
Molecular Action of Polyphenols in Leukaemia and Their Therapeutic Potential


Abstract
1. Introduction
1.1. Leukaemia
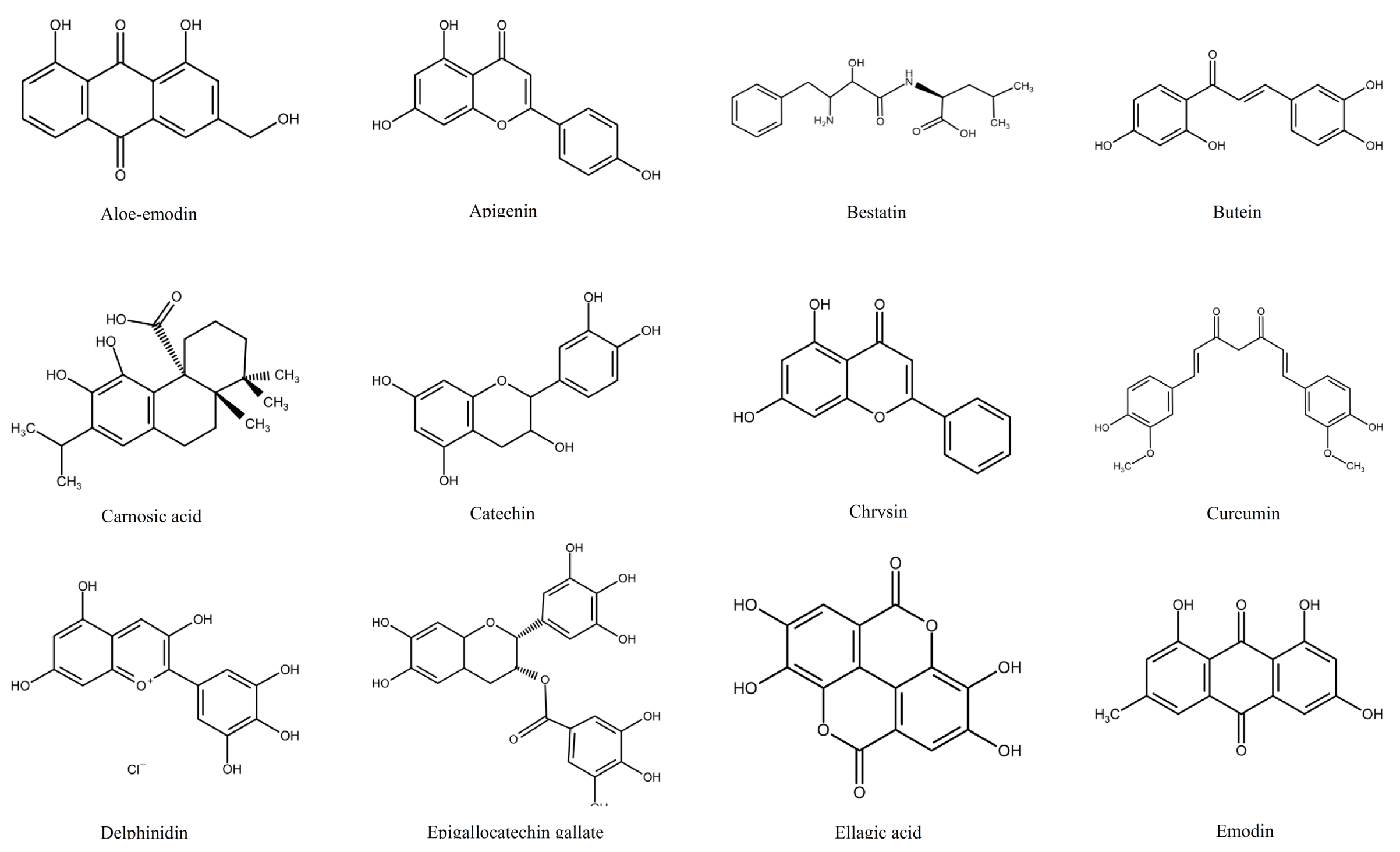
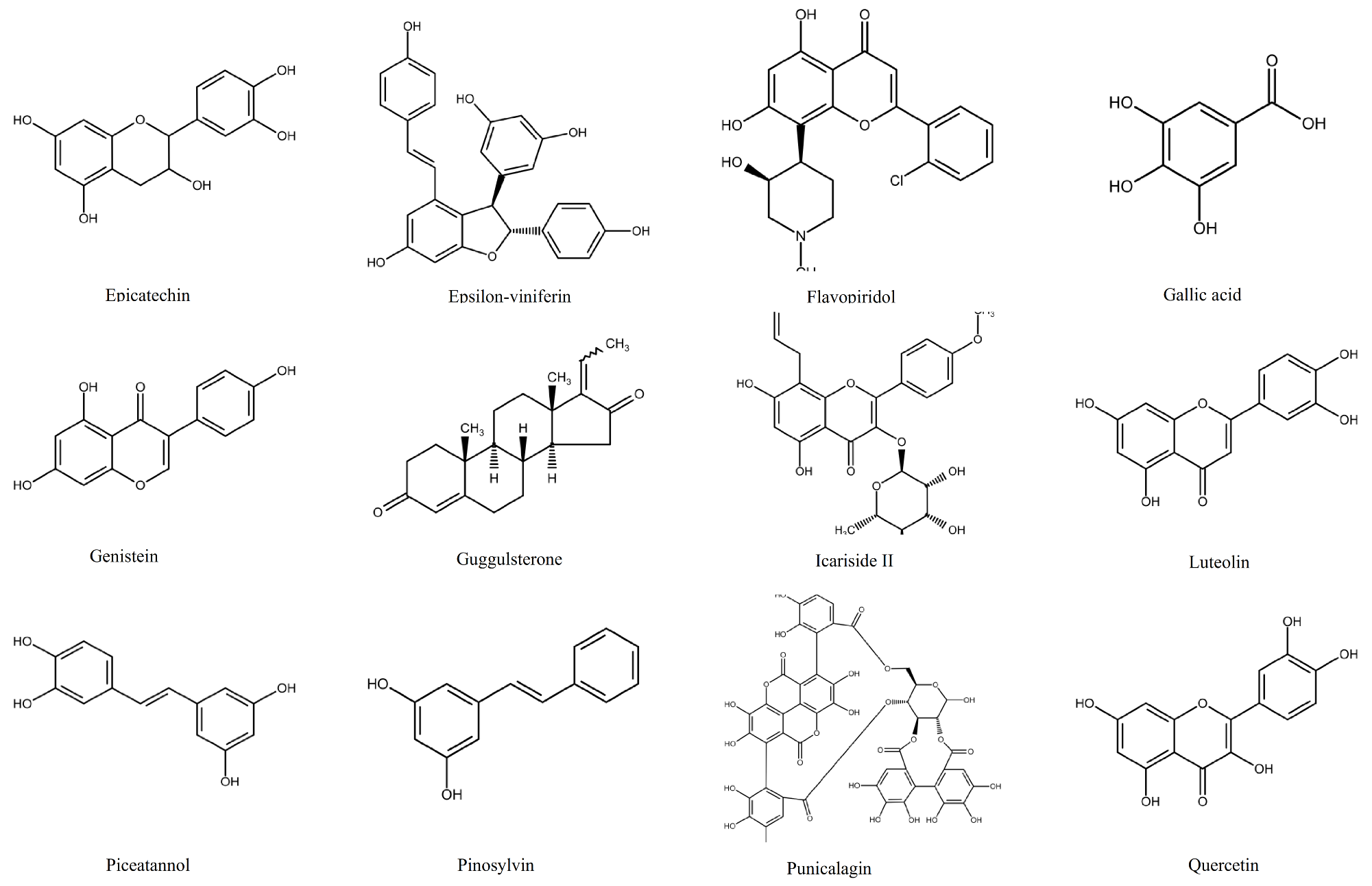
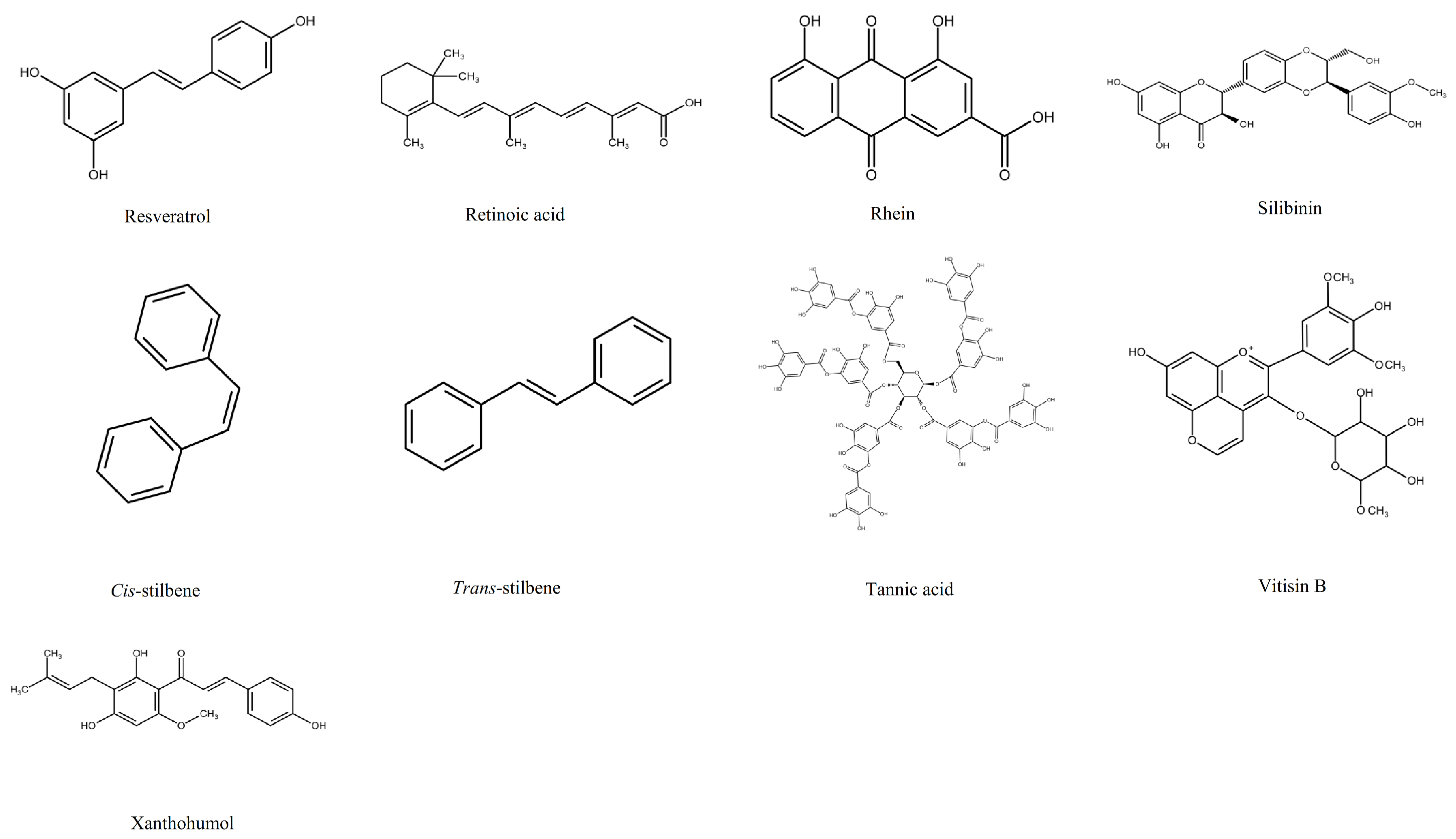
1.2. Polyphenols
2. Molecular Mechanisms of Polyphenols
- 2.1.
- Induction of cellular stress and catabolism: through an increase in reactive oxygen species (ROS) and a decrease in cellular antioxidants such as glutathione (GSH).
- 2.2.
- Modulation of cell metabolic activity.
- 2.3.
- Cell cycle arrest.
- 2.4.
- Induction of cell death via:
- Apoptosis.
- Autophagy.
- 2.5.
- Interaction with chemotherapy agents and the reduction or reversal of multidrug resistance.
2.1. Polyphenols, Cellular Stress and Catabolism
2.2. Polyphenols and Reactive Oxygen Species
2.3. Polyphenols and Metabolic Activity
2.4. Polyphenols and Cell Cycle Arrest
2.5. The Pro-Apoptotic Effect of Polyphenols in Leukaemia
2.5.1. The Extrinsic Pathway
2.5.2. The Intrinsic Pathway
2.5.3. Autophagy
2.5.4. Autophagic Cell Death
2.6. Interaction of Chemotherapy and Multidrug Resistance
3. Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Kampen, K.R. The discovery and early understanding of leukemia. Leuk. Res. 2012, 36, 6–13. [Google Scholar] [CrossRef]
- Jakobsen, N.A.; Vyas, P. From genomics to targeted treatment in haematological malignancies: A focus on acute myeloid leukaemia. Clin. Med. 2018, 18, 47. [Google Scholar] [CrossRef] [PubMed]
- Licht, J.D.; Sternberg, D.W. The Molecular Pathology of Acute Myeloid Leukemia. Hematology 2005, 2005, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E. Über die Bedeutung des Knochenmarks für die Blutbildung. Zent. Med. Wiss. 1868, 44, 122. [Google Scholar]
- Virchow, R. Leukamie, Gesammelte Abhandlungen zur Wissenschaftlichen Medicin; Meidinger: Frankfurt, Germany, 1862; pp. 190–212. [Google Scholar]
- Rafiq, S.; Raza, M.H.; Younas, M.; Naeem, F.; Adeeb, R.; Iqbal, J.; Anwar, P.; Sajid, U.; Manzoor, H.M. Molecular Targets of Curcumin and Future Therapeutic Role in Leukemia. J. Biosci. Med. 2018, 6, 33–50. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Huang, A.; Cheng, H.; Lin, T.; Chen, W.; Lin, J.; Lin, J.; Lu, C.; Chiang, J.; Hsu, S.; Wu, P.; et al. Epigallocatechin gallate (EGCG), influences a murine WEHI-3 leukemia model in vivo through enhancing phagocytosis of macrophages and populations of T-and B-cells. Vivo 2013, 27, 627–634. [Google Scholar]
