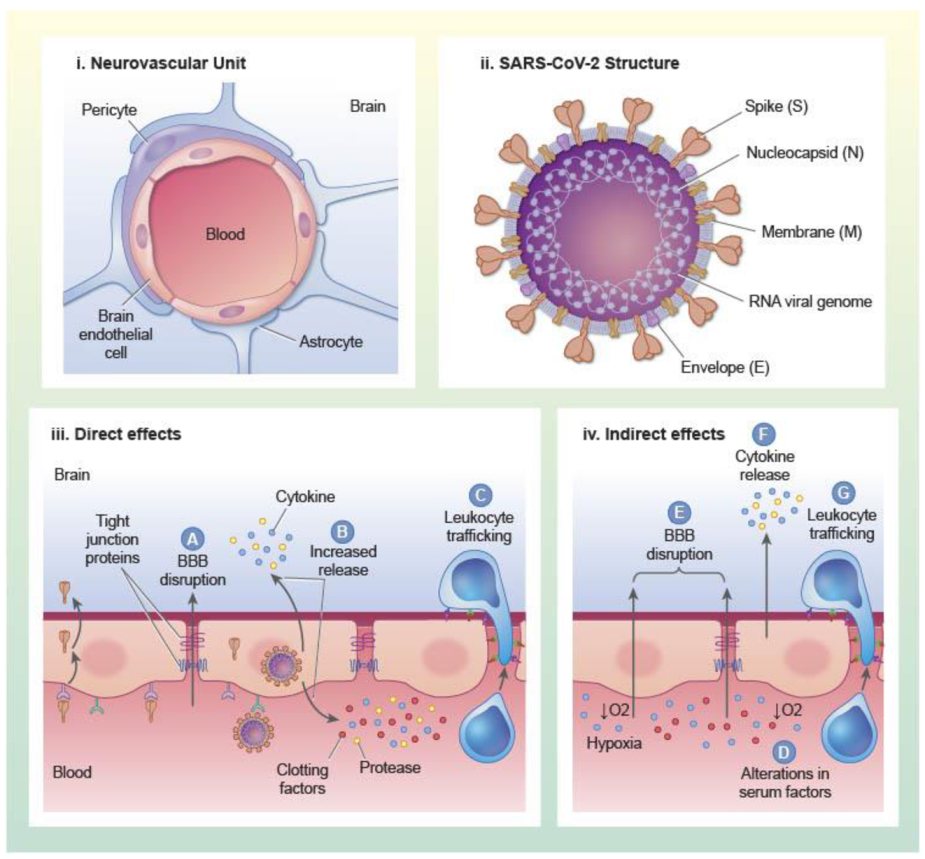
1.1. Structure and Functions of the BBB
1.2. Neurological Complications of SARS-CoV-2 Infection and BBB Involvement
2. Mechanisms of SARS-CoV-2 Infection and Tissue Tropism
2.1. Virus Structure
2.2. Host Receptors and Other Factors That Mediate SARS-CoV-2 Entry into Cells
2.3. Tropism of SARS-CoV-2: Evidence for Direct Infection of the CNS and Implications for Disease
3. Mechanisms of BBB Dysfunction in SARS-CoV-2 Infection
3.1. Direct Interactions of SARS-CoV-2 with Brain Endothelial Cells and Other Constituents of the NVU
3.2. Indirect Effects of SARS-CoV-2 Infection on the BBB
3.2.1. Inflammation
3.2.2. Clotting and Thrombosis
3.2.3. Hypoxia
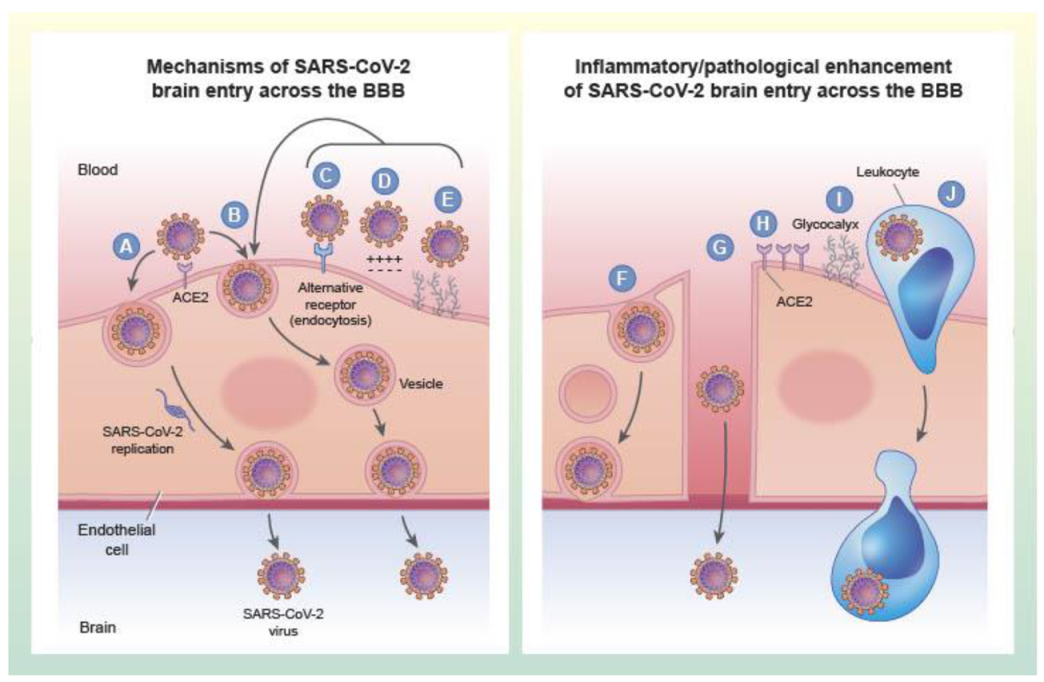
4. Mechanisms of SARS-CoV-2 Transit across the Vascular BBB: Lessons from Neurotropic Viruses
4.1. Viral Entry by Retrograde Nerve Transmission
4.2. Viral Entry across Brain Barriers
4.3. BBB Disruption Plays a Selective Role in Viral Entry
4.4. Entry of Free Virus at the Brain Barriers Is Mediated by Vesicular Pathways
4.5. Inflammation Enhances Uptake of Viruses by Brain
4.6. The Viral Attachment Protein May Bind to a Site on the Receptor Not Used by the Physiological Ligand
4.7. Some Viruses Bind to More Than One Seemingly Unrelated Receptor
4.8. Mechanisms of SARS-CoV-2 Entry into Brain
5. Co-Morbidities That Could Influence SARS-CoV-2 Entry into the Brain
5.1. Type I Interferons
5.2. Apolipoprotein E
5.3. Disease States That May Increase Risk of SARS-CoV-2 Entry into Brain
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Varatharaj, A.; Thomas, N.; Ellul, M.A.; Davies, N.W.S.; Pollak, T.A.; Tenorio, E.L.; Sultan, M.; Easton, A.; Breen, G.; Zandi, M.; et al. Neurological and neuropsychiatric complications of COVID-19 in 153 patients: A UK-wide surveillance study. Lancet Psychiatry 2020, 7, 875–882. [Google Scholar] [CrossRef]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef]
- Luissint, A.C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tietz, S.; Engelhardt, B. Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 2015, 209, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Berndt, P.; Winkler, L.; Cording, J.; Breitkreuz-Korff, O.; Rex, A.; Dithmer, S.; Rausch, V.; Blasig, R.; Richter, M.; Sporbert, A.; et al. Tight junction proteins at the blood-brain barrier: Far more than claudin-5. Cell Mol. Life Sci. 2019, 76, 1987–2002. [Google Scholar] [CrossRef]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Andreone, B.J.; Chow, B.W.; Tata, A.; Lacoste, B.; Ben-Zvi, A.; Bullock, K.; Deik, A.A.; Ginty, D.D.; Clish, C.B.; Gu, C. Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron 2017, 94, 581–594. [Google Scholar] [CrossRef] [Green Version]
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 2014, 509, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begley, D.J. ABC transporters and the blood-brain barrier. Curr. Pharm. Des. 2004, 10, 1295–1312. [Google Scholar] [CrossRef] [PubMed]
- Agundez, J.A.; Jimenez-Jimenez, F.J.; Alonso-Navarro, H.; Garcia-Martin, E. Drug and xenobiotic biotransformation in the blood-brain barrier: A neglected issue. Front. Cell Neurosci. 2014, 8, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuwelt, E.; Abbott, N.J.; Abrey, L.; Banks, W.A.; Blakley, B.; Davis, T.; Engelhardt, B.; Grammas, P.; Nedergaard, M.; Nutt, J.; et al. Strategies to advance translational research into brain barriers. Lancet Neurol. 2008, 7, 84–96. [Google Scholar] [CrossRef]
- Erickson, M.A.; Banks, W.A. Neuroimmune Axes of the Blood-Brain Barriers and Blood-Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions. Pharmacol. Rev. 2018, 70, 278–314. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef]
- Berthiaume, A.A.; Grant, R.I.; McDowell, K.P.; Underly, R.G.; Hartmann, D.A.; Levy, M.; Bhat, N.R.; Shih, A.Y. Dynamic Remodeling of Pericytes In Vivo Maintains Capillary Coverage in the Adult Mouse Brain. Cell Rep. 2018, 22, 8–16. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Ronnback, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Reynolds, J.P.; Chen, Y.; Gourine, A.V.; Rusakov, D.A.; Attwell, D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 2016, 19, 1619–1627. [Google Scholar] [CrossRef] [Green Version]
- Giacomelli, A.; Pezzati, L.; Conti, F.; Bernacchia, D.; Siano, M.; Oreni, L.; Rusconi, S.; Gervasoni, C.; Ridolfo, A.L.; Rizzardini, G.; et al. Self-reported Olfactory and Taste Disorders in Patients With Severe Acute Respiratory Coronavirus 2 Infection: A Cross-sectional Study. Clin. Infect. Dis. 2020, 71, 889–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Erausquin, G.A.; Snyder, H.; Carrillo, M.; Hosseini, A.A.; Brugha, T.S.; Seshadri, S. CNS SARS-CoV-2 Consortium. The chronic neuropsychiatric sequelae of COVID-19: The need for a prospective study of viral impact on brain functioning. Alzheimers Dement. 2021. [Google Scholar] [CrossRef]
- Zubair, A.S.; McAlpine, L.S.; Gardin, T.; Farhadian, S.; Kuruvilla, D.E.; Spudich, S. Neuropathogenesis and Neurologic Manifestations of the Coronaviruses in the Age of Coronavirus Disease 2019: A Review. JAMA Neurol. 2020, 77, 1018–1027. [Google Scholar] [CrossRef]
- Boscolo-Rizzo, P.; Borsetto, D.; Fabbris, C.; Spinato, G.; Frezza, D.; Menegaldo, A.; Mularoni, F.; Gaudioso, P.; Cazzador, D.; Marciani, S.; et al. Evolution of Altered Sense of Smell or Taste in Patients With Mildly Symptomatic COVID-19. JAMA Otolaryngol. Head Neck Surg. 2020, 146, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients With Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Mu, J.; Guo, J.; Lu, L.; Liu, D.; Luo, J.; Li, N.; Liu, J.; Yang, D.; Gao, H.; et al. New onset neurologic events in people with COVID-19 in 3 regions in China. Neurology 2020, 95, e1479–e1487. [Google Scholar] [CrossRef]
- The, L. Facing up to long COVID. Lancet 2020, 396, 1861. [Google Scholar] [CrossRef]
