
A Holistic Evolutionary and 3D Pharmacophore Modelling Study Provides Insights into the Metabolism, Function, and Substrate Selectivity of the Human Monocarboxylate Transporter 4 (hMCT4)

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Evolutionary Study and MCT4 Specific Conserved Motifs Elucidation
2.2. Three-Dimensional Homology Modelling of MCT4
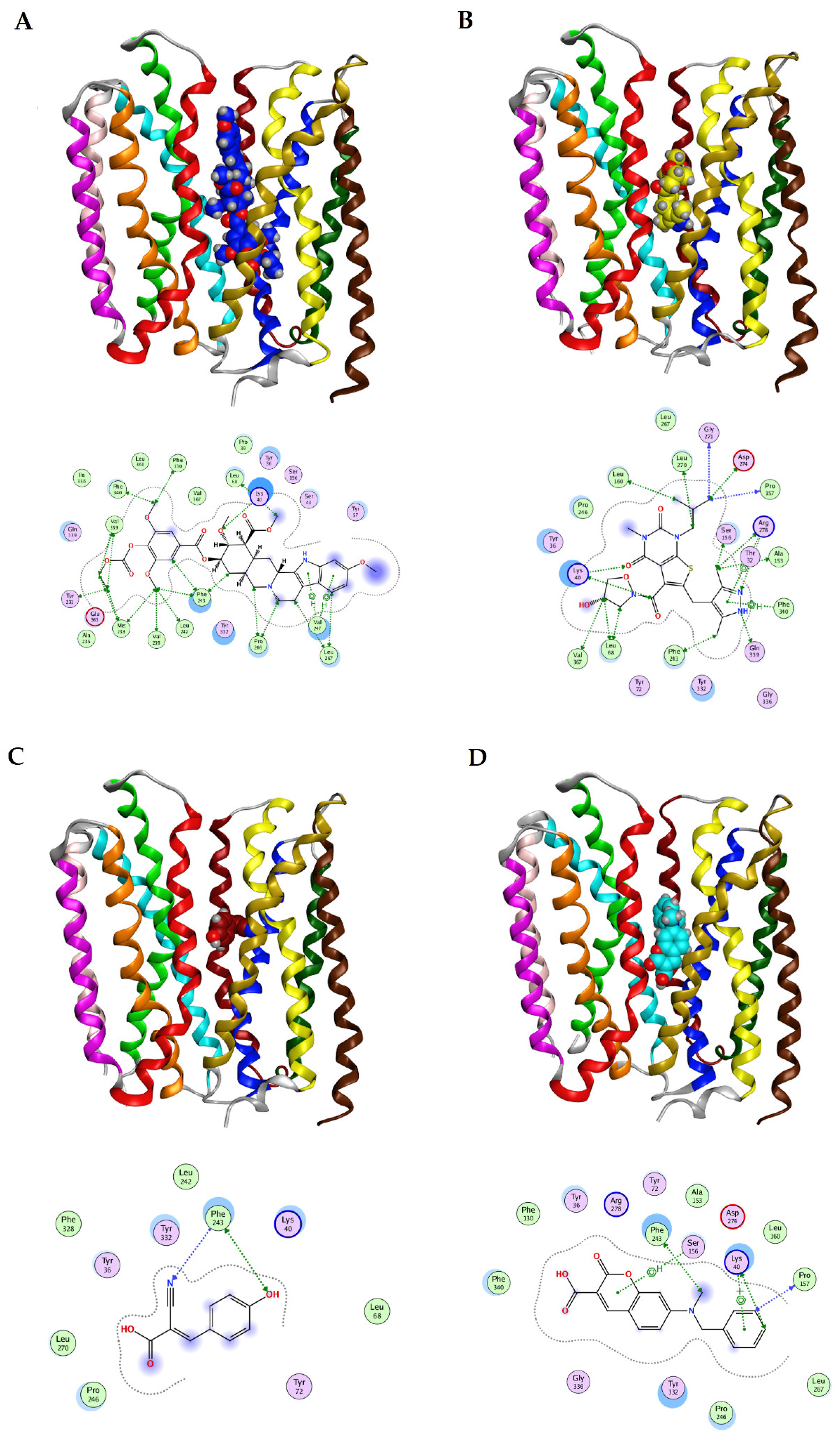
2.3. MCT4 Specific Multimodal Pharmacophore Design
3. Discussion
4. Materials and Methods
4.1. Phylogenetic Analysis and Conserved Motif Identification
4.2. Molecular Modelling and Homology Modelling
4.3. Energy Minimization and Molecular Dynamics Simulations
4.4. Pharmacophore Elucidation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Castro Sant’ Anna, C.; Junior, A.G.F.; Soares, P.; Tuji, F.; Paschoal, E.; Chaves, L.C.; Burbano, R.R. Molecular biology as a tool for the treatment of cancer. Clin. Exp. Med. 2018, 18, 457–464. [Google Scholar] [CrossRef]
- Tashima, T. Effective cancer therapy based on selective drug delivery into cells across their membrane using receptor-mediated endocytosis. Bioorg. Med. Chem. Lett. 2018, 28, 3015–3024. [Google Scholar] [CrossRef]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of cancer pharmacological treatments at the turn of the third millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, A.; Kamphorst, J.J.; Markert, E.K.; Schug, Z.T.; Tardito, S.; Gottlieb, E. Cancer metabolism at a glance. J. Cell Sci. 2016, 129, 3367–3373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66. [Google Scholar] [CrossRef]
- Al Tameemi, W.; Dale, T.P.; Al-Jumaily, R.M.K.; Forsyth, N.R. Hypoxia-modified cancer cell metabolism. Front. Cell Dev. Biol. 2019, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwanaga, T.; Kishimoto, A. Cellular distributions of monocarboxylate transporters: A review. Biomed. Res. 2015, 36, 279–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halestrap, A.P.; Wilson, M.C. The monocarboxylate transporter family—Role and regulation. IUBMB Life 2011, 64, 109–119. [Google Scholar] [CrossRef]
- Li, Z.; Wu, Q.; Sun, S.; Wu, J.; Li, J.; Zhang, Y.; Wang, C.; Yuan, J.; Sun, S. Monocarboxylate transporters in breast cancer and adipose tissue are novel biomarkers and potential therapeutic targets. Biochem. Biophys. Res. Commun. 2018, 501, 962–967. [Google Scholar] [CrossRef]
- Pérez-Escuredo, J.; Van Hée, V.F.; Sboarina, M.; Falces, J.; Payen, V.L.; Pellerin, L.; Sonveaux, P. Monocarboxylate transporters in the brain and in cancer. Biochim. Biophys. Acta 2016, 1863, 2481–2497. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Baeza, Y.; Sandoval, P.Y.; Alarcón, R.; Galaz, A.; Cortés-Molina, F.; Alegría, K.; Baeza-Lehnert, F.; Arce-Molina, R.; Guequén, A.; Flores, C.A.; et al. Monocarboxylate transporter 4 (MCT4) is a high affinity transporter capable of exporting lactate in high-lactate microenvironments. J. Biol. Chem. 2019, 294, 20135–20147. [Google Scholar] [CrossRef] [PubMed]
