
The Impact of Modern Technologies on Molecular Diagnostic Success Rates, with a Focus on Inherited Retinal Dystrophy and Hearing Loss

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Identification of Genes Associated with Hearing Loss and Retinal Dystrophy
2.1. Linkage Analysis
2.2. Homozygosity Mapping
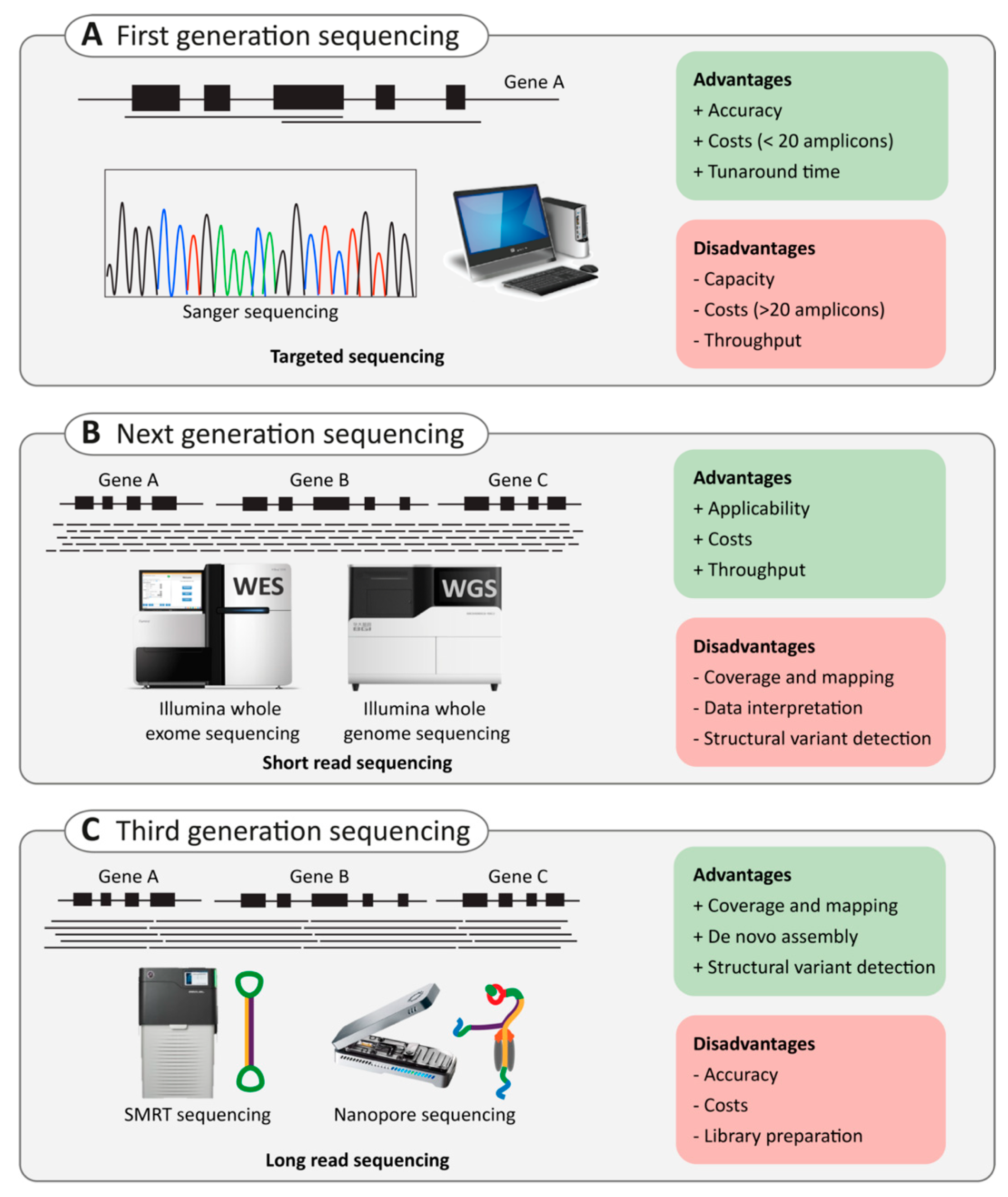
2.3. Next-Generation Sequencing
2.3.1. Targeted Capture Sequencing
2.3.2. Whole-Exome Sequencing Versus Whole-Genome Sequencing
2.4. Third-Generation Sequencing
2.4.1. Single-Molecule Real-Time (SMRT) Sequencing
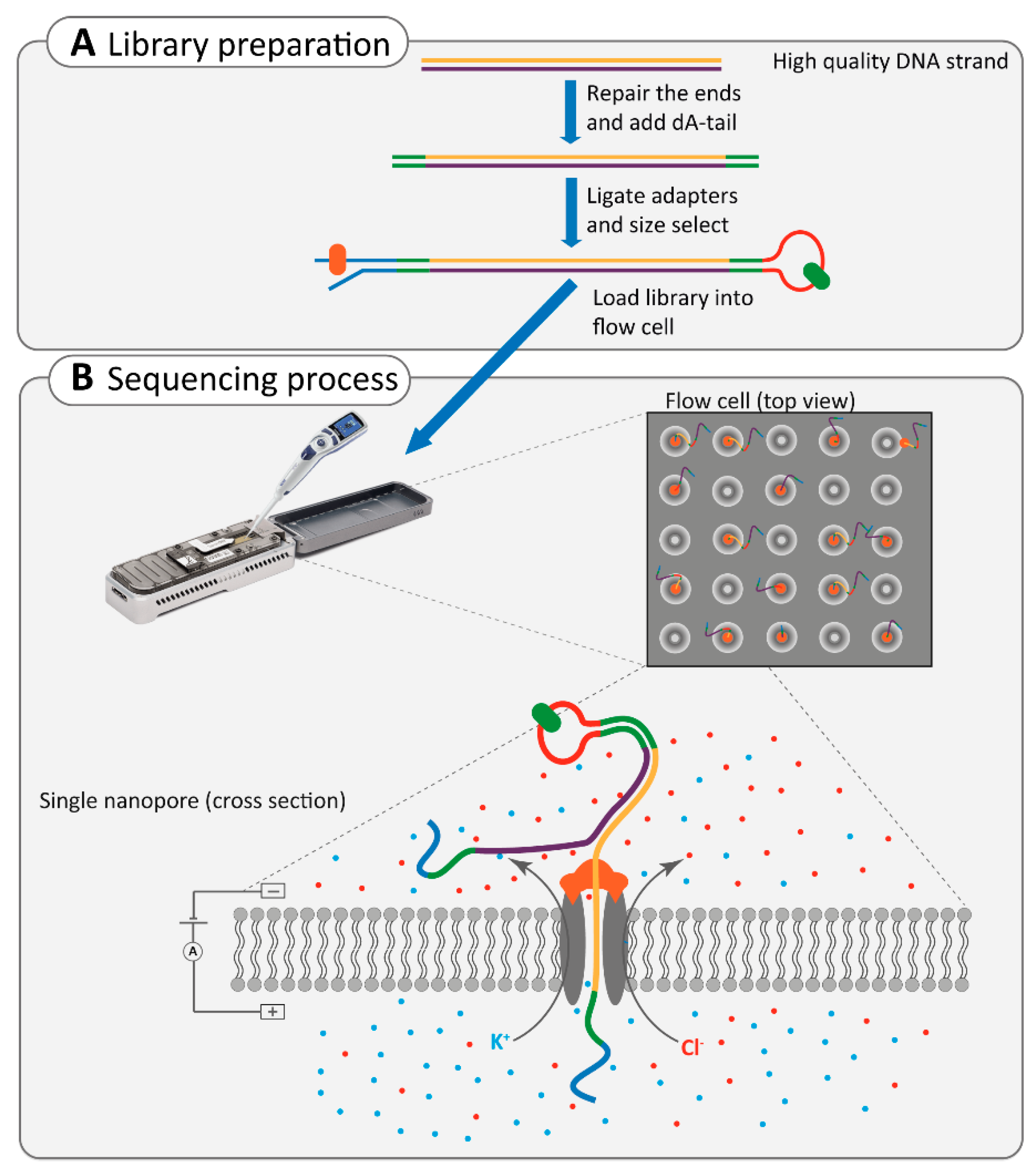
2.4.2. Nanopore Sequencing
2.5. Application of Third-Generation Sequencing in Inherited HL and RD
3. Variant Interpretation
3.1. Literature and Database Use
3.2. Computational and Predictive Data
3.2.1. Null Variants