- Nath, A. Long-Haul COVID. Neurology 2020, 95, 559–560. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Manjunath, K.; Ranjan, R.K.; Kaushik, S.; Kumar, S.; Verma, V. COVID-19 pandemic: Insights into structure, function, and hACE2 receptor recognition by SARS-CoV-2. PLoS Pathog. 2020, 16, e1008762. [Google Scholar] [CrossRef]
- McBride, R.; van Zyl, M.; Fielding, B.C. The coronavirus nucleocapsid is a multifunctional protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Voss, D.; Kern, A.; Traggiai, E.; Eickmann, M.; Stadler, K.; Lanzavecchia, A.; Becker, S. Characterization of severe acute respiratory syndrome coronavirus membrane protein. FEBS Lett. 2006, 580, 968–973. [Google Scholar] [CrossRef] [Green Version]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [Green Version]
- Astuti, I. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 183, 1735. [Google Scholar] [CrossRef]
- Walsh, E.E.; Frenck, R.W., Jr.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2020, 7, eabe5575. [Google Scholar] [CrossRef] [PubMed]
- Rabi, F.A.; Al Zoubi, M.S.; Kasasbeh, G.A.; Salameh, D.M.; Al-Nasser, A.D. SARS-CoV-2 and Coronavirus Disease 2019: What We Know So Far. Pathogens 2020, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Zmora, P.; Gierer, S.; Heurich, A.; Pohlmann, S. Proteolytic activation of the SARS-coronavirus spike protein: Cutting enzymes at the cutting edge of antiviral research. Antivir. Res. 2013, 100, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Anton-Plagaro, C.; Shoemark, D.K.; Simon-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef]
- Shilts, J.; Crozier, T.W.M.; Greenwood, E.J.D.; Lehner, P.J.; Wright, G.J. No evidence for basigin/CD147 as a direct SARS-CoV-2 spike binding receptor. Sci. Rep. 2021, 11, 413. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.N.; Richards, S.J.; Guy, C.S.; Congdon, T.R.; Hasan, M.; Zwetsloot, A.J.; Gallo, A.; Lewandowski, J.R.; Stansfeld, P.J.; Straube, A.; et al. The SARS-COV-2 Spike Protein Binds Sialic Acids and Enables Rapid Detection in a Lateral Flow Point of Care Diagnostic Device. ACS Cent. Sci. 2020, 6, 2046–2052. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Trypsteen, W.; Van Cleemput, J.; Snippenberg, W.V.; Gerlo, S.; Vandekerckhove, L. On the whereabouts of SARS-CoV-2 in the human body: A systematic review. PLoS Pathog. 2020, 16, e1009037. [Google Scholar] [CrossRef]
- Bao, L.; Deng, W.; Huang, B.; Gao, H.; Liu, J.; Ren, L.; Wei, Q.; Yu, P.; Xu, Y.; Qi, F.; et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 2020, 583, 830–833. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e907. [Google Scholar] [CrossRef] [PubMed]
- Nascimento Conde, J.; Schutt, W.R.; Gorbunova, E.E.; Mackow, E.R. Recombinant ACE2 Expression Is Required for SARS-CoV-2 To Infect Primary Human Endothelial Cells and Induce Inflammatory and Procoagulative Responses. mBio 2020, 11, eabe5575. [Google Scholar] [CrossRef]
- Placantonakis, D.G.; Aguero-Rosenfeld, M.; Flaifel, A.; Colavito, J.; Inglima, K.; Zagzag, D.; Snuderl, M.; Louie, E.; Frontera, J.A.; Lewis, A. SARS-CoV-2 Is Not Detected in the Cerebrospinal Fluid of Encephalopathic COVID-19 Patients. Front. Neurol. 2020, 11, 587384. [Google Scholar] [CrossRef]
- Lersy, F.; Benotmane, I.; Helms, J.; Collange, O.; Schenck, M.; Brisset, J.C.; Chammas, A.; Willaume, T.; Lefebvre, N.; Solis, M.; et al. Cerebrospinal fluid features in COVID-19 patients with neurologic manifestations: Correlation with brain MRI findings in 58 patients. J. Infect. Dis. 2020, 223, 600–609. [Google Scholar] [CrossRef]
- Matschke, J.; Lutgehetmann, M.; Hagel, C.; Sperhake, J.P.; Schroder, A.S.; Edler, C.; Mushumba, H.; Fitzek, A.; Allweiss, L.; Dandri, M.; et al. Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet Neurol. 2020, 19, 919–929. [Google Scholar] [CrossRef]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Prado, A.V.; Skriabine, S.; Lu, P.; Weizman, O.E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Solomon, I.H.; Normandin, E.; Bhattacharyya, S.; Mukerji, S.S.; Keller, K.; Ali, A.S.; Adams, G.; Hornick, J.L.; Padera, R.F., Jr.; Sabeti, P. Neuropathological Features of Covid-19. N. Engl. J. Med. 2020, 383, 989–992. [Google Scholar] [CrossRef]
- Lee, M.H.; Perl, D.P.; Nair, G.; Li, W.; Maric, D.; Murray, H.; Dodd, S.J.; Koretsky, A.P.; Watts, J.A.; Cheung, V.; et al. Microvascular Injury in the Brains of Patients with Covid-19. N. Engl. J. Med. 2021, 384, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Virhammar, J.; Kumlien, E.; Fallmar, D.; Frithiof, R.; Jackmann, S.; Skold, M.K.; Kadir, M.; Frick, J.; Lindeberg, J.; Olivero-Reinius, H.; et al. Acute necrotizing encephalopathy with SARS-CoV-2 RNA confirmed in cerebrospinal fluid. Neurology 2020, 95, 445–449. [Google Scholar] [CrossRef]
- Lau, K.K.; Yu, W.C.; Chu, C.M.; Lau, S.T.; Sheng, B.; Yuen, K.Y. Possible central nervous system infection by SARS coronavirus. Emerg. Infect. Dis. 2004, 10, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.S.; Bailey, A.L.; Kafai, N.M.; Nair, S.; McCune, B.T.; Yu, J.; Fox, J.M.; Chen, R.E.; Earnest, J.T.; Keeler, S.P.; et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020, 21, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Brann, D.H.; Tsukahara, T.; Weinreb, C.; Lipovsek, M.; Van den Berge, K.; Gong, B.; Chance, R.; Macaulay, I.C.; Chou, H.J.; Fletcher, R.B.; et al. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci. Adv. 2020, 6, eabc5801. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Ji, W.; Yang, H.; Chen, S.; Zhang, W.; Duan, G. Endothelial activation and dysfunction in COVID-19: From basic mechanisms to potential therapeutic approaches. Signal Transduct. Target. Ther. 2020, 5, 293. [Google Scholar] [CrossRef] [PubMed]
- Netland, J.; Meyerholz, D.K.; Moore, S.; Cassell, M.; Perlman, S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J. Virol. 2008, 82, 7264–7275. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.H.; Chen, Q.; Gu, H.J.; Yang, G.; Wang, Y.X.; Huang, X.Y.; Liu, S.S.; Zhang, N.N.; Li, X.F.; Xiong, R.; et al. A Mouse Model of SARS-CoV-2 Infection and Pathogenesis. Cell Host Microbe 2020, 28, 124–133. [Google Scholar] [CrossRef]