- Sonveaux, P.; Végran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942. [Google Scholar] [CrossRef] [Green Version]
- Pereira-Vieira, J.; Azevedo-Silva, J.; Preto, A.; Casal, M.; Queirós, O. MCT1, MCT4 and CD147 expression and 3-bromopyruvate toxicity in colorectal cancer cells are modulated by the extracellular conditions. Biol. Chem. 2019, 400, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Maciejewski, H.; Bourdin, M.; Féasson, L.; Dubouchaud, H.; Denis, C.; Freund, H.; Messonnier, L.A. Muscle MCT4 content is correlated with the lactate removal ability during recovery following all-out supramaximal exercise in highly trained rowers. Front. Physiol. 2016, 7, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1α-dependent mechanism*. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [Green Version]
- Baltazar, F.; Pinheiro, C.; Morais-Santos, F.; Azevedo-Silva, J.; Queirós, O.; Preto, A.; Casal, M. Monocarboxylate transporters as targets and mediators in cancer therapy response. Histol. Histopathol. 2014, 29, 1511–1524. [Google Scholar] [PubMed]
- Ekberg, H.; Qi, Z.; Pahlman, C.; Veress, B.; Bundick, R.V.; Craggs, R.I.; Holness, E.; Edwards, S.; Murray, C.M.; Ferguson, D.; et al. The specific monocarboxylate transporter-1 (MCT-1) inhibitor, AR-C117977, induces donor-specific suppression, reducing acute and chronic allograft rejection in the rat. Transplantation 2007, 84, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Ovens, M.J.; Davies, A.J.; Wilson, M.C.; Murray, C.M.; Halestrap, A.P. AR-C155858 is a potent inhibitor of monocarboxylate transporters MCT1 and MCT2 that binds to an intracellular site involving transmembrane helices 7–10. Biochem. J. 2010, 425, 523–530. [Google Scholar] [CrossRef]
- Le Floch, R.; Chiche, J.; Marchiq, I.; Naiken, T.; Ilc, K.; Murray, C.M.; Critchlow, S.E.; Roux, D.; Simon, M.-P.; Pouysségur, J. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 16663–16668. [Google Scholar] [CrossRef] [Green Version]
- Draoui, N.; Schicke, O.; Seront, E.; Bouzin, C.; Sonveaux, P.; Riant, O.; Feron, O. Antitumor activity of 7-aminocarboxycoumarin derivatives, a new class of potent inhibitors of lactate influx but not efflux. Mol. Cancer Ther. 2014, 13, 1410–1418. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.-M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual inhibition of the lactate transporters MCT1 and MCT4 is synthetic lethal with metformin due to NAD+ depletion in cancer cells. Cell Rep. 2018, 25, 3047–3058.e4. [Google Scholar] [CrossRef] [Green Version]