- Chen, P.; Wang, B.; Pan, B.; Guo, W. Resveratrol-4-O-D-(2′-galloyl)-glucopyranoside exerts an anticancer effect on leukemia cells via inducing apoptosis. Mol. Med. Rep. 2016, 13, 2281–2286. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Gerson, S. Antimetabolite agent combinations in the treatment of cancer. US Patent Application No. 15,357,423, 16 March 2017. [Google Scholar]
- Moreno, C.; Greil, R.; Demirkan, F.; Tedeschi, A.; Anz, B.; Larratt, L.; Simkovic, M.; Samoilova, O.; Novak, J.; Ben-Yehuda, D.; et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 43–56. [Google Scholar] [CrossRef]
- Melo, J.V.; Chuah, C. Novel Agents in CML Therapy: Tyrosine Kinase Inhibitors and Beyond. Hematology 2008, 2008, 427–435. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef]
- Bitencourt, R.; Zalcberg, I.; Louro, I.D. Imatinib resistance: A review of alternative inhibitors in chronic myeloid leukemia. Rev. Bras. Hematol. Hemoter. 2011, 33, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.; Lee, H.J. The roles of polyphenols in cancer chemoprevention. BioFactors 2006, 26, 105–121. [Google Scholar] [CrossRef]
- Cas, M.D.; Ghidoni, R. Cancer Prevention and Therapy with Polyphenols: Sphingolipid-Mediated Mechanisms. Nutriments 2018, 10, 940. [Google Scholar] [CrossRef] [PubMed]
- Cipolletti, M.; Solar Fernandez, V.; Montalesi, E.; Marino, M.; Fiocchetti, M. Beyond the antioxidant activity of dietary poly-phenols in cancer: The modulation of estrogen receptors (ers) signaling. Int. J. Mol. Sci. 2018, 19, 2624. [Google Scholar] [CrossRef]
- Mojzer, E.B.; Hrnčič, M.K.; Škerget, M.; Knez, Ž.; Bren, U. Polyphenols: Extraction Methods, Antioxidative Action, Bioavailability and Anticarcinogenic Effects. Molecules 2016, 21, 901. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, C.; Russo, M.; Bilotto, S.; Tedesco, I.; Laratta, B.; Russo, G.L. Dietary polyphenols in cancer prevention: The ex-ample of the flavonoid quercetin in leukemia. Ann. NY Acad. Sci. 2012, 1259, 95–103. [Google Scholar] [CrossRef]
- Rasouli, H.; Farzaei, M.H.; Khodarahmi, R. Polyphenols and their benefits: A review. Int. J. Food Prop. 2017, 20, 1–42. [Google Scholar] [CrossRef]
- Sak, K.; Everaus, H. Established Human Cell Lines as Models to Study Anti-leukemic Effects of Flavonoids. Curr. Genom. 2016, 18, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Perveen, S.; Al-Taweel, A.M. Phenolic compounds from the natural sources and their cytotoxicity. In Phenolic Compounds Natural Sources, Importance and Applications; IntechOpen: London, UK, 2017; Volume 29. [Google Scholar]
- Mahbub, A.; Le Maitre, C.; Haywood-Small, S.; Cross, N.; Jordan-Mahy, N. Dietary polyphenols influence antimetabolite agents: Methotrexate, 6-mercaptopurine and 5-fluorouracil in leukemia cell lines. Oncotarget 2017, 8, 104877–104893. [Google Scholar] [CrossRef]
