
Exploiting the Role of Hypoxia-Inducible Factor 1 and Pseudohypoxia in the Myelodysplastic Syndrome Pathophysiology

 , ,
, ,  , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
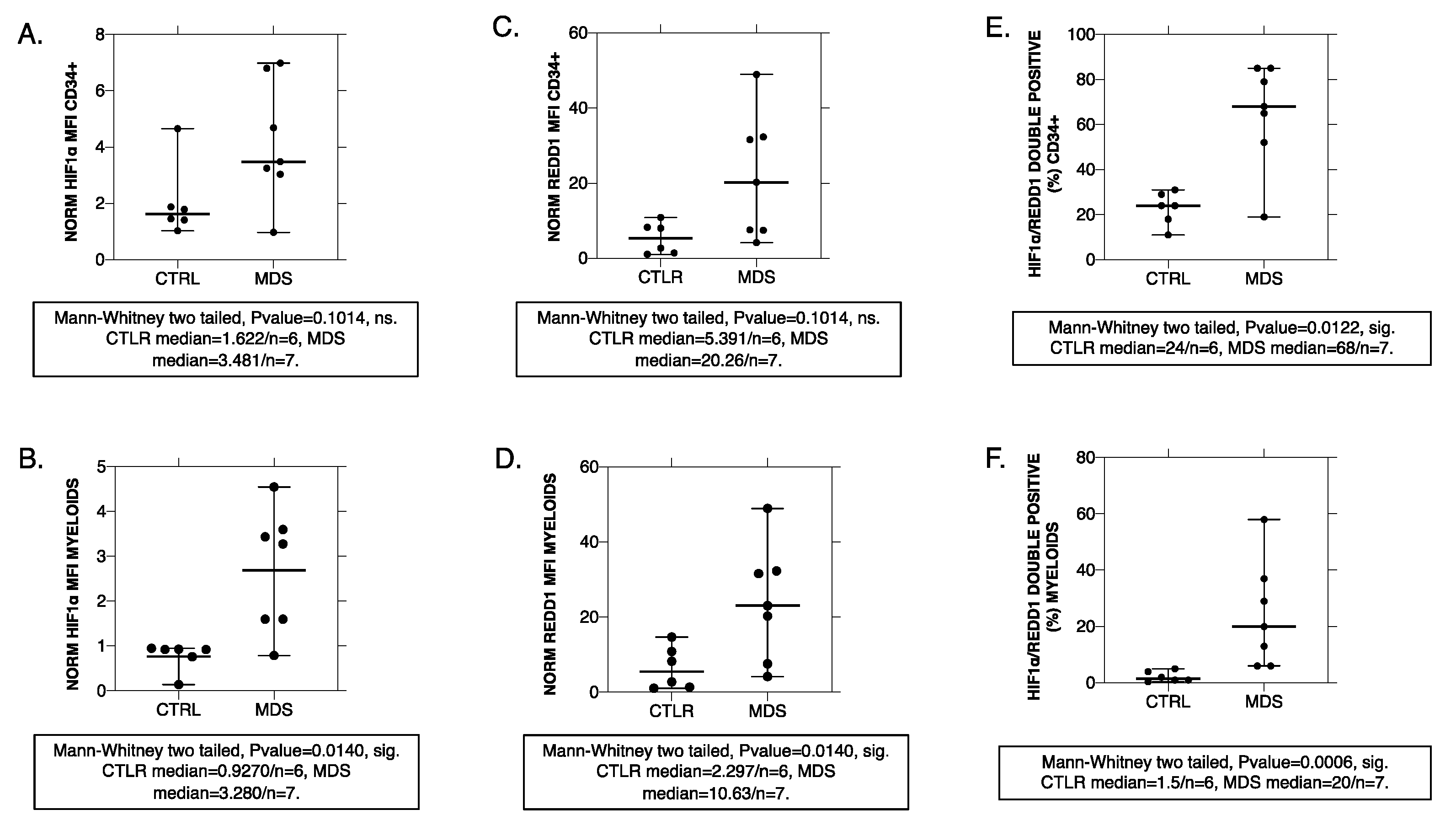
2.1. Human MDS CD34+ and Myeloid-Lineage Cells Display an Activated HIF-1 Profile with Increased REDD1 Protein Levels
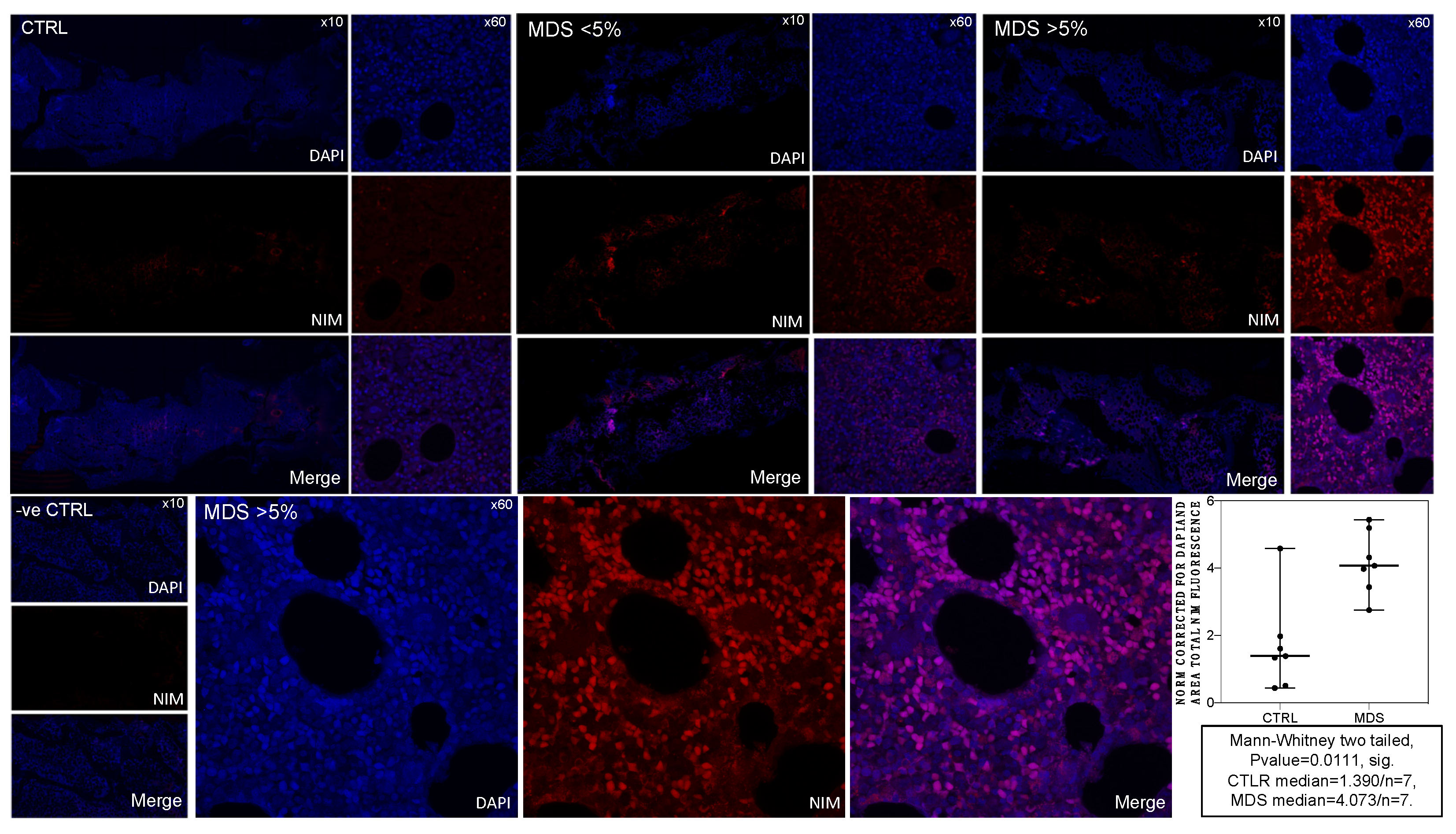
2.2. MDS BM Shows Increased Intracellular NIM-IDC Accumulation That Positively Correlates with MDS Severity
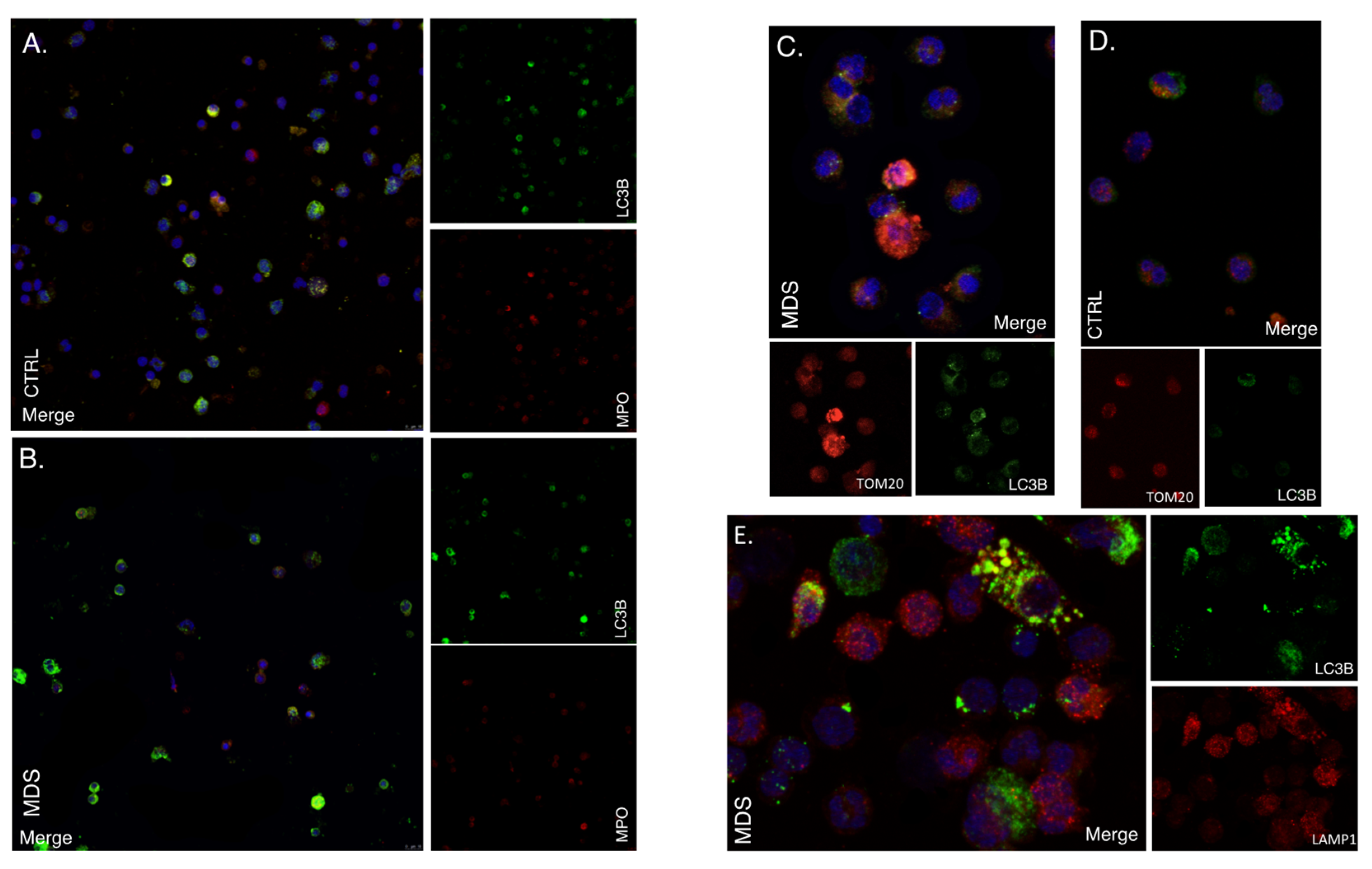
2.3. Mitophagy and Autophagic Cell Death Are Prominently Featured in the MDS Myeloid Lineage; Their Severity Increasing with the Intra BM Blast Counts
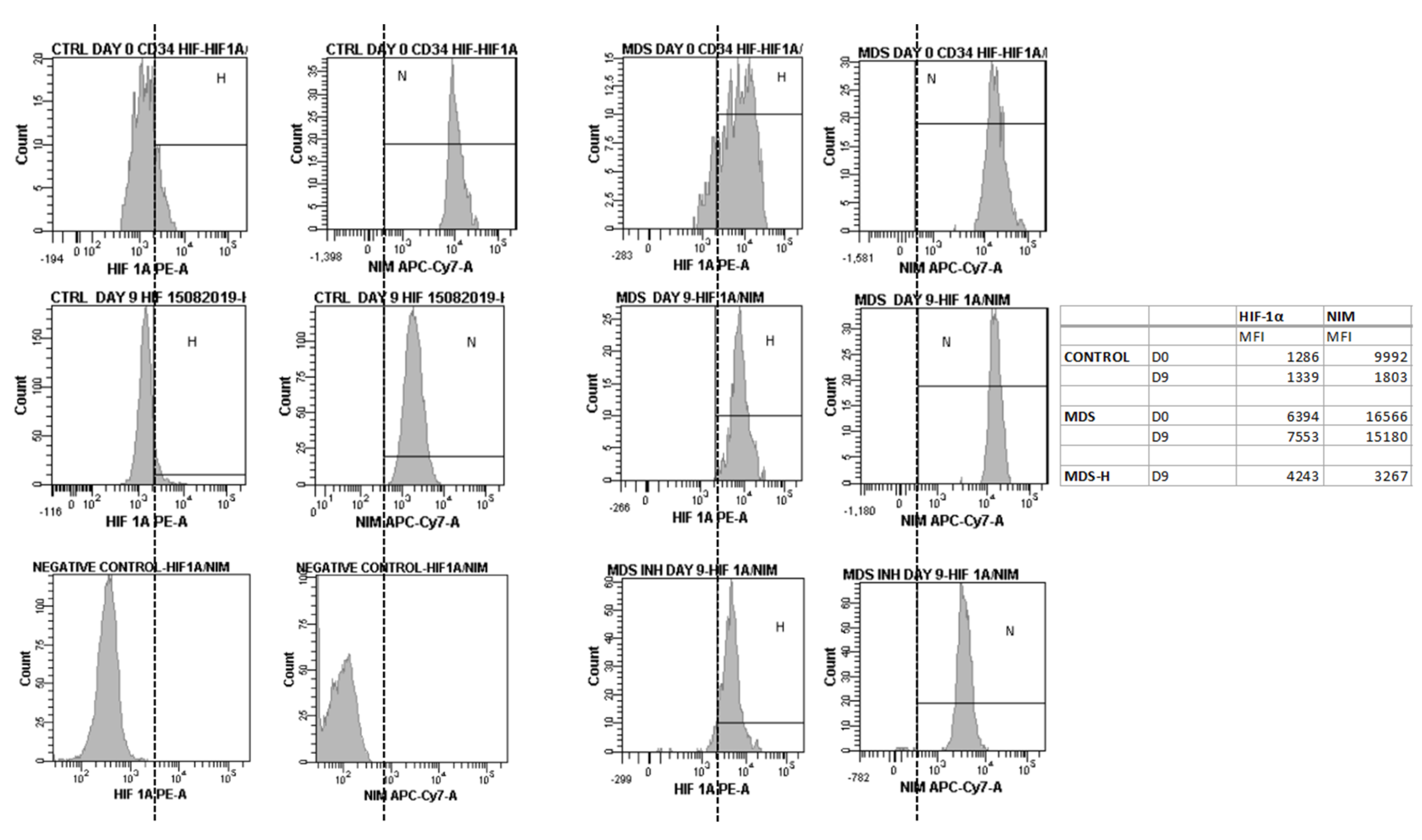
2.4. HIF-1α Inhibition Enhances the Proliferative and Differentiating Capacity of MDS BM CD34+ Cells
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. BM Cell Isolation
4.3. BM Cell Isolation RNA Isolation, cDNA Synthesis and qRT-PCR
4.4. Flow Cytometry
4.5. NIM Dye-Conjugate Synthesis
4.6. NIM Staining and Immunofluorescence in BM Biopsies
4.7. Immunofluoresence
4.8. Electron Microscopy
4.9. CD34+ Cell Cultures
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, W.K.; Koeffler, H.P. Myelodysplastic syndrome. Annu. Rev. Med. 2005, 56, 1–16. [Google Scholar] [CrossRef]
- Hsu, J.; Reilly, A.; Hayes, B.J.; Clough, C.A.; Konnick, E.Q.; Torok-Storb, B.; Gulsuner, S.; Wu, D.; Becker, P.S.; Keel, S.B.; et al. Reprogramming identifies functionally distinct stages of clonal evolution in myelodysplastic syndromes. Blood 2019, 134, 186–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossner, M.; Jann, J.C.; Wittig, J.; Nolte, F.; Fey, S.; Nowak, V.; Obländer, J.; Pressler, J.; Palme, I.; Xanthopoulos, C.; et al. Mutational hierarchies in myelodysplastic syndromes dynamically adapt and evolve upon therapy response and failure. Blood 2016, 128, 1246–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627, quiz 3699. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acuna-Hidalgo, R.; Sengul, H.; Steehouwer, M.; van de Vorst, M.; Vermeulen, S.H.; Kiemeney, L.; Veltman, J.A.; Gilissen, C.; Hoischen, A. Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am. J. Hum. Genet. 2017, 101, 50–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masala, E.; Valencia-Martinez, A.; Pillozzi, S.; Rondelli, T.; Brogi, A.; Sanna, A.; Gozzini, A.; Arcangeli, A.; Sbarba, P.D.; Santini, V. Severe hypoxia selects hematopoietic progenitors with stem cell potential from primary Myelodysplastic syndrome bone marrow cell cultures. Oncotarget 2018, 9, 10561–10571. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.E.; Conlon, J.P.; Yang, X.; Sanchez, P.V.; Carroll, M. Enhanced growth of myelodysplastic colonies in hypoxic conditions. Exp. Hematol. 2007, 35, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Bonora, M.; Ito, K. Metabolism as master of hematopoietic stem cell fate. Int. J. Hematol. 2019, 109, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Kohli, L.; Passegué, E. Surviving change: The metabolic journey of hematopoietic stem cells. Trends Cell Biol. 2014, 24, 479–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem. Cell 2011, 9, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.C.; Sadek, H.A. Hypoxia and metabolic properties of hematopoietic stem cells. Antioxid. Redox Signal. 2014, 20, 1891–1901. [Google Scholar] [CrossRef] [Green Version]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem. Cell 2010, 7, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Shen, S.M.; Zhao, X.Y.; Chen, G.Q. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int. J. Biochem. Mol. Biol. 2012, 3, 165–178. [Google Scholar]