3.2.2. Missense, Synonymous, Indel, and Intronic Variants
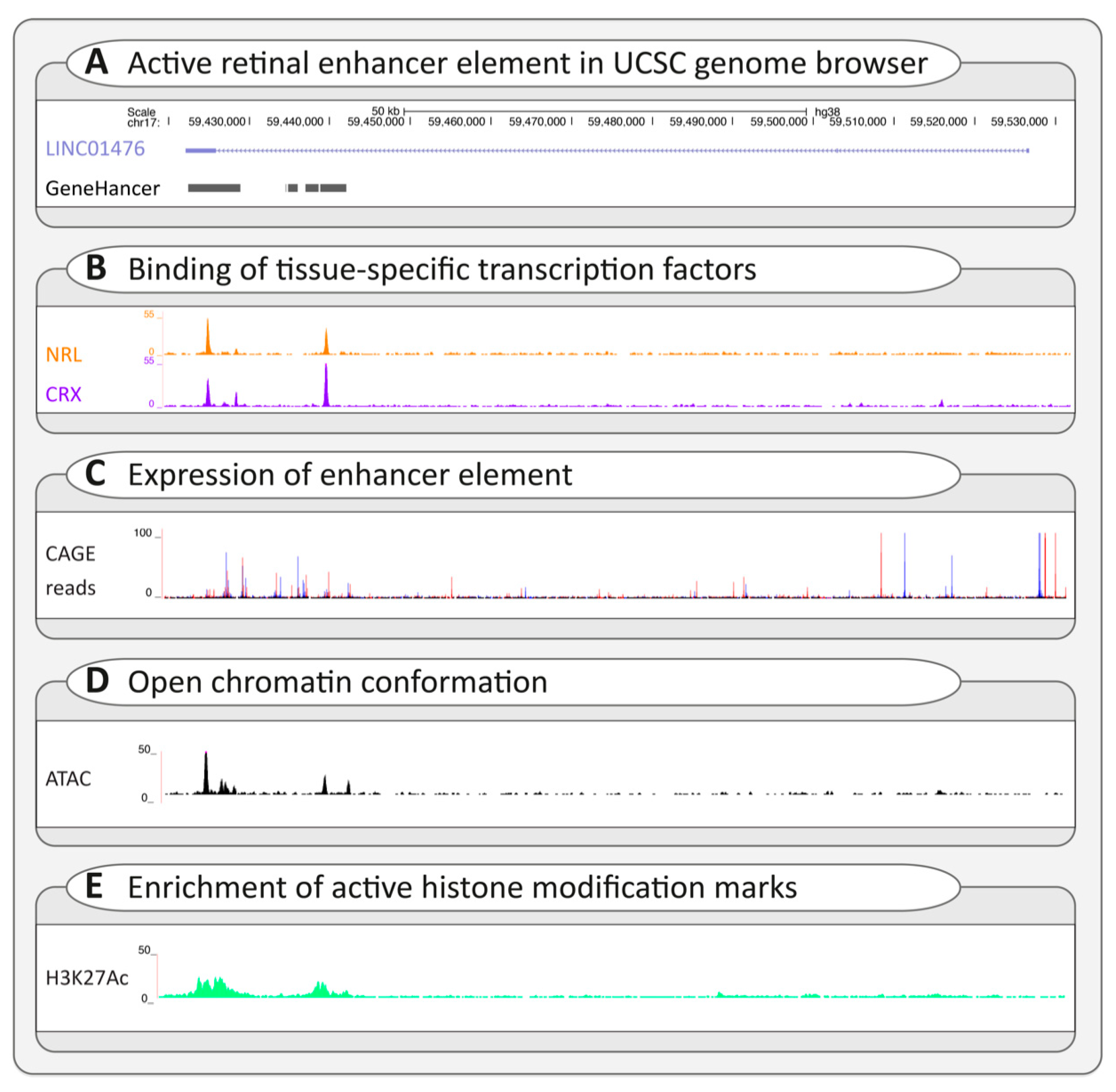
3.2.3. Regulatory Variants

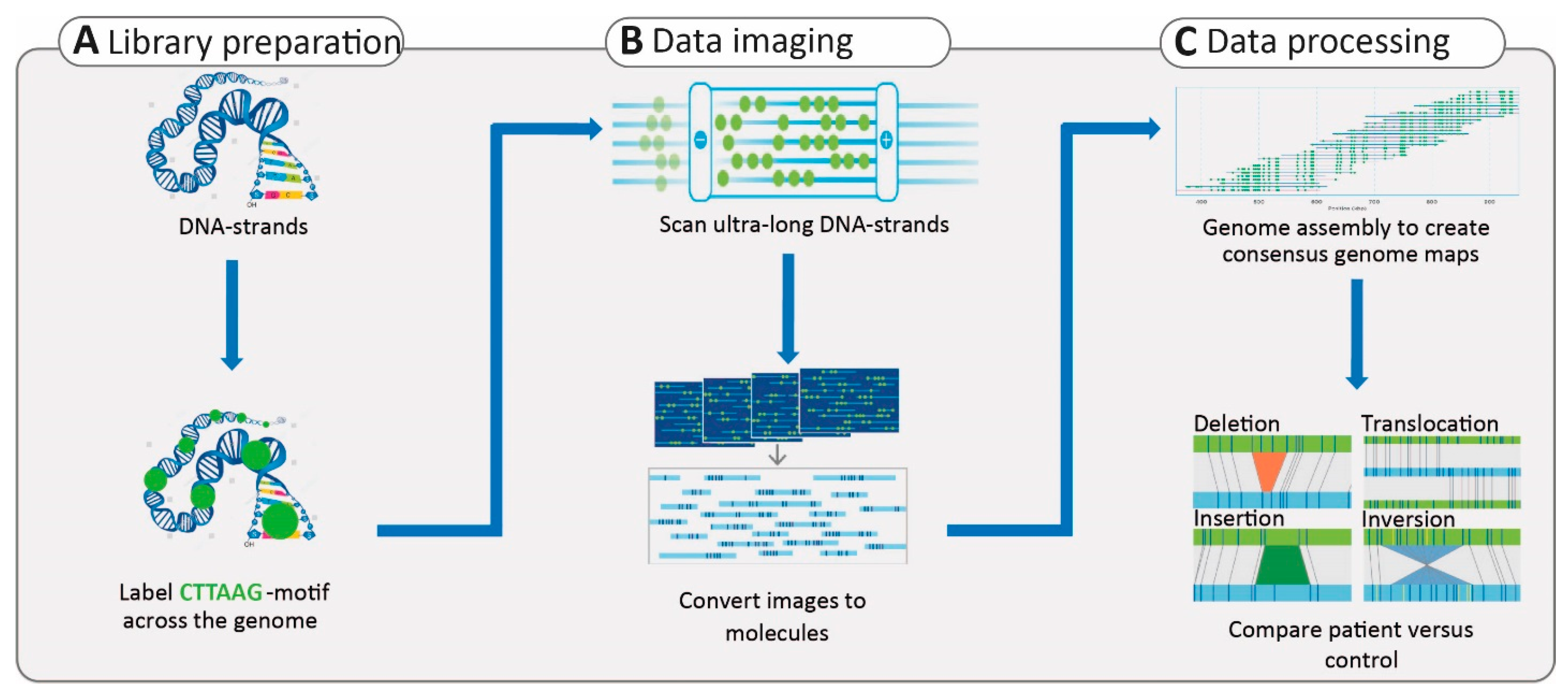
3.2.4. Structural Variants
3.3. Segregation Analysis
3.4. Functional Evaluation of Variants
4. Future Developments
4.1. Development of New Technologies
4.2. Multiomic Approaches
5. Conclusions and Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van Camp, G.; Smith, R. Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org/ (accessed on 4 March 2021).
- RetNet. Available online: https://sph.uth.edu/RetNet/ (accessed on 4 March 2021).
- World Health Organisation. Deafness and Hearing Loss: Key Facts 2019 Update. Available online: https://www.who.int/news-room/fact-sheets/detail/deafness-and-hearing-loss (accessed on 14 December 2020).