- Mahbub, A.A.; Le Maitre, C.L.; Haywood-Small, S.L.; Cross, N.; Jordanmahy, N. Polyphenols act synergistically with doxorubicin and etoposide in leukaemia cell lines. Cell Death Discov. 2015, 1, 15043. [Google Scholar] [CrossRef]
- Duraj, J.; Bodo, J.; Sulikova, M.; Rauko, P.; Sedlak, J. Diverse resveratrol sensitization to apoptosis induced by anticancer drugs in sensitive and resistant leukemia cells. Neoplasma 2006, 53, 384–392. [Google Scholar]
- Mahbub, A.A.; Le Maitre, C.L.; Haywood-Small, S.L.; Cross, N.A.; Jordan-Mahy, N. Glutathione is key to the synergistic en-hancement of doxorubicin and etoposide by polyphenols in leukaemia cell lines. Cell Death Discov. 2015, 6, e2028. [Google Scholar] [CrossRef]
- Li, S.-Z.; Qiao, S.-F.; Zhang, J.-H.; Li, K. Quercetin Increase the Chemosensitivity of Breast Cancer Cells to Doxorubicin Via PTEN/Akt Pathway. Anti-Cancer Agents Med. Chem. 2015, 15, 1185–1189. [Google Scholar] [CrossRef] [PubMed]
- Kuhar, M.; Imran, S.; Singh, N. Curcumin and quercetin combined with cisplatin to induce apoptosis in human laryngeal carcinoma Hep-2 cells through the mitochondrial pathway. J. Cancer Mol. 2007, 3, 121–128. [Google Scholar]
- Ismail, S.; Haris, K.; Abdul Ghani, A.R.I.; Abdullah, J.M.; Johan, M.F.; Mohamed Yusoff, A.A. Enhanced in-duction of cell cycle arrest and apoptosis via the mitochondrial membrane potential disruption in human U87 malignant glioma cells by aloe emodin. J. Asian Nat. Prod. Res. 2013, 15, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, M.; Li, D.; You, J. Apigenin inhibits glioma cell growth through promoting microRNA-16 and suppression of BCL-2 and nuclear factor-κB/MMP 9. Mol. Med. Rep. 2016, 14, 2352–2358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Feng, J.; Cui, L.; Zhang, Y.; Li, W.; Li, C.; Shi, N.; Chen, Y.; Kong, W. Investigation Into Efficiency of a Novel Glycol Chitosan–Bestatin Conjugate to Protect Thymopoietin Oligopeptides From Enzymatic Degradation. J. Pharm. Sci. 2016, 105, 828–837. [Google Scholar] [CrossRef]
- Semwal, R.B.; Semwal, D.K.; Combrinck, S.; Viljoen, A. Butein: From ancient traditional remedy to modern nutraceutical. Phytochem. Lett. 2015, 11, 188–201. [Google Scholar] [CrossRef]
- Liu, P.; Dong, J. Protective effects of carnosic acid against mitochondria-mediated injury in H9c2 cardiomyocytes induced by hypoxia/reoxygenation. Exp. Med. 2017, 14, 5629–5634. [Google Scholar] [CrossRef]