- Yan, N. Structural biology of the major facilitator superfamily transporters. Annu. Rev. Biophys. 2015, 44, 257–283. [Google Scholar] [CrossRef] [PubMed]
- Bosshart, P.D.; Kalbermatter, D.; Bonetti, S.; Fotiadis, D. Mechanistic basis of L-lactate transport in the SLC16 solute carrier family. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nat. Cell Biol. 2014, 510, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Jin, Q.; Xu, L.; Li, N.; Meng, Y.; Chang, S.; Zheng, X.; Wang, J.; Chen, Y.; Neculai, D.; et al. Cooperative transport mechanism of human monocarboxylate transporter 2. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Baek, G.; Tse, Y.F.; Hu, Z.; Cox, D.; Buboltz, N.; McCue, P.; Yeo, C.J.; White, M.A.; DeBerardinis, R.J.; Knudsen, E.S.; et al. MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep. 2014, 9, 2233–2249. [Google Scholar] [CrossRef] [Green Version]
- Bartels, C.C. Syrosingopine: A new rauwolfia preparation. N. Engl. J. Med. 1959, 261, 785–788. [Google Scholar] [CrossRef]
- Jones, R.S.; E Morris, M. Monocarboxylate transporters: Therapeutic targets and prognostic factors in disease. Clin. Pharmacol. Ther. 2016, 100, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.A.; Bell, N.; Blair, H.; Sikka, A.; Thomas, H.; Phillips, N.; Nakjang, S.; Miwa, S.; Crossland, R.; Rand, V.; et al. Inhibition of monocarboxyate transporter 1 by AZD3965 as a novel therapeutic approach for diffuse large B-cell lymphoma and Burkitt lymphoma. Haematologica 2017, 102, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Pierre, K.; Pellerin, L. Monocarboxylate transporters in the central nervous system: Distribution, regulation and function. J. Neurochem. 2005, 94, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Khammanivong, A.; Saha, J.; Spartz, A.K.; Sorenson, B.S.; Bush, A.G.; Korpela, D.M.; Gopalakrishnan, R.; Jonnalagadda, S.; Mereddy, V.R.; O’Brien, T.D.; et al. A novel MCT1 and MCT4 dual inhibitor reduces mitochondrial metabolism and inhibits tumour growth of feline oral squamous cell carcinoma. Vet. Comp. Oncol. 2020, 18, 324–341. [Google Scholar] [CrossRef]
- McNeillis, R.; Greystoke, A.; Walton, J.; Bacon, C.; Keun, H.; Siskos, A.; Petrides, G.; Leech, N.; Jenkinson, F.; Bowron, A.; et al. A case of malignant hyperlactaemic acidosis appearing upon treatment with the mono-carboxylase transporter 1 inhibitor AZD3965. Br. J. Cancer 2020, 122, 1141–1145. [Google Scholar] [CrossRef]
- Puri, S.; Juvale, K. Monocarboxylate transporter 1 and 4 inhibitors as potential therapeutics for treating solid tumours: A review with structure-activity relationship insights. Eur. J. Med. Chem. 2020, 199, 112393. [Google Scholar] [CrossRef] [PubMed]