- Mener, D.J.; Betz, J.; Genther, D.J.; Chen, D.; Lin, F.R. Hearing loss and depression in older adults. J. Am. Geriatr. Soc. 2013, 61, 1627–1629. [Google Scholar] [CrossRef]
- Sung, Y.K.; Li, L.; Blake, C.; Betz, J.; Lin, F.R. Association of Hearing Loss and Loneliness in Older Adults. J. Aging Health 2016, 28, 979–994. [Google Scholar] [CrossRef]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J.H. Hereditary Hearing Loss and Deafness Overview. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Zazo Seco, C.; Wesdorp, M.; Feenstra, I.; Pfundt, R.; Hehir-Kwa, J.Y.; Lelieveld, S.H.; Castelein, S.; Gilissen, C.; de Wijs, I.J.; Admiraal, R.J.; et al. The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in The Netherlands. Eur. J. Hum. Genet. 2017, 25, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Haer-Wigman, L.; van Zelst-Stams, W.A.; Pfundt, R.; van den Born, L.I.; Klaver, C.C.; Verheij, J.B.; Hoyng, C.B.; Breuning, M.H.; Boon, C.J.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Wesdorp, M.; Murillo-Cuesta, S.; Peters, T.; Celaya, A.M.; Oonk, A.; Schraders, M.; Oostrik, J.; Gomez-Rosas, E.; Beynon, A.J.; Hartel, B.P.; et al. MPZL2, Encoding the Epithelial Junctional Protein Myelin Protein Zero-like 2, Is Essential for Hearing in Man and Mouse. Am. J. Hum. Genet. 2018, 103, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Pierrache, L.H.M.; Kimchi, A.; Ratnapriya, R.; Roberts, L.; Astuti, G.D.N.; Obolensky, A.; Beryozkin, A.; Tjon-Fo-Sang, M.J.H.; Schuil, J.; Klaver, C.C.W.; et al. Whole-Exome Sequencing Identifies Biallelic IDH3A Variants as a Cause of Retinitis Pigmentosa Accompanied by Pseudocoloboma. Ophthalmology 2017, 124, 992–1003. [Google Scholar] [CrossRef]
- McClellan, J.; King, M.C. Genetic heterogeneity in human disease. Cell 2010, 141, 210–217. [Google Scholar] [CrossRef]
- Dawn Teare, M.; Barrett, J.H. Genetic linkage studies. Lancet 2005, 366, 1036–1044. [Google Scholar] [CrossRef]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef]
- Cremers, F.P.M.; van de Pol, D.J.; van Kerkhoff, L.P.; Wieringa, B.; Ropers, H.H. Cloning of a gene that is rearranged in patients with choroideraemia. Nature 1990, 347, 674–677. [Google Scholar] [CrossRef]
- Dryja, T.P.; McGee, T.L.; Reichel, E.; Hahn, L.B.; Cowley, G.S.; Yandell, D.W.; Sandberg, M.A.; Berson, E.L. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 1990, 343, 364–366. [Google Scholar] [CrossRef]
- de Kok, Y.J.; van der Maarel, S.M.; Bitner-Glindzicz, M.; Huber, I.; Monaco, A.P.; Malcolm, S.; Pembrey, M.E.; Ropers, H.H.; Cremers, F.P.M. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science 1995, 267, 685–688. [Google Scholar] [CrossRef]
- Broadgate, S.; Yu, J.; Downes, S.M.; Halford, S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Prog. Retin. Eye Res. 2017, 59, 53–96. [Google Scholar] [CrossRef] [PubMed]
- Morton, N.E. Sequential tests for the detection of linkage. Am. J. Hum. Genet. 1955, 7, 277–318. [Google Scholar]
- Kruglyak, L.; Daly, M.J.; Reeve-Daly, M.P.; Lander, E.S. Parametric and nonparametric linkage analysis: A unified multipoint approach. Am. J. Hum. Genet. 1996, 58, 1347–1363. [Google Scholar]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Vona, B.; Nanda, I.; Hofrichter, M.A.; Shehata-Dieler, W.; Haaf, T. Non-syndromic hearing loss gene identification: A brief history and glimpse into the future. Mol. Cell Probes 2015, 29, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Seelow, D.; Schuelke, M.; Hildebrandt, F.; Nürnberg, P. HomozygosityMapper--an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009, 37, W593–W599. [Google Scholar] [CrossRef] [PubMed]
- Woods, C.G.; Cox, J.; Springell, K.; Hampshire, D.J.; Mohamed, M.D.; McKibbin, M.; Stern, R.; Raymond, F.L.; Sandford, R.; Malik Sharif, S.; et al. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am. J. Hum. Genet 2006, 78, 889–896. [Google Scholar] [CrossRef]
- Collin, R.W.; van den Born, L.I.; Klevering, B.J.; de Castro-Miró, M.; Littink, K.W.; Arimadyo, K.; Azam, M.; Yazar, V.; Zonneveld, M.N.; Paun, C.C.; et al. High-resolution homozygosity mapping is a powerful tool to detect novel mutations causative of autosomal recessive RP in the Dutch population. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2227–2239. [Google Scholar] [CrossRef]
- Schraders, M.; Lee, K.; Oostrik, J.; Huygen, P.L.; Ali, G.; Hoefsloot, L.H.; Veltman, J.A.; Cremers, F.P.M.; Basit, S.; Ansar, M.; et al. Homozygosity mapping reveals mutations of GRXCR1 as a cause of autosomal-recessive nonsyndromic hearing impairment. Am. J. Hum. Genet. 2010, 86, 138–147. [Google Scholar] [CrossRef]
- Collin, R.W.J.; Littink, K.W.; Klevering, B.J.; van den Born, L.I.; Koenekoop, R.K.; Zonneveld, M.N.; Blokland, E.A.W.; Strom, T.M.; Hoyng, C.B.; den Hollander, A.I.; et al. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am. J. Hum. Genet. 2008, 83, 594–603. [Google Scholar] [CrossRef]
- Messchaert, M.; Haer-Wigman, L.; Khan, M.I.; Cremers, F.P.M.; Collin, R.W.J. EYS mutation update: In silico assessment of 271 reported and 26 novel variants in patients with retinitis pigmentosa. Hum. Mutat. 2018, 39, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Thiadens, A.A.; den Hollander, A.I.; Roosing, S.; Nabuurs, S.B.; Zekveld-Vroon, R.C.; Collin, R.W.; De Baere, E.; Koenekoop, R.K.; van Schooneveld, M.J.; Strom, T.M.; et al. Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am. J. Hum. Genet. 2009, 85, 240–247. [Google Scholar] [CrossRef]
- Schraders, M.; Ruiz-Palmero, L.; Kalay, E.; Oostrik, J.; del Castillo, F.J.; Sezgin, O.; Beynon, A.J.; Strom, T.M.; Pennings, R.J.; Zazo Seco, C.; et al. Mutations of the gene encoding otogelin are a cause of autosomal-recessive nonsyndromic moderate hearing impairment. Am. J. Hum. Genet. 2012, 91, 883–889. [Google Scholar] [CrossRef]
- Friedman, T.B.; Liang, Y.; Weber, J.L.; Hinnant, J.T.; Barber, T.D.; Winata, S.; Arhya, I.N.; Asher, J.H., Jr. A gene for congenital, recessive deafness DFNB3 maps to the pericentromeric region of chromosome 17. Nat. Genet. 1995, 9, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Waterston, R.H.; Lander, E.S.; Sulston, J.E. On the sequencing of the human genome. Proc. Natl. Acad. Sci. USA 2002, 99, 3712–3716. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J. Human genome project. Ann. Chir. Gynaecol. 2001, 90, 3. [Google Scholar] [PubMed]
- Venter, J.C.; Smith, H.O.; Hood, L. A new strategy for genome sequencing. Nature 1996, 381, 364–366. [Google Scholar] [CrossRef]
- Schloss, J.A. How to get genomes at one ten-thousandth the cost. Nat. Biotechnol. 2008, 26, 1113–1115. [Google Scholar] [CrossRef]