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A.; Bullock, T.A.; McGary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potula, R.; et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol. Dis. 2020, 146, 105131. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Erickson, M.A.; Wilson, M.L.; Banks, W.A. In vitro modeling of blood-brain barrier and interface functions in neuroimmune communication. Fluids Barriers CNS 2020, 17, 26. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zhou, W.; Yan, X.; Guo, T.; Wang, B.; Xia, H.; Ye, L.; Xiong, J.; Jiang, Z.; Liu, Y.; et al. Prognostic Value of C-Reactive Protein in Patients With Coronavirus 2019. Clin. Infect. Dis. 2020, 71, 2174–2179. [Google Scholar] [CrossRef]
- Li, H.; Xiang, X.; Ren, H.; Xu, L.; Zhao, L.; Chen, X.; Long, H.; Wang, Q.; Wu, Q. Serum Amyloid A is a biomarker of severe Coronavirus Disease and poor prognosis. J. Infect. 2020, 80, 646–655. [Google Scholar] [CrossRef]
- Perrin, P.; Collongues, N.; Baloglu, S.; Bedo, D.; Bassand, X.; Lavaux, T.; Gautier-Vargas, G.; Keller, N.; Kremer, S.; Fafi-Kremer, S.; et al. Cytokine release syndrome-associated encephalopathy in patients with COVID-19. Eur J. Neurol. 2021, 28, 248–258. [Google Scholar] [CrossRef]
- Pilotto, A.; Masciocchi, S.; Volonghi, I.; De Giuli, V.; Caprioli, F.; Mariotto, S.; Ferrari, S.; Bozzetti, S.; Imarisio, A.; Risi, B.; et al. SARS-CoV-2 encephalitis is a cytokine release syndrome: Evidences from cerebrospinal fluid analyses. Clin. Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Garcia, M.A.; Barreras, P.V.; Lewis, A.; Pinilla, G.; Sokoll, L.J.; Kickler, T.; Mostafa, H.; Caturegli, M.; Moghekar, A.; Fitzgerald, K.C.; et al. Cerebrospinal fluid in COVID-19 neurological complications: No cytokine storm or neuroinflammation. medRxiv 2021. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hsuchou, H.; Kastin, A.J.; Mishra, P.K.; Pan, W. C-reactive protein increases BBB permeability: Implications for obesity and neuroinflammation. Cell Physiol. Biochem. 2012, 30, 1109–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A.; Freed, E.O.; Wolf, K.M.; Robinson, S.M.; Franko, M.; Kumar, V.B. Transport of human immunodeficiency virus type 1 pseudoviruses across the blood-brain barrier: Role of envelope proteins and adsorptive endocytosis. J. Virol. 2001, 75, 4681–4691. [Google Scholar] [CrossRef] [Green Version]
- Tandon, M.; Kataria, S.; Patel, J.; Mehta, T.R.; Daimee, M.; Patel, V.; Prasad, A.; Chowdhary, A.A.; Jaiswal, S.; Sriwastava, S. A Comprehensive Systematic Review of CSF analysis that defines Neurological Manifestations of COVID-19. Int. J. Infect. Dis. 2021, 104, 390–397. [Google Scholar] [CrossRef]
- Engelhardt, B.; Coisne, C. Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids Barriers CNS 2011, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallmann, R.; Zhang, X.; Di Russo, J.; Li, L.; Song, J.; Hannocks, M.J.; Sorokin, L. The regulation of immune cell trafficking by the extracellular matrix. Curr. Opin. Cell Biol. 2015, 36, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Cardot-Leccia, N.; Hubiche, T.; Dellamonica, J.; Burel-Vandenbos, F.; Passeron, T. Pericyte alteration sheds light on micro-vasculopathy in COVID-19 infection. Intensive Care Med. 2020, 46, 1777–1778. [Google Scholar] [CrossRef]
- Grant, M.C.; Geoghegan, L.; Arbyn, M.; Mohammed, Z.; McGuinness, L.; Clarke, E.L.; Wade, R.G. The prevalence of symptoms in 24,410 adults infected by the novel coronavirus (SARS-CoV-2; COVID-19): A systematic review and meta-analysis of 148 studies from 9 countries. PLoS ONE 2020, 15, e0234765. [Google Scholar] [CrossRef] [PubMed]
- Engstrom, L.; Ruud, J.; Eskilsson, A.; Larsson, A.; Mackerlova, L.; Kugelberg, U.; Qian, H.; Vasilache, A.M.; Larsson, P.; Engblom, D.; et al. Lipopolysaccharide-induced fever depends on prostaglandin E2 production specifically in brain endothelial cells. Endocrinology 2012, 153, 4849–4861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, M.; Klawonn, A.M.; Nilsson, A.; Singh, A.K.; Zajdel, J.; Wilhelms, D.B.; Lazarus, M.; Lofberg, A.; Jaarola, M.; Kugelberg, U.O.; et al. Prostaglandin-dependent modulation of dopaminergic neurotransmission elicits inflammation-induced aversion in mice. J. Clin. Investig. 2016, 126, 695–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A.; Farr, S.A.; Morley, J.E. Entry of blood-borne cytokines into the central nervous system: Effects on cognitive processes. Neuroimmunomodulation 2002, 10, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R. Cytokine, sickness behavior, and depression. Neurol. Clin. 2006, 24, 441–460. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, J.C.; Lawson, M.A.; Andre, C.; Moreau, M.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry 2009, 14, 511–522. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.K.; Wing, E.E.; Banks, W.A.; Dantzer, R. Leucine competes with kynurenine for blood-to-brain transport and prevents lipopolysaccharide-induced depression-like behavior in mice. Mol. Psychiatry 2019, 24, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Oxley, T.J.; Mocco, J.; Majidi, S.; Kellner, C.P.; Shoirah, H.; Singh, I.P.; De Leacy, R.A.; Shigematsu, T.; Ladner, T.R.; Yaeger, K.A.; et al. Large-Vessel Stroke as a Presenting Feature of Covid-19 in the Young. N. Engl. J. Med. 2020, 382, e60. [Google Scholar] [CrossRef]
- Nicholson, P.; Alshafai, L.; Krings, T. Neuroimaging Findings in Patients with COVID-19. AJNR Am. J. Neuroradiol. 2020, 41, 1380–1383. [Google Scholar] [CrossRef] [PubMed]
- Batra, A.; Clark, J.R.; LaHaye, K.; Shlobin, N.A.; Hoffman, S.C.; Orban, Z.S.; Colton, K.; Dematte, J.E.; Sorond, F.A.; Koralnik, I.J.; et al. Transcranial Doppler Ultrasound Evidence of Active Cerebral Embolization in COVID-19. J. Stroke Cerebrovasc. Dis. 2021, 30, 105542. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T.; Erkan, D.; Winakur, J.; James, J.A. Emerging evidence of a COVID-19 thrombotic syndrome has treatment implications. Nat. Rev. Rheumatol. 2020, 16, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Shatzel, J.J.; Taylor, J.A. Syndromes of Thrombotic Microangiopathy. Med. Clin. N. Am. 2017, 101, 395–415. [Google Scholar] [CrossRef] [PubMed]