- Lu, H.; Forbes, R.A.; Verma, A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J. Biol. Chem. 2002, 277, 23111–23115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem. J. 2007, 405, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Suda, T. Hyperactivated mitophagy in hematopoietic stem cells. Nat. Immunol. 2018, 19, 2–3. [Google Scholar] [CrossRef]
- Shoshani, T.; Faerman, A.; Mett, I.; Zelin, E.; Tenne, T.; Gorodin, S.; Moshel, Y.; Elbaz, S.; Budanov, A.; Chajut, A.; et al. Identification of a novel hypoxia-inducible factor 1-responsive gene, RTP801, involved in apoptosis. Mol. Cell. Biol. 2002, 22, 2283–2293. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Yokota, A.; Harada, H.; Huang, G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1α in cancer. Cancer Sci. 2019, 110, 1510–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iommarini, L.; Porcelli, A.M.; Gasparre, G.; Kurelac, I. Non-Canonical Mechanisms Regulating Hypoxia-Inducible Factor 1 Alpha in Cancer. Front. Oncol. 2017, 7, 286. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishi-Itakura, C.; Buss, F. The Use of Correlative Light-Electron Microscopy (CLEM) to Study PINK1/Parkin-Mediated Mitophagy. Methods Mol. Biol. 2018, 1759, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, M.P.; Lin, Y.; Maas, M.L.; Van Keulen, V.P.; Johnston, P.B.; Peikert, T.; Gastineau, D.A.; Dietz, A.B. A method for identification and analysis of non-overlapping myeloid immunophenotypes in humans. PLoS ONE 2015, 10, e0121546. [Google Scholar] [CrossRef] [PubMed]