- Vona, B.; Müller, M.; Dofek, S.; Holderried, M.; Löwenheim, H.; Tropitzsch, A. A Big Data Perspective on the Genomics of Hearing Loss. Laryngorhinootologie 2019, 98, S32–S81. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.E.; Myers, R.M. Advancements in Next-Generation Sequencing. Annu. Rev. Genom. Hum. Genet. 2016, 17, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Fung, S.; Hunkapiller, M.W.; Hunkapiller, T.J.; Hood, L.E. The synthesis of oligonucleotides containing an aliphatic amino group at the 5' terminus: Synthesis of fluorescent DNA primers for use in DNA sequence analysis. Nucleic Acids Res. 1985, 13, 2399–2412. [Google Scholar] [CrossRef]
- Smith, L.M.; Sanders, J.Z.; Kaiser, R.J.; Hughes, P.; Dodd, C.; Connell, C.R.; Heiner, C.; Kent, S.B.; Hood, L.E. Fluorescence detection in automated DNA sequence analysis. Nature 1986, 321, 674–679. [Google Scholar] [CrossRef]
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific enzymatic amplification of DNA in vitro: The polymerase chain reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51 Pt 1, 263–273. [Google Scholar] [CrossRef]
- Buermans, H.P.; den Dunnen, J.T. Next generation sequencing technology: Advances and applications. Biochim. Biophys. Acta 2014, 1842, 1932–1941. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Park, G.; Gim, J.; Kim, A.R.; Kim, B.J.; Kim, H.S.; Park, J.H.; Park, T.; Oh, S.H.; Han, K.H.; et al. Diagnostic application of targeted resequencing for familial nonsyndromic hearing loss. PLoS ONE 2013, 8, e68692. [Google Scholar] [CrossRef] [PubMed]
- Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations. Genes (Basel) 2017, 8, 304. [Google Scholar] [CrossRef]
- Khan, M.; Cornelis, S.S.; Pozo-Valero, M.D.; Whelan, L.; Runhart, E.H.; Mishra, K.; Bults, F.; AlSwaiti, Y.; AlTalbishi, A.; De Baere, E.; et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genet. Med. 2020, 22, 1235–1246. [Google Scholar] [CrossRef]
- Lin, X.; Tang, W.; Ahmad, S.; Lu, J.; Colby, C.C.; Zhu, J.; Yu, Q. Applications of targeted gene capture and next-generation sequencing technologies in studies of human deafness and other genetic disabilities. Hear. Res. 2012, 288, 67–76. [Google Scholar] [CrossRef]
- Choi, M.; Scholl, U.I.; Ji, W.; Liu, T.; Tikhonova, I.R.; Zumbo, P.; Nayir, A.; Bakkaloğlu, A.; Ozen, S.; Sanjad, S.; et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc. Natl. Acad. Sci. USA 2009, 106, 19096–19101. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Ball, E.V.; Howells, K.; Phillips, A.D.; Mort, M.; Cooper, D.N. The Human Gene Mutation Database: Providing a comprehensive central mutation database for molecular diagnostics and personalized genomics. Hum. Genom. 2009, 4, 69–72. [Google Scholar] [CrossRef]
- Stranneheim, H.; Wedell, A. Exome and genome sequencing: A revolution for the discovery and diagnosis of monogenic disorders. J. Intern. Med. 2016, 279, 3–15. [Google Scholar] [CrossRef]
- Tucker, T.; Marra, M.; Friedman, J.M. Massively parallel sequencing: The next big thing in genetic medicine. Am. J. Hum. Genet. 2009, 85, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Guan, L.; Shen, T.; Zhang, J.; Xiao, X.; Jiang, H.; Li, S.; Yang, J.; Jia, X.; Yin, Y.; et al. Mutations of 60 known causative genes in 157 families with retinitis pigmentosa based on exome sequencing. Hum. Genet. 2014, 133, 1255–1271. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Bahr, A.; Bähr, L.; Fleischhauer, J.; Zinkernagel, M.S.; Winkler, N.; Barthelmes, D.; Berger, L.; Gerth-Kahlert, C.; Neidhardt, J.; et al. Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci. Rep. 2016, 6, 28755. [Google Scholar] [CrossRef] [PubMed]
- Abu-Safieh, L.; Alrashed, M.; Anazi, S.; Alkuraya, H.; Khan, A.O.; Al-Owain, M.; Al-Zahrani, J.; Al-Abdi, L.; Hashem, M.; Al-Tarimi, S.; et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013, 23, 236–247. [Google Scholar] [CrossRef]
- Sang, S.; Ling, J.; Liu, X.; Mei, L.; Cai, X.; Li, T.; Li, W.; Li, M.; Wen, J.; Liu, X.; et al. Proband Whole-Exome Sequencing Identified Genes Responsible for Autosomal Recessive Non-Syndromic Hearing Loss in 33 Chinese Nuclear Families. Front. Genet. 2019, 10, 639. [Google Scholar] [CrossRef]
- Morton, C.C.; Nance, W.E. Newborn hearing screening--a silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; Williams, S.G.; Sergouniotis, P.I.; O'Sullivan, J.; Lamb, J.A.; Perveen, R.; Hall, G.; Newman, W.G.; et al. Whole Genome Sequencing Increases Molecular Diagnostic Yield Compared with Current Diagnostic Testing for Inherited Retinal Disease. Ophthalmology 2016, 123, 1143–1150. [Google Scholar] [CrossRef]
- Barbitoff, Y.A.; Polev, D.E.; Glotov, A.S.; Serebryakova, E.A.; Shcherbakova, I.V.; Kiselev, A.M.; Kostareva, A.A.; Glotov, O.S.; Predeus, A.V. Systematic dissection of biases in whole-exome and whole-genome sequencing reveals major determinants of coding sequence coverage. Sci. Rep. 2020, 10, 2057. [Google Scholar] [CrossRef] [PubMed]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef] [PubMed]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Wetterstrand, K. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). Available online: www.genome.gov/sequencingcostsdata (accessed on 14 December 2020).
- Méjécase, C.; Malka, S.; Guan, Z.; Slater, A.; Arno, G.; Moosajee, M. Practical guide to genetic screening for inherited eye diseases. Ther. Adv. Ophthalmol. 2020, 12, 2515841420954592. [Google Scholar] [CrossRef]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Magi, A.; Semeraro, R.; Mingrino, A.; Giusti, B.; D'Aurizio, R. Nanopore sequencing data analysis: State of the art, applications and challenges. Brief. Bioinform. 2018, 19, 1256–1272. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Schadt, E.E.; Turner, S.; Kasarskis, A. A window into third-generation sequencing. Hum. Mol. Genet. 2010, 19, R227–R240. [Google Scholar] [CrossRef] [PubMed]
- Travers, K.J.; Chin, C.S.; Rank, D.R.; Eid, J.S.; Turner, S.W. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res. 2010, 38, e159. [Google Scholar] [CrossRef]
- Ardui, S.; Ameur, A.; Vermeesch, J.R.; Hestand, M.S. Single molecule real-time (SMRT) sequencing comes of age: Applications and utilities for medical diagnostics. Nucleic Acids Res. 2018, 46, 2159–2168. [Google Scholar] [CrossRef]
- Xu, M.; Fujita, D.; Hanagata, N. Perspectives and challenges of emerging single-molecule DNA sequencing technologies. Small 2009, 5, 2638–2649. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M.; et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2015, 517, 608–611. [Google Scholar] [CrossRef]
- Seo, J.S.; Rhie, A.; Kim, J.; Lee, S.; Sohn, M.H.; Kim, C.U.; Hastie, A.; Cao, H.; Yun, J.Y.; Kim, J.; et al. De novo assembly and phasing of a Korean human genome. Nature 2016, 538, 243–247. [Google Scholar] [CrossRef]
- Levene, M.J.; Korlach, J.; Turner, S.W.; Foquet, M.; Craighead, H.G.; Webb, W.W. Zero-mode waveguides for single-molecule analysis at high concentrations. Science 2003, 299, 682–686. [Google Scholar] [CrossRef]