- Diorio, C.; McNerney, K.O.; Lambert, M.; Paessler, M.; Anderson, E.M.; Henrickson, S.E.; Chase, J.; Liebling, E.J.; Burudpakdee, C.; Lee, J.H.; et al. Evidence of thrombotic microangiopathy in children with SARS-CoV-2 across the spectrum of clinical presentations. Blood Adv. 2020, 4, 6051–6063. [Google Scholar] [CrossRef] [PubMed]
- Bellander, B.M.; Olafsson, I.H.; Ghatan, P.H.; Bro Skejo, H.P.; Hansson, L.O.; Wanecek, M.; Svensson, M.A. Secondary insults following traumatic brain injury enhance complement activation in the human brain and release of the tissue damage marker S100B. Acta Neurochir. 2011, 153, 90–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsberg, P.J.; Ohman, J.; Lehto, T.; Karjalainen-Lindsberg, M.L.; Paetau, A.; Wuorimaa, T.; Carpen, O.; Kaste, M.; Meri, S. Complement activation in the central nervous system following blood-brain barrier damage in man. Ann. Neurol. 1996, 40, 587–596. [Google Scholar] [CrossRef]
- Lee, K.R.; Kawai, N.; Kim, S.; Sagher, O.; Hoff, J.T. Mechanisms of edema formation after intracerebral hemorrhage: Effects of thrombin on cerebral blood flow, blood-brain barrier permeability, and cell survival in a rat model. J. Neurosurg. 1997, 86, 272–278. [Google Scholar] [CrossRef]
- Liu, D.Z.; Ander, B.P.; Xu, H.; Shen, Y.; Kaur, P.; Deng, W.; Sharp, F.R. Blood-brain barrier breakdown and repair by Src after thrombin-induced injury. Ann. Neurol. 2010, 67, 526–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, N.; Roberts, A.M.; Dean, W.L.; Tyagi, S.C.; Lominadze, D. Fibrinogen induces endothelial cell permeability. Mol. Cell Biochem. 2008, 307, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Patibandla, P.K.; Tyagi, N.; Dean, W.L.; Tyagi, S.C.; Roberts, A.M.; Lominadze, D. Fibrinogen induces alterations of endothelial cell tight junction proteins. J. Cell Physiol. 2009, 221, 195–203. [Google Scholar] [CrossRef]
- Su, E.J.; Fredriksson, L.; Geyer, M.; Folestad, E.; Cale, J.; Andrae, J.; Gao, Y.; Pietras, K.; Mann, K.; Yepes, M.; et al. Activation of PDGF-CC by tissue plasminogen activator impairs blood-brain barrier integrity during ischemic stroke. Nat. Med. 2008, 14, 731–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yepes, M.; Sandkvist, M.; Moore, E.G.; Bugge, T.H.; Strickland, D.K.; Lawrence, D.A. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J. Clin. Investig. 2003, 112, 1533–1540. [Google Scholar] [CrossRef]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Yang, L.; Liu, R.; Liu, F.; Wu, K.L.; Li, J.; Liu, X.H.; Zhu, C.L. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin. Chem. Lab. Med. 2020, 58, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobin, M.J.; Laghi, F.; Jubran, A. Why COVID-19 Silent Hypoxemia Is Baffling to Physicians. Am. J. Respir. Crit. Care Med. 2020, 202, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Covassin, N.; Fan, Z.; Singh, P.; Gao, W.; Li, G.; Kara, T.; Somers, V.K. Association Between Hypoxemia and Mortality in Patients With COVID-19. Mayo Clin. Proc. 2020, 95, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Kashani, K.B. Hypoxia in COVID-19: Sign of Severity or Cause for Poor Outcomes. Mayo Clin. Proc. 2020, 95, 1094–1096. [Google Scholar] [CrossRef]
- Rello, J.; Storti, E.; Belliato, M.; Serrano, R. Clinical phenotypes of SARS-CoV-2: Implications for clinicians and researchers. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef]
- Yang, Y.; Rosenberg, G.A. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef] [Green Version]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood-brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Yang, Y.; Rosenberg, G.A. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience 2009, 158, 983–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mark, K.S.; Davis, T.P. Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1485–1494. [Google Scholar] [CrossRef] [Green Version]
- Josko, J.; Knefel, K. The role of vascular endothelial growth factor in cerebral oedema formation. Folia Neuropathol. 2003, 41, 161–166. [Google Scholar]
- Plateel, M.; Teissier, E.; Cecchelli, R. Hypoxia dramatically increases the nonspecific transport of blood-borne proteins to the brain. J. Neurochem. 1997, 68, 874–877. [Google Scholar] [CrossRef]
- Cipolla, M.J.; Crete, R.; Vitullo, L.; Rix, R.D. Transcellular transport as a mechanism of blood-brain barrier disruption during stroke. Front. Biosci. 2004, 9, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Nzou, G.; Wicks, R.T.; VanOstrand, N.R.; Mekky, G.A.; Seale, S.A.; El-Taibany, A.; Wicks, E.E.; Nechtman, C.M.; Marrotte, E.J.; Makani, V.S.; et al. Multicellular 3D Neurovascular Unit Model for Assessing Hypoxia and Neuroinflammation Induced Blood-Brain Barrier Dysfunction. Sci. Rep. 2020, 10, 9766. [Google Scholar] [CrossRef]
- Chen, X.; Sadowska, G.B.; Zhang, J.; Kim, J.E.; Cummings, E.E.; Bodge, C.A.; Lim, Y.P.; Makeyev, O.; Besio, W.G.; Gaitanis, J.; et al. Neutralizing anti-interleukin-1beta antibodies modulate fetal blood-brain barrier function after ischemia. Neurobiol. Dis. 2015, 73, 118–129. [Google Scholar] [CrossRef] [Green Version]
- Patra, A.; Chen, X.; Sadowska, G.B.; Zhang, J.; Lim, Y.P.; Padbury, J.F.; Banks, W.A.; Stonestreet, B.S. Neutralizing anti-interleukin-1beta antibodies reduce ischemia-related interleukin-1beta transport across the blood-brain barrier in fetal sheep. Neuroscience 2017, 346, 113–125. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Hovanesian, V.; Naqvi, S.; Lim, Y.P.; Tucker, R.; Donahue, J.E.; Stopa, E.G.; Stonestreet, B.S. Systemic infusions of anti-interleukin-1beta neutralizing antibodies reduce short-term brain injury after cerebral ischemia in the ovine fetus. Brain Behav. Immun. 2018, 67, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochim Biophys Acta 2011, 1813, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Comerford, K.M.; Wallace, T.J.; Karhausen, J.; Louis, N.A.; Montalto, M.C.; Colgan, S.P. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar] [PubMed]