- Nunn, A.; Linder, K.; Strauss, H.W. Nitroimidazoles and imaging hypoxia. Eur. J. Nucl. Med. 1995, 22, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Takanuki, K.; Igarashi, T.; Hata, K.; Hori, H.; Shibata, T.; Kitagawa, H.; Satoh, T.; Inayama, S. Glutathione depletion by 2-nitroimidazole-1-acetohydroxamic acid as a radiosensitizer in hypoxic rat hepatocytes. Biochem. Int. 1988, 17, 155–162. [Google Scholar]
- Liu, K.; Zhu, H.L. Nitroimidazoles as anti-tumor agents. Anticancer Agents Med. Chem. 2011, 11, 687–691. [Google Scholar] [CrossRef]
- Krohn, K.A.; Link, J.M.; Mason, R.P. Molecular imaging of hypoxia. J. Nucl. Med. 2008, 49 (Suppl. 2), 129s–148s. [Google Scholar] [CrossRef] [Green Version]
- Westrate, L.M.; Drocco, J.A.; Martin, K.R.; Hlavacek, W.S.; MacKeigan, J.P. Mitochondrial morphological features are associated with fission and fusion events. PLoS ONE 2014, 9, e95265. [Google Scholar] [CrossRef] [Green Version]
- Michaela, F.; Emmanuel, G. Apoptotic pathways to death in myelodysplastic syndromes. Haematologica 2008, 93, 1288–1292. [Google Scholar] [CrossRef] [Green Version]
- Poulaki, A.; Katsila, T.; Stergiou, I.E.; Giannouli, S.; Gόmez-Tamayo, J.C.; Piperaki, E.T.; Kambas, K.; Dimitrakopoulou, A.; Patrinos, G.P.; Tzioufas, A.G.; et al. Bioenergetic Profiling of the Differentiating Human MDS Myeloid Lineage with Low and High Bone Marrow Blast Counts. Cancers 2020, 12, 3520. [Google Scholar] [CrossRef]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Riffelmacher, T.; Simon, A.-K. Mechanistic roles of autophagy in hematopoietic differentiation. FEBS J. 2017, 284, 1008–1020. [Google Scholar] [CrossRef] [Green Version]