- Isemura, M.; Miyoshi, N.; Pervin, M.; Suzuki, T.; Unno, K.; Nakamura, Y. Green tea catechins for well-being and therapy: Prospects and opportunities. Bot. Targets 2015, 5, 85–96. [Google Scholar] [CrossRef]
- Jiang, Y.; Gong, F.-L.; Zhao, G.-B.; Li, J. Chrysin Suppressed Inflammatory Responses and the Inducible Nitric Oxide Synthase Pathway after Spinal Cord Injury in Rats. Int. J. Mol. Sci. 2014, 15, 12270–12279. [Google Scholar] [CrossRef]
- Kawamori, T.; Lubet, R.; Steele, V.E.; Kelloff, G.J.; Kaskey, R.B.; Rao, C.V.; Reddy, B.S. Chemopreventive effect of curcumin, a naturally occurring anti-inflammatory agent, during the promotion/progression stages of colon cancer. Cancer Res. 1999, 59, 597–601. [Google Scholar]
- Masheta, D.Q.; Al-Azzawi, S.K. Antioxidant and Anti-Inflammatory Effects of Delphinidin on Glial Cells and Lack of Effect on Secretase Enzyme. IOP Conf. Ser. Mater. Sci. Eng. 2018, 454, 012061. [Google Scholar] [CrossRef]
- Eng, Q.Y.; Thanikachalam, P.V.; Ramamurthy, S. Molecular understanding of Epigallocatechin gallate (EGCG) in cardiovas-cular and metabolic diseases. J. Ethnopharmacol. 2018, 210, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Cheshomi, H.; Bahrami, A.R.; Matin, M.M. Ellagic acid and human cancers: A systems pharmacology and docking study to identify principal hub genes and main mechanisms of action. Mol. Divers. 2021, 25, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Meier, N.; Meier, B.; Peter, S.; Wolfram, E. In-Silico UHPLC Method Optimization for Aglycones in the Herbal Laxatives Aloe barbadensis Mill., Cassia angustifolia Vahl Pods, Rhamnus frangula L.; Bark, Rhamnus purshianus DC. Bark, and Rheum palmatum L. Roots. Molecules 2017, 22, 1838. [Google Scholar] [CrossRef] [PubMed]
- Hosseinimehr, S.J.; Rostamnezad, M.; Ghafari, R.V. Epicatechin Enhances Anti-Proliferative Effect of Bleomycin in Ovarian Cancer Cell. Res. Mol. Med. 2013, 1, 24–27. [Google Scholar] [CrossRef]
- Mattio, L.M.; Marengo, M.; Parravicini, C.; Eberini, I.; Dallavalle, S.; Bonomi, F.; Iametti, S.; Pinto, A. Inhibition of Pancreatic α-amylase by Resveratrol Derivatives: Biological Activity and Molecular Modelling Evidence for Cooperativity between Viniferin Enantiomers. Molecules 2019, 24, 3225. [Google Scholar] [CrossRef]