- Morey, M.; Fernández-Marmiesse, A.; Castiñeiras, D.; Fraga, J.M.; Couce, M.L.; Cocho, J.A. A glimpse into past, present, and future DNA sequencing. Mol. Genet. Metab. 2013, 110, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, D.; Heron, A.J.; Mikhailova, E.; Maglia, G.; Bayley, H. Single-nucleotide discrimination in immobilized DNA oligonucleotides with a biological nanopore. Proc. Natl. Acad. Sci. USA 2009, 106, 7702–7707. [Google Scholar] [CrossRef]
- Kono, N.; Arakawa, K. Nanopore sequencing: Review of potential applications in functional genomics. Dev. Growth Differ. 2019, 61, 316–326. [Google Scholar] [CrossRef]
- Branton, D.; Deamer, D.W.; Marziali, A.; Bayley, H.; Benner, S.A.; Butler, T.; Di Ventra, M.; Garaj, S.; Hibbs, A.; Huang, X.; et al. The potential and challenges of nanopore sequencing. Nat. Biotechnol. 2008, 26, 1146–1153. [Google Scholar] [CrossRef]
- Ip, C.L.C.; Loose, M.; Tyson, J.R.; de Cesare, M.; Brown, B.L.; Jain, M.; Leggett, R.M.; Eccles, D.A.; Zalunin, V.; Urban, J.M.; et al. MinION Analysis and Reference Consortium: Phase 1 data release and analysis. F1000Resrearch 2015, 4, 1075. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef]
- Mardis, E.R. DNA sequencing technologies: 2006–2016. Nat. Protoc. 2017, 12, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Ebbert, M.T.W.; Jensen, T.D.; Jansen-West, K.; Sens, J.P.; Reddy, J.S.; Ridge, P.G.; Kauwe, J.S.K.; Belzil, V.; Pregent, L.; Carrasquillo, M.M.; et al. Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. Genome Biol. 2019, 20, 97. [Google Scholar] [CrossRef] [PubMed]
- Reiner, J.; Pisani, L.; Qiao, W.; Singh, R.; Yang, Y.; Shi, L.; Khan, W.A.; Sebra, R.; Cohen, N.; Babu, A.; et al. Cytogenomic identification and long-read single molecule real-time (SMRT) sequencing of a Bardet-Biedl Syndrome 9 (BBS9) deletion. NPJ Genom. Med. 2018, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Bedoni, N.; Quinodoz, M.; Pinelli, M.; Cappuccio, G.; Torella, A.; Nigro, V.; Testa, F.; Simonelli, F.; Corton, M.; Lualdi, S.; et al. An Alu-mediated duplication in NMNAT1, involved in NAD biosynthesis, causes a novel syndrome, SHILCA, affecting multiple tissues and organs. Hum. Mol. Genet. 2020, 29, 2250–2260. [Google Scholar] [CrossRef]
- Laurent, S.; Gehrig, C.; Nouspikel, T.; Amr, S.S.; Oza, A.; Murphy, E.; Vannier, A.; Béna, F.S.; Carminho-Rodrigues, M.T.; Blouin, J.L.; et al. Molecular characterization of pathogenic OTOA gene conversions in hearing loss patients. Hum. Mutat. 2021. [Google Scholar] [CrossRef]
- Hafford-Tear, N.J.; Tsai, Y.C.; Sadan, A.N.; Sanchez-Pintado, B.; Zarouchlioti, C.; Maher, G.J.; Liskova, P.; Tuft, S.J.; Hardcastle, A.J.; Clark, T.A.; et al. CRISPR/Cas9-targeted enrichment and long-read sequencing of the Fuchs endothelial corneal dystrophy-associated TCF4 triplet repeat. Genet. Med. 2019, 21, 2092–2102. [Google Scholar] [CrossRef]
- Edwards, H.S.; Krishnakumar, R.; Sinha, A.; Bird, S.W.; Patel, K.D.; Bartsch, M.S. Real-Time Selective Sequencing with RUBRIC: Read Until with Basecall and Reference-Informed Criteria. Sci. Rep. 2019, 9, 11475. [Google Scholar] [CrossRef] [PubMed]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Eichler, E.E. Genetic Variation, Comparative Genomics, and the Diagnosis of Disease. N. Engl. J. Med. 2019, 381, 64–74. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Tucci, S.; Akey, J.M. The long walk to African genomics. Genome Biol. 2019, 20, 130. [Google Scholar] [CrossRef]
- Choudhury, A.; Aron, S.; Botigué, L.R.; Sengupta, D.; Botha, G.; Bensellak, T.; Wells, G.; Kumuthini, J.; Shriner, D.; Fakim, Y.J.; et al. High-depth African genomes inform human migration and health. Nature 2020, 586, 741–748. [Google Scholar] [CrossRef]
- Boomsma, D.I.; Wijmenga, C.; Slagboom, E.P.; Swertz, M.A.; Karssen, L.C.; Abdellaoui, A.; Ye, K.; Guryev, V.; Vermaat, M.; van Dijk, F.; et al. The Genome of the Netherlands: Design, and project goals. Eur. J. Hum. Genet. 2014, 22, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.; Min, J.L.; Huang, J.; Crooks, L.; Memari, Y.; McCarthy, S.; Perry, J.R.; Xu, C.; Futema, M.; Lawson, D.; et al. The UK10K project identifies rare variants in health and disease. Nature 2015, 526, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Erikson, G.A.; Bodian, D.L.; Rueda, M.; Molparia, B.; Scott, E.R.; Scott-Van Zeeland, A.A.; Topol, S.E.; Wineinger, N.E.; Niederhuber, J.E.; Topol, E.J.; et al. Whole-Genome Sequencing of a Healthy Aging Cohort. Cell 2016, 165, 1002–1011. [Google Scholar] [CrossRef]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen--the Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.; Taschner, P.E.; Schaafsma, G.C.; Celli, J.; Laros, J.F.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef]
- Azaiez, H.; Booth, K.T.; Ephraim, S.S.; Crone, B.; Black-Ziegelbein, E.A.; Marini, R.J.; Shearer, A.E.; Sloan-Heggen, C.M.; Kolbe, D.; Casavant, T.; et al. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 2018, 103, 484–497. [Google Scholar] [CrossRef]
- Cremers, F.P.M.; den Dunnen, J.T.; Ajmal, M.; Hussain, A.; Preising, M.N.; Daiger, S.P.; Qamar, R. Comprehensive registration of DNA sequence variants associated with inherited retinal diseases in Leiden Open Variation Databases. Hum. Mutat. 2014, 35, 147–148. [Google Scholar] [CrossRef][Green Version]
- Cornelis, S.S.; Bax, N.M.; Zernant, J.; Allikmets, R.; Fritsche, L.G.; den Dunnen, J.T.; Ajmal, M.; Hoyng, C.B.; Cremers, F.P. In Silico Functional Meta-Analysis of 5,962 ABCA4 Variants in 3,928 Retinal Dystrophy Cases. Hum. Mutat. 2017, 38, 400–408. [Google Scholar] [CrossRef]
- Baux, D.; Blanchet, C.; Hamel, C.; Meunier, I.; Larrieu, L.; Faugère, V.; Vaché, C.; Castorina, P.; Puech, B.; Bonneau, D.; et al. Enrichment of LOVD-USHbases with 152 USH2A genotypes defines an extensive mutational spectrum and highlights missense hotspots. Hum. Mutat. 2014, 35, 1179–1186. [Google Scholar] [CrossRef]
- Astuti, G.D.; Bertelsen, M.; Preising, M.N.; Ajmal, M.; Lorenz, B.; Faradz, S.M.; Qamar, R.; Collin, R.W.; Rosenberg, T.; Cremers, F.P. Comprehensive genotyping reveals RPE65 as the most frequently mutated gene in Leber congenital amaurosis in Denmark. Eur. J. Hum. Genet. 2016, 24, 1071–1079. [Google Scholar] [CrossRef]
- Mackay, D.S.; Borman, A.D.; Sui, R.; van den Born, L.I.; Berson, E.L.; Ocaka, L.A.; Davidson, A.E.; Heckenlively, J.R.; Branham, K.; Ren, H.; et al. Screening of a large cohort of leber congenital amaurosis and retinitis pigmentosa patients identifies novel LCA5 mutations and new genotype-phenotype correlations. Hum. Mutat. 2013, 34, 1537–1546. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Thomas, H.B.; Rowlands, C.; Arno, G.; Beaman, G.; Gomes-Silva, B.; Campbell, C.; Gossan, N.; Hardcastle, C.; Webb, K.; et al. Functional and in-silico interrogation of rare genomic variants impacting RNA splicing for the diagnosis of genomic disorders. BioRxiv 2019, 781088. [Google Scholar] [CrossRef]