- Riffelmacher, T.; Clarke, A.; Richter, F.C.; Stranks, A.; Pandey, S.; Danielli, S.; Hublitz, P.; Yu, Z.; Johnson, E.; Schwerd, T.; et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 2017, 47, 466–480.e465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Levine, B. Autosis and autophagic cell death: The dark side of autophagy. Cell Death Differ. 2015, 22, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Zhang, Y.; Yokota, A.; Yan, X.; Liu, J.; Choi, K.; Li, B.; Sashida, G.; Peng, Y.; Xu, Z.; et al. Pathobiological Pseudohypoxia as a Putative Mechanism Underlying Myelodysplastic Syndromes. Cancer Discov. 2018, 8, 1438–1457. [Google Scholar] [CrossRef] [Green Version]
- Agani, F.; Jiang, B.H. Oxygen-independent regulation of HIF-1: Novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr Cancer Drug Targets 2013, 13, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Braccini, L.; Ciraolo, E.; Martini, M.; Pirali, T.; Germena, G.; Rolfo, K.; Hirsch, E. PI3K keeps the balance between metabolism and cancer. Adv. Biol. Regul. 2012, 52, 389–405. [Google Scholar] [CrossRef]
- Nepstad, I.; Hatfield, K.J.; Grønningsæter, I.S.; Reikvam, H. The PI3K-Akt-mTOR Signaling Pathway in Human Acute Myeloid Leukemia (AML) Cells. Int. J. Mol. Sci. 2020, 21, 2907. [Google Scholar] [CrossRef] [Green Version]
- Nyåkern, M.; Tazzari, P.L.; Finelli, C.; Bosi, C.; Follo, M.Y.; Grafone, T.; Piccaluga, P.P.; Martinelli, G.; Cocco, L.; Martelli, A.M. Frequent elevation of Akt kinase phosphorylation in blood marrow and peripheral blood mononuclear cells from high-risk myelodysplastic syndrome patients. Leukemia 2006, 20, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Follo, M.Y.; Mongiorgi, S.; Bosi, C.; Cappellini, A.; Finelli, C.; Chiarini, F.; Papa, V.; Libra, M.; Martinelli, G.; Cocco, L.; et al. The Akt/Mammalian Target of Rapamycin Signal Transduction Pathway Is Activated in High-Risk Myelodysplastic Syndromes and Influences Cell Survival and Proliferation. Cancer Res. 2007, 67, 4287–4294. [Google Scholar] [CrossRef] [Green Version]