- DeMars, K.M.; Yang, C.; Hawkins, K.E.; McCrea, A.O.; Siwarski, D.M.; Candelario-Jalil, E. Spatiotemporal Changes in P-glycoprotein Levels in Brain and Peripheral Tissues Following Ischemic Stroke in Rats. J. Exp. Neurosci. 2017, 11, 1179069517701741. [Google Scholar] [CrossRef]
- Stanimirovic, D.; Satoh, K. Inflammatory mediators of cerebral endothelium: A role in ischemic brain inflammation. Brain Pathol. 2000, 10, 113–126. [Google Scholar] [CrossRef]
- Colgan, S.P.; Furuta, G.T.; Taylor, C.T. Hypoxia and Innate Immunity: Keeping Up with the HIFsters. Annu. Rev. Immunol. 2020, 38, 341–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, B.M.; Rall, G.F.; Schnell, M.J. Everything You Always Wanted to Know About Rabies Virus (But Were Afraid to Ask). Annu. Rev. Virol. 2015, 2, 451–471. [Google Scholar] [CrossRef] [PubMed]
- Enquist, L.W.; Husak, P.J.; Banfield, B.W.; Smith, G.A. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virus Res. 1998, 51, 237–347. [Google Scholar] [CrossRef]
- Morrison, L.A.; Sidman, R.L.; Fields, B.N. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc. Natl. Acad. Sci. USA 1991, 88, 3852–3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlman, S.; Evans, G.; Afifi, A. Effect of olfactory bulb ablation on spread of a neurotropic coronavirus into the mouse brain. J. Exp. Med. 1990, 172, 1127–1132. [Google Scholar] [CrossRef] [Green Version]
- Bodian, D. Viremia in experimental poliomyelitis. I. General aspects of infection after intravascular inoculation with strains of high and of low invasiveness. Am. J. Hyg. 1954, 60, 339–357. [Google Scholar] [PubMed]
- Bragg, D.C.; Childers, T.A.; Tompkins, M.B.; Tompkins, W.A.; Meeker, R.B. Infection of the choroid plexus by feline immunodeficiency virus. J. Neurovirol. 2002, 8, 211–224. [Google Scholar] [CrossRef]
- Cabirac, G.F.; Soike, K.F.; Zhang, J.Y.; Hoel, K.; Butunoi, C.; Cai, G.Y.; Johnson, S.; Murray, R.S. Entry of coronavirus into primate CNS following peripheral infection. Microb. Pathog. 1994, 16, 349–357. [Google Scholar] [CrossRef]
- Niu, J.; Shen, L.; Huang, B.; Ye, F.; Zhao, L.; Wang, H.; Deng, Y.; Tan, W. Non-invasive bioluminescence imaging of HCoV-OC43 infection and therapy in the central nervous system of live mice. Antivir. Res. 2020, 173, 104646. [Google Scholar] [CrossRef]
- Watanabe, R.; Kakizaki, M.; Ikehara, Y.; Togayachi, A. Formation of fibroblastic reticular network in the brain after infection with neurovirulent murine coronavirus. Neuropathology 2016, 36, 513–526. [Google Scholar] [CrossRef]
- Hong, S.; Banks, W.A. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav. Immun. 2014, 45, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.; Chan, K.H.; Choi, G.K.; To, K.K.; Tse, H.; Cai, J.P.; Yeung, M.L.; Cheng, V.C.; Chen, H.; Che, X.Y.; et al. Differential cell line susceptibility to the emerging novel human betacoronavirus 2c EMC/2012: Implications for disease pathogenesis and clinical manifestation. J. Infect. Dis. 2013, 207, 1743–1752. [Google Scholar] [CrossRef] [Green Version]
- Wolburg, H.; Wolburg-Buchholz, K.; Engelhardt, B. Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol. 2005, 109, 181–190. [Google Scholar] [CrossRef]
- Greenwood, J.; Heasman, S.J.; Alvarez, J.I.; Prat, A.; Lyck, R.; Engelhardt, B. Review: Leucocyte-endothelial cell crosstalk at the blood-brain barrier: A prerequisite for successful immune cell entry to the brain. Neuropathol. Appl. Neurobiol. 2011, 37, 24–39. [Google Scholar] [CrossRef]
- Banks, W.A.; Niehoff, M.L.; Ponzio, N.M.; Erickson, M.A.; Zalcman, S.S. Pharmacokinetics and modeling of immune cell trafficking: Quantifying differential influences of target tissues versus lymphocytes in SJL and lippolysaccaride-treated mice. J. Neuroinflammation 2012, 9, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persidsky, Y.; Stins, M.; Way, D.; Witte, M.H.; Weinand, M.; Kim, K.S.; Bock, P.; Gendelman, H.E.; Fiala, M. A model for monocyte migration through the blood-brain barrier during HIV-1 encephalitis. J. Immunol. 1997, 158, 3499–3510. [Google Scholar]
- Koskiniemi, M.; Vaheri, A.; Taskinen, E. Cerebrospinal fluid alterations in herpes simplex virus encephalitis. Rev. Infect. Dis. 1984, 6, 608–618. [Google Scholar] [CrossRef]
- Rapoport, S.I. Osmotic opening of the blood-brain barrier: Principles, mechanism, and therapeutic applications. Cell Mol. Neurobiol. 2000, 20, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M. The entry of enveloped viruses into cells by endocytosis. Biochem. J. 1984, 218, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klasse, P.J.; Bron, R.; Marsh, M. Mechanisms of enveloped virus entry into animal cells. Adv. Drug Deliv. Rev. 1998, 34, 65–91. [Google Scholar] [CrossRef]
- Broadwell, R.D. Transcytosis of macromolecules through the blood-brain barrier: A cell biological perspective and critical appraisal. ACTA Neuropathol. (Berlin) 1989, 79, 117–128. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Brennan, J.M.; Vallance, K.L. Adsorptive endocytosis of HIV-1gp120 by blood-brain barrier is enhanced by lipopolysaccharide. Exp. Neurol. 1999, 156, 165–171. [Google Scholar] [CrossRef]
- Dohgu, S.; Fleegal-DeMotta, M.A.; Banks, W.A. Lipopolysaccharide-enhanced transcellular transport of HIV-1 across the blood-brain barrier is mediated by luminal microvessel IL-6 and GM-CSF. J. Neuroinflammation 2011, 8, 167. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, S.H.; Fan, S.; Dykstra, H.; Reichenbach, N.; Del Valle, L.; Potula, R.; Phipps, R.P.; Maggirwar, S.B.; Persidsky, Y. Dyad of CD40/CD40 ligand fosters neuroinflammation at the blood-brain barrier and is regulated via JNK signaling: Implications for HIV-1 encephalitis. J. Neurosci. 2010, 30, 9454–9464. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Lo, Y.; Chapagain, M.; Lum, S.; Kumar, M.; Gurjav, U.; Luo, H.; Nakatsuka, A.; Nerurkar, V.R. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier. Virology 2009, 385, 425–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clé, M.; Desmetz, C.; Barthelemy, J.; Martin, M.F.; Constant, O.; Maarifi, G.; Foulongne, V.; Bolloré, K.; Glasson, Y.; De Bock, F.; et al. Zika Virus Infection Promotes Local Inflammation, Cell Adhesion Molecule Upregulation, and Leukocyte Recruitment at the Blood-Brain Barrier. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Martínez-Palomo, A.; Ordoñez, G.; Pineda, B. Varicella-zoster virus in cerebrospinal fluid at relapses of multiple sclerosis. Ann. Neurol. 2008, 63, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Akerstrom, V. HIV-1 protein gp120 crosses the blood-brain barrier: Role of adsorptive endocytosis. Life Sci. 1997, 61, PL119–PL125. [Google Scholar] [CrossRef]