- Chohan, T.A.; Qayyum, A.; Rehman, K.; Tariq, M.; Akash, M.S.H. An insight into the emerging role of cyclin-dependent kinase inhibitors as potential therapeutic agents for the treatment of advanced cancers. Biomed. Pharm. 2018, 107, 1326–1341. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.; Je, I.-G.; Shin, T.-Y.; Kim, S.-H.; Seo, S.-Y. Synthesis of Gallic Acid Analogs as Histamine and Pro-Inflammatory Cytokine Inhibitors for Treatment of Mast Cell-Mediated Allergic Inflammation. Molecules 2017, 22, 898. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Zhang, H.; Zeng, X.; Huang, L.; Wang, Z.; Liu, G.; Wu, Y.; Yang, C. Fabrication of genistein-loaded biodegradable TPGS-b-PCL nanoparticles for improved therapeutic effects in cervical cancer cells. Int. J. Nanomed. 2015, 10, 2461–2473. [Google Scholar] [CrossRef] [PubMed]
- Kalariya, N.M.; Shoeb, M.; Reddy, A.B.M.; Zhang, M.; Van Kuijk, F.J.G.M.; Ramana, K.V. Prevention of Endotoxin-Induced Uveitis in Rats by Plant Sterol Guggulsterone. Investig. Opthalmology Vis. Sci. 2010, 51, 5105–5113. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, J.; Guan, M.; Wong, P.F.; Yu, H.; Dong, J.; Xu, J. Icariside II potentiates paclitaxel-induced apoptosis in human melano-ma A375 cells by inhibiting TLR4 signaling pathway. Food Chem. Toxicol. 2012, 50, 3019–3024. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Rauf, A.; Abu-Izneid, T.; Nadeem, M.; Shariati, M.A.; Khan, I.A.; Imran, A.; Orhan, I.E.; Rizwan, M.; Atif, M.; et al. Luteolin, a flavonoid, as an anticancer agent: A review. Biomed. Pharm. 2019, 112, 108612. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.-J.; Kim, H.-H.; Kim, E.-J.; Katakura, Y.; Lee, W.-S.; Kim, G.-S.; Ryu, C.-H. Piceatannol inhibits mast cell-mediated allergic inflammation. Int. J. Mol. Med. 2013, 31, 951–958. [Google Scholar] [CrossRef]
- Nascimento, M.D.; Santana, A.; Maranhão, C.A.; Oliveira, L.S.; Bieber, L. Phenolic extractives and natural resistance of wood. In Biodegradation: Life of Science; IntechOpen: Rijeka, Croatia, 2013; Volume 801. [Google Scholar]
- Li, J.; Wang, G.; Hou, C.; Li, J.; Luo, Y.; Li, B. Punicalagin and ellagic acid from pomegranate peel induce apoptosis and in-hibits proliferation in human HepG2 hepatoma cells through targeting mitochondria. Food Agric. Immunol. 2019, 30, 897–912. [Google Scholar] [CrossRef]
- Wu, C.W.; Nakamoto, Y.; Hisatome, T.; Yoshida, S.; Miyazaki, H. Resveratrol and its dimers ε-viniferin and δ-viniferin in red wine protect vascular endothelial cells by a similar mechanism with different potency and efficacy. Kaohsiung J. Med. Sci. 2020, 36, 535–542. [Google Scholar] [CrossRef]
- Crotti, S.; Posocco, B.; Marangon, E.; Nitti, N.; Toffoli, G.; Agostini, M. Mass spectrometry in the pharmacokinetic studies of anticancer natural products. Mass Spectrom. Rev. 2015, 36, 213–251. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, Z.; Huang, P.; He, L.; Ling, Y. Design, synthesis and biological evaluation of rhein derivatives as anticancer agents. MedChemComm 2016, 7, 1812–1818. [Google Scholar] [CrossRef]