- Hanany, M.; Sharon, D. Allele frequency analysis of variants reported to cause autosomal dominant inherited retinal diseases question the involvement of 19% of genes and 10% of reported pathogenic variants. J. Med. Genet. 2019, 56, 536–542. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef]
- McHugh, R.K.; Friedman, R.A. Genetics of hearing loss: Allelism and modifier genes produce a phenotypic continuum. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2006, 288, 370–381. [Google Scholar] [CrossRef]
- Cremers, F.P.M.; Lee, W.; Collin, R.W.J.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef] [PubMed]
- Runhart, E.H.; Khan, M.; Cornelis, S.S.; Roosing, S.; Del Pozo-Valero, M.; Lamey, T.M.; Liskova, P.; Roberts, L.; Stöhr, H.; Klaver, C.C.W.; et al. Association of Sex With Frequent and Mild ABCA4 Alleles in Stargardt Disease. JAMA Ophthalmol. 2020, 138, 1035–1042. [Google Scholar] [CrossRef] [PubMed]
- Runhart, E.H.; Valkenburg, D.; Cornelis, S.S.; Khan, M.; Sangermano, R.; Albert, S.; Bax, N.M.; Astuti, G.D.N.; Gilissen, C.; Pott, J.R.; et al. Late-Onset Stargardt Disease Due to Mild, Deep-Intronic ABCA4 Alleles. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4249–4256. [Google Scholar] [CrossRef]
- Astuto, L.M.; Bork, J.M.; Weston, M.D.; Askew, J.W.; Fields, R.R.; Orten, D.J.; Ohliger, S.J.; Riazuddin, S.; Morell, R.J.; Khan, S.; et al. CDH23 Mutation and Phenotype Heterogeneity: A Profile of 107 Diverse Families with Usher Syndrome and Nonsyndromic Deafness. Am. J. Hum. Genet. 2002, 71, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Becirovic, E.; Ebermann, I.; Nagy, D.; Zrenner, E.; Seeliger, M.W.; Bolz, H.J. Usher syndrome type 1 due to missense mutations on both CDH23 alleles: Investigation of mRNA splicing. Hum. Mutat. 2008, 29, 452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cheng, J.; Zhou, Q.; Khan, M.A.; Fu, J.; Duan, C.; Sun, S.; Lv, H.; Fu, J. Targeted Next-Generation Sequencing Identified Novel Compound Heterozygous Variants in the CDH23 Gene Causing Usher Syndrome Type ID in a Chinese Patient. Front. Genet. 2020, 11, 422. [Google Scholar] [CrossRef] [PubMed]
- Molina-Ramírez, L.P.; Lenassi, E.; Ellingford, J.M.; Sergouniotis, P.I.; Ramsden, S.C.; Bruce, I.A.; Black, G.C.M. Establishing Genotype-phenotype Correlation in USH2A-related Disorders to Personalize Audiological Surveillance and Rehabilitation. Otol. Neurotol. 2020, 41, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Rivolta, C.; Sweklo, E.A.; Berson, E.L.; Dryja, T.P. Missense mutation in the USH2A gene: Association with recessive retinitis pigmentosa without hearing loss. Am. J. Hum. Genet. 2000, 66, 1975–1978. [Google Scholar] [CrossRef]
- Tatour, Y.; Ben-Yosef, T. Syndromic Inherited Retinal Diseases: Genetic, Clinical and Diagnostic Aspects. Diagnostics (Basel) 2020, 10, 779. [Google Scholar] [CrossRef]
- Gettelfinger, J.D.; Dahl, J.P. Syndromic Hearing Loss: A Brief Review of Common Presentations and Genetics. J. Pediatr. Genet. 2018, 7, 1–8. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Shen, J.; Scheffer, D.I.; Kwan, K.Y.; Corey, D.P. SHIELD: An integrative gene expression database for inner ear research. Database (Oxford) 2015, 2015, bav071. [Google Scholar] [CrossRef]
- gEAR Portal. Available online: https://umgear.org/ (accessed on 14 December 2020).
- Ratnapriya, R.; Sosina, O.A.; Starostik, M.R.; Kwicklis, M.; Kapphahn, R.J.; Fritsche, L.G.; Walton, A.; Arvanitis, M.; Gieser, L.; Pietraszkiewicz, A.; et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat. Genet. 2019, 51, 606–610. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Sobreira, N.; Schiettecatte, F.; Valle, D.; Hamosh, A. GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015, 36, 928–930. [Google Scholar] [CrossRef]
- The European Retinal Disease Consortium (ERDC). Available online: www.ERDC.info (accessed on 5 February 2021).
- Di Stazio, M.; Morgan, A.; Brumat, M.; Bassani, S.; Dell'Orco, D.; Marino, V.; Garagnani, P.; Giuliani, C.; Gasparini, P.; Girotto, G. New age-related hearing loss candidate genes in humans: An ongoing challenge. Gene 2020, 742, 144561. [Google Scholar] [CrossRef]
- Astuti, G.D.N.; van den Born, L.I.; Khan, M.I.; Hamel, C.P.; Bocquet, B.; Manes, G.; Quinodoz, M.; Ali, M.; Toomes, C.; McKibbin, M.; et al. Identification of Inherited Retinal Disease-Associated Genetic Variants in 11 Candidate Genes. Genes (Basel) 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alföldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Littink, K.W.; Pott, J.W.; Collin, R.W.; Kroes, H.Y.; Verheij, J.B.; Blokland, E.A.; de Castro Miró, M.; Hoyng, C.B.; Klaver, C.C.; Koenekoop, R.K.; et al. A novel nonsense mutation in CEP290 induces exon skipping and leads to a relatively mild retinal phenotype. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3646–3652. [Google Scholar] [CrossRef]
- Roosing, S.; Cremers, F.P.M.; Riemslag, F.C.C.; Zonneveld-Vrieling, M.N.; Talsma, H.E.; Klessens-Godfroy, F.J.M.; den Hollander, A.I.; van den Born, L.I. A Rare Form of Retinal Dystrophy Caused by Hypomorphic Nonsense Mutations in CEP290. Genes (Basel) 2017, 8, 208. [Google Scholar] [CrossRef] [PubMed]
- DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Bowser, M.J.; Hynes, E.; Grant, A.R.; Siegert, R.K.; Oza, A.M.; Gonzalez, M.A.; Amr, S.S.; et al. Curating Clinically Relevant Transcripts for the Interpretation of Sequence Variants. J. Mol. Diagn. 2018, 20, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O'Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Grantham, R. Amino Acid Difference Formula to Help Explain Protein Evolution. Science 1974, 185, 862. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361. [Google Scholar] [CrossRef] [PubMed]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Vaser, R.; Adusumalli, S.; Leng, S.N.; Sikic, M.; Ng, P.C. SIFT missense predictions for genomes. Nature Protocols 2015, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Sangermano, R.; Khan, M.; Cornelis, S.S.; Richelle, V.; Albert, S.; Garanto, A.; Elmelik, D.; Qamar, R.; Lugtenberg, D.; van den Born, L.I.; et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2018, 28, 100–110. [Google Scholar] [CrossRef]
- Collin, R.W.; de Heer, A.M.; Oostrik, J.; Pauw, R.J.; Plantinga, R.F.; Huygen, P.L.; Admiraal, R.; de Brouwer, A.P.; Strom, T.M.; Cremers, C.W.; et al. Mid-frequency DFNA8/12 hearing loss caused by a synonymous TECTA mutation that affects an exonic splice enhancer. Eur. J. Hum. Genet. 2008, 16, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Riepe, T.; Khan, M.; Roosing, S.; Cremers, F.P.M.; ‘t Hoen, P. Benchmarking deep learning splice prediction tools using functional splice assays. Authorea Prepr. 2020. [Google Scholar] [CrossRef]
- Rowlands, C.F.; Baralle, D.; Ellingford, J.M. Machine Learning Approaches for the Prioritization of Genomic Variants Impacting Pre-mRNA Splicing. Cells 2019, 8, 513. [Google Scholar] [CrossRef] [PubMed]