- Faenza, I.; Matteucci, A.; Manzoli, L.; Billi, A.M.; Aluigi, M.; Peruzzi, D.; Vitale, M.; Castorina, S.; Suh, P.G.; Cocco, L. A role for nuclear phospholipase Cbeta 1 in cell cycle control. J. Biol. Chem. 2000, 275, 30520–30524. [Google Scholar] [CrossRef] [Green Version]
- Manzoli, L.; Billi, A.M.; Gilmour, R.S.; Martelli, A.M.; Matteucci, A.; Rubbini, S.; Weber, G.; Cocco, L. Phosphoinositide signaling in nuclei of Friend cells: Tiazofurin down-regulates phospholipase C beta 1. Cancer Res. 1995, 55, 2978–2980. [Google Scholar] [PubMed]
- Follo, M.Y.; Mongiorgi, S.; Clissa, C.; Paolini, S.; Martinelli, G.; Martelli, A.M.; Fioravanti, G.; Manzoli, L.; Finelli, C.; Cocco, L. Activation of nuclear inositide signalling pathways during erythropoietin therapy in low-risk MDS patients. Leukemia 2012, 26, 2474–2482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Follo, M.Y.; Finelli, C.; Mongiorgi, S.; Clissa, C.; Chiarini, F.; Ramazzotti, G.; Paolini, S.; Martinelli, G.; Martelli, A.M.; Cocco, L. Synergistic induction of PI-PLCβ1 signaling by azacitidine and valproic acid in high-risk myelodysplastic syndromes. Leukemia 2011, 25, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Pavlik, C.; Biswal, N.C.; Gaenzler, F.C.; Morton, M.D.; Kuhn, L.T.; Claffey, K.P.; Zhu, Q.; Smith, M.B. Synthesis and fluorescent characteristics of imidazole–indocyanine green conjugates. Dyes Pigment. 2011, 89, 9–15. [Google Scholar] [CrossRef]