- Ezhilarasan, D. Lead compounds with the potentials for the treatment of chronic liver diseases. Phytochem. Lead Compd. New Drug Discov. 2020, 195–210. [Google Scholar] [CrossRef]
- Wonanke, A.D.D.; Ferguson, J.L.; Fitchett, C.M.; Crittenden, D.L.; Gulbrandsen, J.L.; Fitchett, C.M. Predicting the Outcome of Photocyclisation Reactions: A Joint Experimental and Computational Investigation. Chem. Asian J. 2018, 14, 1293–1303. [Google Scholar] [CrossRef]
- Takemoto, Y.; Ajiro, H.; Akashi, M. Hydrogen-Bonded Multilayer Films Based on Poly(N-vinylamide) Derivatives and Tannic Acid. Langmuir 2015, 31, 6863–6869. [Google Scholar] [CrossRef]
- He, F.; Liang, N.; Mu, L.; Pan, Q.; Wang, J.; Reeves, M.J.; Duan, C.Q. Anthocyanins and their variation in red wines, I.I. An-thocya-nin derived pigments and their color evolution. Molecules 2012, 17, 1483–1519. [Google Scholar] [CrossRef]
- Pacurari, M.; Brown, H.; Rieland, A. Hop-derived Xanthohumol Induces HL-60 Leukemia Cells Death. Int. J. Biochem. Res. Rev. 2020, 29, 61–72. [Google Scholar] [CrossRef][Green Version]
- Wendel-Hansen, V.; Sällström, J.; De Campos-Lima, P.O.; Kjellström, G.; Sandlund, A.; Siegbahn, A.; Carlsson, M.; Nilsson, K.; Rosén, A. Epstein-Barr virus (EBV) can immortalize B-cll cells activated by cytokines. Leukemia 1994, 8, 476–484. [Google Scholar]
- Gokbulut, A.A.; Apohan, E.; Baran, Y. Resveratrol and quercetin-induced apoptosis of human 232B4 chronic lymphocytic leukemia cells by activation of caspase-3 and cell cycle arrest. Hematology 2013, 18, 144–150. [Google Scholar] [CrossRef]
- Hoogendoorn, M.; Wolbers, J.O.; Smit, W.M.; Schaafsma, M.R.; Barge, R.M.Y.; Willemze, R.; Falkenburg, J.H.F. Generation of B-cell chronic lymphocytic leukemia (B-CLL)-reactive T-cell lines and clones from HLA class I-matched donors using modified B-CLL cells as stimulators: Implications for adoptive immunotherapy. Leukemia 2004, 18, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Angelo, L.S.; Kurzrock, R. Turmeric and green tea: A recipe for B-Chronic Lymphocytic Leukemia. Clin. cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 1123. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Kay, N.E.; Secreto, C.R.; Shanafelt, T.D. Curcumin inhibits prosurvival pathways in chronic lymphocytic leu-kemia B cells and may overcome their stromal protection in combination with EGCG. Clin. cancer Res. 2009, 15, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Quiney, C.; Dauzonne, D.; Kern, C.; Fourneron, J.