- Cherry, T.J.; Yang, M.G.; Harmin, D.A.; Tao, P.; Timms, A.E.; Bauwens, M.; Allikmets, R.; Jones, E.M.; Chen, R.; De Baere, E.; et al. Mapping the cis-regulatory architecture of the human retina reveals noncoding genetic variation in disease. Proc. Natl. Acad. Sci. USA 2020, 117, 9001–9012. [Google Scholar] [CrossRef]
- Van der Lee, R.; Correard, S.; Wasserman, W.W. Deregulated Regulators: Disease-Causing cis Variants in Transcription Factor Genes. Trends Genet. 2020, 36, 523–539. [Google Scholar] [CrossRef]
- Lupiáñez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef] [PubMed]
- De Kok, Y.J.M.; Vossenaar, E.R.; Cremers, C.W.R.J.; Dahl, N.; Laporte, J.; Jia Hu, L.; Lacombe, D.; Fischel-Ghodsian, N.; Friedman, R.A.; Parnes, L.S.; et al. Identification of a Hot Spot for Microdeletions in Patients with X-linked Deafness Type 3 (DFN3) 900 kb Proximal to the DFN3 gene POU3F4. Hum. Mol. Genet. 1996, 5, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, S.; Voesenek, K.; de la Calle-Mustienes, E.; Robert-Moreno, A.; Kokotas, H.; Grigoriadou, M.; Economides, J.; Van Camp, G.; Hilgert, N.; Moreno, F.; et al. Multiple enhancers located in a 1-Mb region upstream of POU3F4 promote expression during inner ear development and may be required for hearing. Hum. Genet. 2010, 128, 411–419. [Google Scholar] [CrossRef][Green Version]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020, 48, D87–D92. [Google Scholar] [CrossRef] [PubMed]
- Perez-Cervantes, C.; Smith, L.A.; Nadadur, R.D.; Hughes, A.E.O.; Wang, S.; Corbo, J.C.; Cepko, C.; Lonfat, N.; Moskowitz, I.P. Enhancer transcription identifies cis-regulatory elements for photoreceptor cell types. Development 2020, 147. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [PubMed]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017, 2017. [Google Scholar] [CrossRef]
- Gao, T.; Qian, J. EnhancerAtlas 2.0: An updated resource with enhancer annotation in 586 tissue/cell types across nine species. Nucleic Acids Res. 2020, 48, D58–D64. [Google Scholar] [CrossRef] [PubMed]
- de Bruijn, S.E.; Fiorentino, A.; Ottaviani, D.; Fanucchi, S.; Melo, U.S.; Corral-Serrano, J.C.; Mulders, T.; Georgiou, M.; Rivolta, C.; Pontikos, N.; et al. Structural Variants Create New Topological-Associated Domains and Ectopic Retinal Enhancer-Gene Contact in Dominant Retinitis Pigmentosa. Am. J. Hum. Genet. 2020, 107, 802–814. [Google Scholar] [CrossRef]
- Lizio, M.; Abugessaisa, I.; Noguchi, S.; Kondo, A.; Hasegawa, A.; Hon, C.C.; de Hoon, M.; Severin, J.; Oki, S.; Hayashizaki, Y.; et al. Update of the FANTOM web resource: Expansion to provide additional transcriptome atlases. Nucleic Acids Res. 2018, 47, D752–D758. [Google Scholar] [CrossRef]
- Brandt, T.; Sack, L.M.; Arjona, D.; Tan, D.; Mei, H.; Cui, H.; Gao, H.; Bean, L.J.H.; Ankala, A.; Del Gaudio, D.; et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet Med. 2020, 22, 336–344. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, M.; Lupiáñez, D.G.; Mundlos, S. Structural variation in the 3D genome. Nat. Rev. Genet. 2018, 19, 453–467. [Google Scholar] [CrossRef]
- Franke, M.; Ibrahim, D.M.; Andrey, G.; Schwarzer, W.; Heinrich, V.; Schöpflin, R.; Kraft, K.; Kempfer, R.; Jerković, I.; Chan, W.L.; et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 2016, 538, 265–269. [Google Scholar] [CrossRef]
- Ibrahim, D.M.; Mundlos, S. Three-dimensional chromatin in disease: What holds us together and what drives us apart? Curr. Opin. Cell Biol. 2020, 64, 1–9. [Google Scholar] [CrossRef]
- Van Schil, K.; Naessens, S.; Van de Sompele, S.; Carron, M.; Aslanidis, A.; Van Cauwenbergh, C.; Kathrin Mayer, A.; Van Heetvelde, M.; Bauwens, M.; Verdin, H.; et al. Mapping the genomic landscape of inherited retinal disease genes prioritizes genes prone to coding and noncoding copy-number variations. Genet. Med. 2018, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Shearer, A.E.; Kolbe, D.L.; Azaiez, H.; Sloan, C.M.; Frees, K.L.; Weaver, A.E.; Clark, E.T.; Nishimura, C.J.; Black-Ziegelbein, E.A.; Smith, R.J. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Nikopoulos, K.; Cisarova, K.; Quinodoz, M.; Koskiniemi-Kuendig, H.; Miyake, N.; Farinelli, P.; Rehman, A.U.; Khan, M.I.; Prunotto, A.; Akiyama, M.; et al. A frequent variant in the Japanese population determines quasi-Mendelian inheritance of rare retinal ciliopathy. Nat. Commun. 2019, 10, 2884. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Liu, X.-Z. Modifiers of hearing impairment in humans and mice. Curr. Genom. 2010, 11, 269–278. [Google Scholar] [CrossRef][Green Version]
- Norman, C.S.; O’Gorman, L.; Gibson, J.; Pengelly, R.J.; Baralle, D.; Ratnayaka, J.A.; Griffiths, H.; Rose-Zerilli, M.; Ranger, M.; Bunyan, D.; et al. Identification of a functionally significant tri-allelic genotype in the Tyrosinase gene (TYR) causing hypomorphic oculocutaneous albinism (OCA1B). Sci. Rep. 2017, 7, 4415. [Google Scholar] [CrossRef]
- Grønskov, K.; Jespersgaard, C.; Bruun, G.H.; Harris, P.; Brøndum-Nielsen, K.; Andresen, B.S.; Rosenberg, T. A pathogenic haplotype, common in Europeans, causes autosomal recessive albinism and uncovers missing heritability in OCA1. Sci. Rep. 2019, 9, 645. [Google Scholar] [CrossRef]
- Green, D.J.; Sallah, S.R.; Ellingford, J.M.; Lovell, S.C.; Sergouniotis, P.I. Variability in Gene Expression is Associated with Incomplete Penetrance in Inherited Eye Disorders. Genes (Basel) 2020, 11, 179. [Google Scholar] [CrossRef]
- Llavona, P.; Pinelli, M.; Mutarelli, M.; Marwah, V.S.; Schimpf-Linzenbold, S.; Thaler, S.; Yoeruek, E.; Vetter, J.; Kohl, S.; Wissinger, B. Allelic Expression Imbalance in the Human Retinal Transcriptome and Potential Impact on Inherited Retinal Diseases. Genes (Basel) 2017, 8, 283. [Google Scholar] [CrossRef]
- Runhart, E.H.; Sangermano, R.; Cornelis, S.S.; Verheij, J.; Plomp, A.S.; Boon, C.J.F.; Lugtenberg, D.; Roosing, S.; Bax, N.M.; Blokland, E.A.W.; et al. The Common ABCA4 Variant p.Asn1868Ile Shows Nonpenetrance and Variable Expression of Stargardt Disease When Present in trans With Severe Variants. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3220–3231. [Google Scholar] [CrossRef]
- Smits, J.J.; van Beelen, E.; Weegerink, N.J.D.; Oostrik, J.; Huygen, P.L.M.; Beynon, A.J.; Lanting, C.P.; Kunst, H.P.M.; Schraders, M.; Kremer, H.; et al. A Novel COCH Mutation Affects the vWFA2 Domain and Leads to a Relatively Mild DFNA9 Phenotype. Otol. Neurotol. 2020. [Google Scholar] [CrossRef]
- Vithana, E.N.; Abu-Safieh, L.; Pelosini, L.; Winchester, E.; Hornan, D.; Bird, A.C.; Hunt, D.M.; Bustin, S.A.; Bhattacharya, S.S. Expression of PRPF31 mRNA in patients with autosomal dominant retinitis pigmentosa: A molecular clue for incomplete penetrance? Investig. Ophthalmol. Vis. Sci. 2003, 44, 4204–4209. [Google Scholar] [CrossRef]
- de Bruijn, S.E.; Smits, J.J.; Liu, C.; Lanting, C.P.; Beynon, A.J.; Blankevoort, J.; Oostrik, J.; Koole, W.; de Vrieze, E.; Cremers, C.W.R.J.; et al. A RIPOR2 in-frame deletion is a frequent and highly penetrant cause of adult-onset hearing loss. J. Med Genet. 2021, 58, 96–104. [Google Scholar] [CrossRef]