- Schweighardt, B.; Atwood, W.J. Virus receptors in the human central nervous system. J. Neurovirol. 2001, 7, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Moses, A.V.; Bloom, F.E.; Pauza, C.D.; Nelson, J.A. Human immunodeficiency virus infection of human brain capillary endothelial cells occurs via a CD4/galactosylceramide-independent mechanism. Proc. Natl. Acad. Sci. USA 1993, 90, 10474–10478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohgu, S.; Ryerse, J.S.; Robinson, S.M.; Banks, W.A. Human immunodeficiency virus-1 uses the mannose-6-phosphate receptor to cross the blood-brain barrier. PLoS ONE 2012, 7, e39565. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gao, J.; Xu, Y.P.; Zhou, T.L.; Jin, Y.Y.; Lou, J.N. Expression of severe acute respiratory syndrome coronavirus receptors, ACE2 and CD209L in different organ derived microvascular endothelial cells. Zhonghua Yi Xue Za Zhi 2007, 87, 833–837. [Google Scholar]
- Rhea, E.M.; Logsdon, A.F.; Hansen, K.M.; Williams, L.M.; Reed, M.J.; Baumann, K.K.; Holden, S.J.; Raber, J.; Banks, W.A.; Erickson, M.A. The S1 protein of SARS-CoV-2 crosses the blood-brain barrier in mice. Nat. Neurosci. 2021, 24, 368–378. [Google Scholar] [CrossRef]
- Bryche, B.; St Albin, A.; Murri, S.; Lacote, S.; Pulido, C.; Ar Gouilh, M.; Lesellier, S.; Servat, A.; Wasniewski, M.; Picard-Meyer, E.; et al. Massive transient damage of the olfactory epithelium associated with infection of sustentacular cells by SARS-CoV-2 in golden Syrian hamsters. Brain Behav. Immun. 2020, 89, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Radzikowska, U.; Ding, M.; Tan, G.; Zhakparov, D.; Peng, Y.; Wawrzyniak, P.; Wang, M.; Li, S.; Morita, H.; Altunbulakli, C.; et al. Distribution of ACE2, CD147, CD26 and other SARS-CoV-2 associated molecules in tissues and immune cells in health and in asthma, COPD, obesity, hypertension, and COVID-19 risk factors. Allergy 2020, 75, 2829–2845. [Google Scholar] [CrossRef] [PubMed]
- Vankadari, N.; Wilce, J.A. Emerging WuHan (COVID-19) coronavirus: Glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg. Microbes Infect. 2020, 9, 601–604. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef]
- Pellegrini, L.; Albecka, A.; Mallery, D.L.; Kellner, M.J.; Paul, D.; Carter, A.P.; James, L.C.; Lancaster, M.A. SARS-CoV-2 Infects the Brain Choroid Plexus and Disrupts the Blood-CSF Barrier in Human Brain Organoids. Cell Stem Cell 2020, 27, 951–961.e955. [Google Scholar] [CrossRef] [PubMed]
- Israelow, B.; Song, E.; Mao, T.; Lu, P.; Meir, A.; Liu, F.; Alfajaro, M.M.; Wei, J.; Dong, H.; Homer, R.J.; et al. Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- De Andrea, M.; Ravera, R.; Gioia, D.; Gariglio, M.; Landolfo, S. The interferon system: An overview. Eur. J. Paediatr. Neurol. 2002, 6 (Suppl. SA), A41–A46. [Google Scholar] [CrossRef] [Green Version]
- Borden, E.C.; Sen, G.C.; Uze, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Meffre, E.; Iwasaki, A. Interferon deficiency can lead to severe COVID. Nature 2020, 587, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Beziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- van den Pol, A.N.; Ding, S.; Robek, M.D. Long-distance interferon signaling within the brain blocks virus spread. J. Virol. 2014, 88, 3695–3704. [Google Scholar] [CrossRef] [Green Version]
- Kraus, J.; Voigt, K.; Schuller, A.M.; Scholz, M.; Kim, K.S.; Schilling, M.; Schabitz, W.R.; Oschmann, P.; Engelhardt, B. Interferon-beta stabilizes barrier characteristics of the blood-brain barrier in four different species in vitro. Mult. Scler. 2008, 14, 843–852. [Google Scholar] [CrossRef] [PubMed]
- Daniels, B.P.; Klein, R.S. Knocking on Closed Doors: Host Interferons Dynamically Regulate Blood-Brain Barrier Function during Viral Infections of the Central Nervous System. PLoS Pathog. 2015, 11, e1005096. [Google Scholar] [CrossRef] [PubMed]
- Finch, C.E.; Kulminski, A.M. The ApoE locus and COVID-19: Are we going where we have been? J. Gerontol. A Biol. Sci. Med. Sci. 2020, 76, e1–e3. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Kuchel, G.A.; Melzer, D. APOE e4 Genotype Predicts Severe COVID-19 in the UK Biobank Community Cohort. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 2231–2232. [Google Scholar] [CrossRef]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E: Structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J. Lipid Res. 2009, 50, S183–S188. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Wu, X.; Tang, H.; Luo, G. Apolipoprotein E mediates attachment of clinical hepatitis C virus to hepatocytes by binding to cell surface heparan sulfate proteoglycan receptors. PLoS ONE 2013, 8, e67982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, R.; Hueging, K.; Brown, R.J.P.; Todt, D.; Joecks, S.; Vondran, F.W.R.; Pietschmann, T. Hepatitis C Virus Strain-Dependent Usage of Apolipoprotein E Modulates Assembly Efficiency and Specific Infectivity of Secreted Virions. J. Virol. 2017, 91, e00422-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.M.; Bhattacharjee, P.S.; Neumann, D.M. Apolipoprotein E alleles can contribute to the pathogenesis of numerous clinical conditions including HSV-1 corneal disease. Exp. Eye Res. 2007, 84, 801–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.R.; Shang, D.; Wilcock, G.K.; Itzhaki, R.F. Alzheimer’s disease, herpes simplex virus type 1, cold sores and apolipoprotein E4. Biochem. Soc. Trans. 1995, 23, 594S. [Google Scholar] [CrossRef] [Green Version]