-D.; Izard, J.-C.; Mohammad, R.M.; Kolb, J.-P.; Billard, C. Flavones and polyphenols inhibit the NO pathway during apoptosis of leukemia B-cells. Leuk. Res. 2004, 28, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Roman, V.; Billard, C.; Kern, C.; Ferry-Dumazet, H.; Izard, J.; Mohammad, R.; Mossalayi, D.M.; Kolb, J.P. Analysis of resvera-trol-induced apoptosis in human B-cell chronic leukaemia. Br. J. Haematol. 2002, 117, 842–851. [Google Scholar] [CrossRef]
- Mahbub, A.A.; Le Maitre, L.C.; Haywood-Small, L.S.; McDougall, J.G.; Cross, A.N.; Jordan-Mahy, N. Differential effects of polyphenols on proliferation and apoptosis in human myeloid and lymphoid leukemia cell lines. Anti-Cancer Agents Med. Chem. 2013, 13, 1601–1613. [Google Scholar] [CrossRef]
- Dahlawi, H.; Jordan-Mahy, N.; Clench, M.; McDougall, G.J.; Maitre, C.L. Polyphenols are responsible for the proapoptotic properties of pomegranate juice on leukemia cell lines. Food Sci. Nutr. 2013, 1, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Galasso, S.; Pacifico, S.; Kretschmer, N.; Pan, S.-P.; Marciano, S.; Piccolella, S.; Monaco, P.; Bauer, R. Influence of seasonal variation on Thymus longicaulis C. Presl chemical composition and its antioxidant and anti-inflammatory properties. Phytochemistry 2014, 107, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Heydarabad, M.Z.; Nikasa, M.; Vatanmakanian, M.; Azimi, A.; Hagh, M.F. Regulatory effect of resveratrol and prednisolone onMDR1gene expression in acute lymphoblastic leukemia cell line (CCRF-CEM): An epigenetic perspective. J. Cell. Biochem. 2018, 119, 4890–4896. [Google Scholar] [CrossRef]
- Mahbub, A.A.; Le Maitre, C.L.; Haywood-Small, S.; Cross, N.A.; Jordan-Mahy, N. Polyphenols enhance the activity of alkyl-ating agents in leukaemia cell lines. Oncotarget 2019, 10, 4570. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, H.; Krstin, S.; Wink, M. Modulation of multidrug resistant in cancer cells by EGCG, tannic acid and curcumin. Phytomedicine 2018, 50, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Strasser-Wozak, E.M.; Hattmannstorfer, R.; Hála, M.; Hartmann, B.L.; Fiegl, M.; Geley, S.; Kofler, R. Splice site mutation in the glucocorticoid receptor gene causes resistance to glucocorticoid-induced apoptosis in a human acute leukemic cell line. Cancer Res. 1995, 55, 348–353. [Google Scholar] [PubMed]
- Bernhard, D.; Tinhofer, I.; Tonko, M.; Hübl, H.; Ausserlechner, M.J.; Greil, R.; Kofler, R.; Csordas, A. Resveratrol causes arrest in the S-phase prior to Fas-independent apoptosis in CEM-C7H2 acute leukemia cells. Cell Death Differ. 2000, 7, 834–842. [Google Scholar] [CrossRef]