- Jie, Z.; Zhang, Y.; Wang, C.; Shen, B.; Guan, X.; Ren, Z.; Ding, X.; Dai, W.; Jiang, Y. Large-scale ex vivo generation of human neutrophils from cord blood CD34+ cells. PLoS ONE 2017, 12, e0180832. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stergiou, I.E.; Kambas, K.; Poulaki, A.; Giannouli, S.; Katsila, T.; Dimitrakopoulou, A.; Vidali, V.; Mouchtouris, V.; Kloukina, I.; Xingi, E.; et al. Exploiting the Role of Hypoxia-Inducible Factor 1 and Pseudohypoxia in the Myelodysplastic Syndrome Pathophysiology. Int. J. Mol. Sci. 2021, 22, 4099. https://doi.org/10.3390/ijms22084099
Stergiou IE, Kambas K, Poulaki A, Giannouli S, Katsila T, Dimitrakopoulou A, Vidali V, Mouchtouris V, Kloukina I, Xingi E, et al. Exploiting the Role of Hypoxia-Inducible Factor 1 and Pseudohypoxia in the Myelodysplastic Syndrome Pathophysiology. International Journal of Molecular Sciences. 2021; 22(8):4099. https://doi.org/10.3390/ijms22084099
Chicago/Turabian StyleStergiou, Ioanna E., Konstantinos Kambas, Aikaterini Poulaki, Stavroula Giannouli, Theodora Katsila, Aglaia Dimitrakopoulou, Veroniki Vidali, Vasileios Mouchtouris, Ismini Kloukina, Evangelia Xingi, and et al. 2021. "Exploiting the Role of Hypoxia-Inducible Factor 1 and Pseudohypoxia in the Myelodysplastic Syndrome Pathophysiology" International Journal of Molecular Sciences 22, no. 8: 4099. https://doi.org/10.3390/ijms22084099
APA StyleStergiou, I. E., Kambas, K., Poulaki, A., Giannouli, S., Katsila, T., Dimitrakopoulou, A., Vidali, V., Mouchtouris, V., Kloukina, I., Xingi, E., Pagakis, S. N., Probert, L., Patrinos, G. P., Ritis, K., Tzioufas, A. G., & Voulgarelis, M. (2021). Exploiting the Role of Hypoxia-Inducible Factor 1 and Pseudohypoxia in the Myelodysplastic Syndrome Pathophysiology. International Journal of Molecular Sciences, 22(8), 4099. https://doi.org/10.3390/ijms22084099