- Coordinators, N.R.; Agarwala, R.; Barrett, T.; Beck, J.; A Benson, D.; Bollin, C.; Bolton, E.; Bourexis, D.; Brister, J.R.; Bryant, S.H.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Molecular Operating Environment (MOE); Chemical Computing Group ULC: Montreal, QC, Canada, 2018.
- The UniProt Consortium UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef] [Green Version]
- Boratyn, G.M.; A Schäffer, A.; Agarwala, R.; Altschul, S.F.; Lipman, D.J.; Madden, T.L. Domain enhanced lookup time accelerated BLAST. Biol. Direct 2012, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Jiang, X.; Zhang, S.; Zhu, A.; Yuan, Y.; Xu, H.; Lei, J.; Yan, C. Structural basis of human monocarboxylate transporter 1 inhibition by anti-cancer drug candidates. Cell 2021, 184, 370–383.e13. [Google Scholar] [CrossRef] [PubMed]
- Labute, P. The generalized Born/volume integral implicit solvent model: Estimation of the free energy of hydration using London dispersion instead of atomic surface area. J. Comput. Chem. 2008, 29, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Tieleman, D.P.; Berendsen, H.J.C. Molecular dynamics simulations of a fully hydrated dipalmitoylphosphatidylcholine bilayer with different macroscopic boundary conditions and parameters. J. Chem. Phys. 1996, 105, 4871–4880. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; A Shoemaker, B.; A Thiessen, P.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papakonstantinou, E.; Vlachakis, D.; Thireou, T.; Vlachoyiannopoulos, P.G.; Eliopoulos, E. A Holistic Evolutionary and 3D Pharmacophore Modelling Study Provides Insights into the Metabolism, Function, and Substrate Selectivity of the Human Monocarboxylate Transporter 4 (hMCT4). Int. J. Mol. Sci. 2021, 22, 2918. https://doi.org/10.3390/ijms22062918
Papakonstantinou E, Vlachakis D, Thireou T, Vlachoyiannopoulos PG, Eliopoulos E. A Holistic Evolutionary and 3D Pharmacophore Modelling Study Provides Insights into the Metabolism, Function, and Substrate Selectivity of the Human Monocarboxylate Transporter 4 (hMCT4). International Journal of Molecular Sciences. 2021; 22(6):2918. https://doi.org/10.3390/ijms22062918
Chicago/Turabian StylePapakonstantinou, Eleni, Dimitrios Vlachakis, Trias Thireou, Panayiotis G. Vlachoyiannopoulos, and Elias Eliopoulos. 2021. "A Holistic Evolutionary and 3D Pharmacophore Modelling Study Provides Insights into the Metabolism, Function, and Substrate Selectivity of the Human Monocarboxylate Transporter 4 (hMCT4)" International Journal of Molecular Sciences 22, no. 6: 2918. https://doi.org/10.3390/ijms22062918