- Burgos, J.S.; Ramirez, C.; Sastre, I.; Bullido, M.J.; Valdivieso, F. ApoE4 is more efficient than E3 in brain access by herpes simplex virus type 1. Neuroreport 2003, 14, 1825–1827. [Google Scholar] [CrossRef]
- Burt, T.D.; Agan, B.K.; Marconi, V.C.; He, W.; Kulkarni, H.; Mold, J.E.; Cavrois, M.; Huang, Y.; Mahley, R.W.; Dolan, M.J.; et al. Apolipoprotein (apo) E4 enhances HIV-1 cell entry in vitro, and the APOE epsilon4/epsilon4 genotype accelerates HIV disease progression. Proc. Natl. Acad. Sci. USA 2008, 105, 8718–8723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, I.; Minihane, A.M.; Huebbe, P.; Nebel, A.; Rimbach, G. Apolipoprotein E genotype and hepatitis C, HIV and herpes simplex disease risk: A literature review. Lipids Health Dis. 2010, 9, 8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wu, L.M.; Wu, J. Cross-talk between apolipoprotein E and cytokines. Mediat. Inflamm. 2011, 2011, 949072. [Google Scholar] [CrossRef] [Green Version]
- Vitek, M.P.; Brown, C.M.; Colton, C.A. APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 2009, 30, 1350–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, K.; Mackman, N.; Curtiss, L.K. Interferon-gamma inhibits macrophage apolipoprotein E production by posttranslational mechanisms. J. Clin. Investig. 1993, 91, 2031–2039. [Google Scholar] [CrossRef] [Green Version]
- Monteiro-Junior, R.S. COVID-19: Thinking about further mental and neurological disorders. Med. Hypotheses 2020, 143, 109894. [Google Scholar] [CrossRef]
- Drucker, D.J. Coronavirus Infections and Type 2 Diabetes-Shared Pathways with Therapeutic Implications. Endocr. Rev. 2020, 41, bnaa011. [Google Scholar] [CrossRef]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1alpha/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 498–499. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; She, Z.G.; Cheng, X.; Qin, J.J.; Zhang, X.J.; Cai, J.; Lei, F.; Wang, H.; Xie, J.; Wang, W.; et al. Association of Blood Glucose Control and Outcomes in Patients with COVID-19 and Pre-existing Type 2 Diabetes. Cell Metab. 2020, 31, 1068–1077.e1063. [Google Scholar] [CrossRef]
- Lim, S.; Bae, J.H.; Kwon, H.S.; Nauck, M.A. COVID-19 and diabetes mellitus: From pathophysiology to clinical management. Nat. Rev. Endocrinol. 2021, 17, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Freeman, C.W.; Masur, J.; Hassankhani, A.; Wolf, R.L.; Levine, J.M.; Mohan, S. COVID-19-Related Disseminated Leukoencephalopathy (CRDL): A Retrospective Study of Findings on Brain MRI. AJR Am. J. Roentgenol. 2020, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W. Hypertension, Diabetes Increase Risk of COVID-19 Neurological Complications. Available online: https://www.diagnosticimaging.com/view/hypertension-diabetes-increase-risk-of-covid-19-neurological-complications (accessed on 14 December 2020).
- Yu, X.; Song, R.; Jiaerken, Y.; Yuan, L.; Huang, P.; Lou, M.; Jiang, Q.; Zhang, M. White matter injury induced by diabetes in acute stroke is clinically relevant: A preliminary study. Diab. Vasc. Dis. Res. 2017, 14, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanek, K.M.; Grieve, S.M.; Brickman, A.M.; Korgaonkar, M.S.; Paul, R.H.; Cohen, R.A.; Gunstad, J.J. Obesity is associated with reduced white matter integrity in otherwise healthy adults. Obesity (Silver Spring) 2011, 19, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Rom, S.; Zuluaga-Ramirez, V.; Gajghate, S.; Seliga, A.; Winfield, M.; Heldt, N.A.; Kolpakov, M.A.; Bashkirova, Y.V.; Sabri, A.K.; Persidsky, Y. Hyperglycemia-Driven Neuroinflammation Compromises BBB Leading to Memory Loss in Both Diabetes Mellitus (DM) Type 1 and Type 2 Mouse Models. Mol. Neurobiol. 2019, 56, 1883–1896. [Google Scholar] [CrossRef]
- O’Donnell, M.J.; Xavier, D.; Liu, L.; Zhang, H.; Chin, S.L.; Rao-Melacini, P.; Rangarajan, S.; Islam, S.; Pais, P.; McQueen, M.J.; et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): A case-control study. Lancet 2010, 376, 112–123. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Tissue Examined | Subject/Cohort Description | Post-Mortem Interval | Method of SARS-CoV-2 Detection in CNS | Detection Information | Associated Pathologies | Ref |
|---|---|---|---|---|---|---|
| CSF | Multiple, systematic review | Not reported | qPCR | Detected in 4/18 subjects | Not reported | [51] |
| Brain Biopsies | Detected in 8/34 subjects | |||||
| Brain tissue | Case report, 74-year-old Hispanic male with Parkinson’s disease; PCR positive NP swab. Febrile, hypotensive, thrombocytopenic, declining SpO2, elevated CRP, D-dimer, ferritin | Not reported | Transmission electron microscopic detection of viral-like particles | Detection of viral-like particles in frontal lobe, inside endothelial cell vesicles, and blebbing from endothelial membrane. Additionally, in neurons. | Vacuolization of neuronal cytoplasm | [55] |
| Brain tissue | qPCR | Detected | ||||
| CSF (post-mortem) | qPCR | Not detected | ||||
| CSF | 13 subjects with severe SARS-CoV-2, NP swab confirmed and presenting with pneumonia, seizures and/or encephalopathy | N/A, subjects were alive | qPCR (LOD 181 copies/mL) | SARS-CoV-2 not detected in CSF, but verified in NP swabs, some taken on same day. | No pleocytosis in CSF except in once case of hemorrhage. 9/11 examined had abnormal MRI/CT, evidence of subcortical hypoxic/ischemic injury | [58] |
| CSF, brain CT/MRI | 58 patients with NP swab confirmed SARS-CoV-2 and neurological manifestations, 47 had acute respiratory failure | N/A, subjects were alive | qPCR (LOD 500 copies/mL) | SARS-CoV-2 detected in 4/58 subjects; 3 below LOD | In CSF: 10 had increased WBCs, 19 had elevated albumin quotient, 21 had elevated IgG, 5–7 had elevated IL-6 and TNF-a. 36/53 subjects evaluated had CT/MRI abnormalities. | [59] |