- Yauy, K.; de Leeuw, N.; Yntema, H.G.; Pfundt, R.; Gilissen, C. Accurate detection of clinically relevant uniparental disomy from exome sequencing data. Genet. Med. 2020, 22, 803–808. [Google Scholar] [CrossRef]
- Fingert, J.H.; Eliason, D.A.; Phillips, N.C.; Lotery, A.J.; Sheffield, V.C.; Stone, E.M. Case of Stargardt disease caused by uniparental isodisomy. Arch. Ophthalmol. 2006, 124, 744–745. [Google Scholar] [CrossRef][Green Version]
- Alvarez, A.; del Castillo, I.; Pera, A.; Villamar, M.; Moreno-Pelayo, M.A.; Rivera, T.; Solanellas, J.; Moreno, F. Uniparental disomy of chromosome 13q causing homozygosity for the 35delG mutation in the gene encoding connexin26 (GJB2) results in prelingual hearing impairment in two unrelated Spanish patients. J. Med. Genet. 2003, 40, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Shen, S.; Cheng, J.; Lv, H.; Fu, J. A case of Usher syndrome type IIA caused by a rare USH2A homozygous frameshift variant with maternal uniparental disomy (UPD) in a Chinese family. J. Cell. Mol. Med. 2020, 24, 7743–7750. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.; Lenarduzzi, S.; Cappellani, S.; Pecile, V.; Morgutti, M.; Orzan, E.; Ghiselli, S.; Ambrosetti, U.; Brumat, M.; Gajendrarao, P.; et al. Genomic Studies in a Large Cohort of Hearing Impaired Italian Patients Revealed Several New Alleles, a Rare Case of Uniparental Disomy (UPD) and the Importance to Search for Copy Number Variations. Front. Genet. 2018, 9, 681. [Google Scholar] [CrossRef]
- Slijkerman, R.W.; Song, F.; Astuti, G.D.; Huynen, M.A.; van Wijk, E.; Stieger, K.; Collin, R.W. The pros and cons of vertebrate animal models for functional and therapeutic research on inherited retinal dystrophies. Prog. Retin. Eye Res. 2015, 48, 137–159. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Vona, B.; Doll, J.; Hofrichter, M.A.H.; Haaf, T.; Varshney, G.K. Small fish, big prospects: Using zebrafish to unravel the mechanisms of hereditary hearing loss. Hear. Res. 2020, 107906. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.C.; Hashino, E.; Nelson, R.F. Progress in Modeling and Targeting Inner Ear Disorders with Pluripotent Stem Cells. Stem Cell Rep. 2020, 14, 996–1008. [Google Scholar] [CrossRef] [PubMed]
- Kruczek, K.; Swaroop, A. Pluripotent stem cell-derived retinal organoids for disease modeling and development of therapies. Stem Cells 2020, 38, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.; Veltman, J.A.; van Kessel, A.G.; Brunner, H.G. Identification of disease genes by whole genome CGH arrays. Hum. Mol. Genet. 2005, 14, R215–R223. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Shu, W.; Li, P. Fluorescence In situ Hybridization: Cell-Based Genetic Diagnostic and Research Applications. Front. Cell Dev. Biol. 2016, 4, 89. [Google Scholar] [CrossRef] [PubMed]
- Hyon, C. Usefulness of CGH-array and SNP-array for the etiological diagnosis of premature ovarian insufficiency. Biol. Aujourdhui 2017, 211, 199–205. [Google Scholar] [CrossRef]
- Mantere, T.; Neveling, K.; Pebrel-Richard, C.; Benoist, M.; van der Zande, G.; Kater-Baats, E.; Baatout, I.; van Beek, R.; Yammine, T.; Oorsprong, M.; et al. Next generation cytogenetics: Genome-imaging enables comprehensive structural variant detection for 100 constitutional chromosomal aberrations in 85 samples. BioRxiv 2020. [Google Scholar] [CrossRef]
- Lee, C.N.; Lin, S.Y.; Lin, C.H.; Shih, J.C.; Lin, T.H.; Su, Y.N. Clinical utility of array comparative genomic hybridisation for prenatal diagnosis: A cohort study of 3171 pregnancies. BJOG 2012, 119, 614–625. [Google Scholar] [CrossRef]
- Yuan, Y.; Chung, C.Y.; Chan, T.F. Advances in optical mapping for genomic research. Comput. Struct. Biotechnol. J. 2020, 18, 2051–2062. [Google Scholar] [CrossRef]
- Chan, S.; Lam, E.; Saghbini, M.; Bocklandt, S.; Hastie, A.; Cao, H.; Holmlin, E.; Borodkin, M. Structural Variation Detection and Analysis Using Bionano Optical Mapping. Methods Mol. Biol. 2018, 1833, 193–203. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, M.; Qian, Y.; Yang, Y.; Sun, Y.; Liu, B.; Wang, L.; Dong, M. Identification of a likely pathogenic structural variation in the LAMA1 gene by Bionano optical mapping. NPJ Genom. Med. 2020, 5, 31. [Google Scholar] [CrossRef]
- Cummings, B.B.; Marshall, J.L.; Tukiainen, T.; Lek, M.; Donkervoort, S.; Foley, A.R.; Bolduc, V.; Waddell, L.B.; Sandaradura, S.A.; O'Grady, G.L.; et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Kremer, L.S.; Bader, D.M.; Mertes, C.; Kopajtich, R.; Pichler, G.; Iuso, A.; Haack, T.B.; Graf, E.; Schwarzmayr, T.; Terrile, C.; et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat. Commun. 2017, 8, 15824. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, K.R.; Sloan, C.A.; Malladi, V.S.; Dreszer, T.R.; Learned, K.; Kirkup, V.M.; Wong, M.C.; Maddren, M.; Fang, R.; Heitner, S.G.; et al. ENCODE data in the UCSC Genome Browser: Year 5 update. Nucleic Acids Res. 2013, 41, D56–D63. [Google Scholar] [CrossRef] [PubMed]
- Ray, T.A.; Cochran, K.; Kozlowski, C.; Wang, J.; Alexander, G.; Cady, M.A.; Spencer, W.J.; Ruzycki, P.A.; Clark, B.S.; Laeremans, A.; et al. Comprehensive identification of mRNA isoforms reveals the diversity of neural cell-surface molecules with roles in retinal development and disease. Nat. Commun. 2020, 11, 3328. [Google Scholar] [CrossRef]
- Single Cell Portal (Broad Institute). Available online: https://singlecell.broadinstitute.org/single_cell (accessed on 14 December 2020).




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Bruijn, S.E.; Fadaie, Z.; Cremers, F.P.M.; Kremer, H.; Roosing, S. The Impact of Modern Technologies on Molecular Diagnostic Success Rates, with a Focus on Inherited Retinal Dystrophy and Hearing Loss. Int. J. Mol. Sci. 2021, 22, 2943. https://doi.org/10.3390/ijms22062943
de Bruijn SE, Fadaie Z, Cremers FPM, Kremer H, Roosing S. The Impact of Modern Technologies on Molecular Diagnostic Success Rates, with a Focus on Inherited Retinal Dystrophy and Hearing Loss. International Journal of Molecular Sciences. 2021; 22(6):2943. https://doi.org/10.3390/ijms22062943
Chicago/Turabian Stylede Bruijn, Suzanne E., Zeinab Fadaie, Frans P. M. Cremers, Hannie Kremer, and Susanne Roosing. 2021. "The Impact of Modern Technologies on Molecular Diagnostic Success Rates, with a Focus on Inherited Retinal Dystrophy and Hearing Loss" International Journal of Molecular Sciences 22, no. 6: 2943. https://doi.org/10.3390/ijms22062943
APA Stylede Bruijn, S. E., Fadaie, Z., Cremers, F. P. M., Kremer, H., & Roosing, S. (2021). The Impact of Modern Technologies on Molecular Diagnostic Success Rates, with a Focus on Inherited Retinal Dystrophy and Hearing Loss. International Journal of Molecular Sciences, 22(6), 2943. https://doi.org/10.3390/ijms22062943