| Post-mortem FFPE and frozen brain tissues | 43 patients confirmed with NP swab, age range 51–94. 40 had chronic medical conditions, 13 had pre-existing neurological disease, 12 died in ICU; deaths were primarily from viral pneumonia | 0–9 days (3.3 mean) | qPCR, S and N histochemistry | 9/23 total had RNA detected; 9 in frozen frontal lobe, 4 in FFPE medulla oblongata. 16/40 had S and/or N detected in medulla oblongata and along cranial nerves; 14/16 S+, 7/16 N+. 21/40 had RNA or protein detected; Of 16 brains with RNA and protein measured, 8 had both, 4 had protein only, 4 had RNA only. | Brain edema (53%), Arteriosclerosis (100%), Gross macroscopic abnormalities (30%), Fresh ischemic lesions of cerebral arteries (14%), no cerebral bleeding/small vessel thrombosis, astrogliosis in olfactory bulb, basal ganglia, brainstem, cerebellum, microgliosis in brainstem and cerebellum, HL-DR in subpial and subependymal regions. | [60] |
| Post-mortem FFPE brain sections | Three subjects who died of severe COVID-19; respiratory failure, on ventilator, PCR positive postmortem lung. All had comorbidities (hypertension, obesity, or kidney transplantation) | Not reported | S1 histochemistry | S1 detected in cortical neurons and endothelial cells; positive viral staining detected around infarcts in one patient | No leukocyte infiltration in regions with S1 staining | [61] |
| Tissue Examined | Subject/Cohort Description | Post-Mortem Interval | Method of SARS-CoV-2 Detection in CNS | Detection Information | Associated Pathologies | Ref |
| FFPE brain tissue sections | 18 subjects with PCR-confirmed COVID-19 age 48–90. Neurologic sequalae: myalgia (3), headache (3), loss of taste (1). Co-morbidities: diabetes (12), hypertension (11), cardiovascular disease (5), hyperlipidemia (5), chronic kidney disease (4), prior stroke (4), dementia (4), anaplastic astrocytoma (1) | 20–102 h | qPCR for SARS-CoV-2 nucleocapsid mRNA and histochemistry for N protein: frontal lobe/olfactory nerve and medulla for all patients; cingulate/corpus collosum, hippocampus, occipital lobe, basal ganglia, thalamus, cerebellum, midbrain, pons were additionally tested in two subjects | Equivocal detection (<5 copies/cm3) in 5/10 and 4/10 sections from the two subjects with 10 regions assessed; in 16 subjects with 2 regions assessed, 5 subjects had > 5 copies/cm3, 8 subjects had equivocal detection, and 3 subjects had no detectable mRNA. N protein not detected. | All subjects had evidence of acute hypoxic changes in the cerebrum and cerebellum, no microscopic abnormalities of olfactory bulb/olfactory tracts, neuronal loss in hippocampus, cerebrum and cerebellum but no thrombi or vasculitis. Perivascular lymphocyte foci detected in 2/18 subjects. | [62] |
| Post-mortem FFPE and frozen brain tissue | 19 patients confirmed with NP swab, age range 5–73 | 5–368 h | qPCR, RNAscope | SARS-CoV-2 not detected | Out of 19: Vascular pathology (11), perivascular infiltrates (13), acute hypoxic/ischemic neuronal damage (6), no path findings (2) | [63] |
| CSF, brain CT/MRI | Case report, 55-year-old previously healthy woman with PCR-confirmed COVID-19, pulmonary ground glass opacities. Found unresponsive in bed without hemodynamic or respiratory issues. | N/A, patient survived and was discharged. | qPCR | CSF was collected on day 9, 12, 14, and 26 from first symptoms, SARS-CoV-2 detected only on day 14 (cycle threshold = 34.29) | CSF day 9: no pleocytosis, but elevated albumin. CSF day 12: no pleocytosis, albumin normal, IgG elevated without autoantibodies. Elevated GFAP, NFL, tau, and IL-6. CSF day 14: further increases in NFL and tau and reductions in GFAP and IL-6, increases in CSF total protein, and appearance of oligoclonal bands. CT and MRI: symmetrical hypodensities in thalami that progressed to midbrain; acute necrotizing encephalitis | [64] |
| Model | Tissue Examined | Method of SARS-CoV-2 Detection | Detection Information | Associated Pathologies | Ref |
|---|---|---|---|---|---|
| K18-hACE2 1.5 × 106 PFU intranasal | Whole brain homogenate | qPCR | Yes- day 2, 4 and 7 post-infection all mice | [61] | |
| Whole brain homogenate | Viral titers | Yes- day 2, 4 and 7 post-infection all mice | |||
| iDISCO cleared brains | Immunolabeling of N protein/ light sheet microscopy | Yes- Forebrain neural cells, sensory cortex, dentate gyrus, globus pallidus, cortical layer IV, not cerebellum, not endothelium day 7 post-infection | Remodeling of vasculature found in proximity to virus | ||
| K18-hACE2 2.5 × 104 PFU intranasal | Whole brain homogenate | qPCR | Yes- day 2, 4, and 7 post-infection all mice | [66] | |
| Whole brain homogenate | Viral titers | Yes- day 7 for 4/10 mice tested, but not on day 2 or 4. | |||
| FFPE Brain sections | qPCR (non-fixed side) | Sections from brains with high or low/no viral load compared for pathologic changes | No/low SARS-CoV-2 brains had minimal/no brain pathology, SARS-CoV-2 infected brains had meningeal inflammation, leukocyte extravasation to parenchyma and microglia activation | ||
| hACE2 humanized mouse 4 × 105 PFU intranasal | Whole brain homogenate | qPCR | Yes- day 6 post-infection all mice, absent in control mice | [70] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erickson, M.A.; Rhea, E.M.; Knopp, R.C.; Banks, W.A. Interactions of SARS-CoV-2 with the Blood–Brain Barrier. Int. J. Mol. Sci. 2021, 22, 2681. https://doi.org/10.3390/ijms22052681
Erickson MA, Rhea EM, Knopp RC, Banks WA. Interactions of SARS-CoV-2 with the Blood–Brain Barrier. International Journal of Molecular Sciences. 2021; 22(5):2681. https://doi.org/10.3390/ijms22052681
Chicago/Turabian StyleErickson, Michelle A., Elizabeth M. Rhea, Rachel C. Knopp, and William A. Banks. 2021. "Interactions of SARS-CoV-2 with the Blood–Brain Barrier" International Journal of Molecular Sciences 22, no. 5: 2681. https://doi.org/10.3390/ijms22052681
APA StyleErickson, M. A., Rhea, E. M., Knopp, R. C., & Banks, W. A. (2021). Interactions of SARS-CoV-2 with the Blood–Brain Barrier. International Journal of Molecular Sciences, 22(5), 2681. https://doi.org/10.3390/ijms22052681