- Dörrie, J.; Gerauer, H.; Wachter, Y.; Zunino, S.J. Resveratrol induces extensive apoptosis by depolarizing mitochondrial membranes and activating caspase-9 in acute lymphoblastic leukemia cells. Cancer Res. 2001, 61, 4731–4739. [Google Scholar] [PubMed]
- Frazzi, R.; Guardi, M. Cellular and Molecular Targets of Resveratrol on Lymphoma and Leukemia Cells. Molecules 2017, 22, 885. [Google Scholar] [CrossRef]
- Jiao, G.E.; Yan, L.; Qiang, L.I.; Xia, G.; Ling, G.U.; Gui, Z.; Zhu, Y.P. Resveratrol induces apoptosis and autophagy in T-cell acute lymphoblastic leukemia cells by inhibiting Akt/mTOR and activating p38-MAPK. Biomed. Environ. Sci. 2013, 26, 902–911. [Google Scholar]
- Gu, L.; Zhang, G.; Zhang, Y. A novel method to establish glucocorticoid resistant acute lymphoblastic leukemia cell lines. J. Exp. Clin. Cancer Res. 2019, 38, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.; Srour, E.F.; Turner, R.; Carey, R.; Maze, R.; Starrett, B.; Kanagala, R.; Pereira, D.; Merchant, P.; Taylor, M.; et al. Characterization of a new cell line (ESKOL) resem-bling hairy-cell leukemia: A model for oncogene regulation and late B-cell differentiation. Leuk. Res. 1991, 15, 733–744. [Google Scholar] [CrossRef]
- Billard, C.; Quiney, C.; Tang, R.; Kern, C.; Ajchenbaum-Cymbalista, F.; Dauzonne, D.; Kolb, J. The Inducible NO Synthase Is Downregulated during Apoptosis of Malignant Cells from B-Cell Chronic Lymphocytic Leukemia Induced by Flavopiridol and Polyphenols. Ann. N. Y. Acad. Sci. 2003, 1010, 381–383. [Google Scholar] [CrossRef] [PubMed]
- Luzi, C.; Brisdelli, F.; Cinque, B.; Cifone, G.; Bozzi, A.; Cifone, M.G. Differential sensitivity to resveratrol-induced apoptosis of human chronic myeloid (K562) and acute lymphoblastic (HSB-2) leukemia cells. Biochem. Pharm. 2004, 68, 2019–2030. [Google Scholar] [CrossRef]
- Kim, N. Butein sensitizes human leukemia cells to apoptosis induced by tumor necrosis factor-related apoptosis inducing ligand (TRAIL). Arch. Pharmacal Res. 2008, 31, 1179–1186. [Google Scholar] [CrossRef]
- Mackenzie, G.G.; Carrasquedo, F.; Delfino, J.M.; Keen, C.L.; Fraga, C.G.; Oteiza, P.I. Epicatechin, catechin, and dimeric pro-cyanidins inhibit PMA-induced NF-κB activation at multiple steps in Jurkat T cells. FASEB J. 2004, 18, 167–169. [Google Scholar] [CrossRef]
- Nam, S.; Smith, D.M.; Dou, Q.P. Ester Bond-containing Tea Polyphenols Potently Inhibit Proteasome Activity in Vitro and in Vivo. J. Biol. Chem. 2001, 276, 13322–13330. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, B.; Cheng, J.; Yang, L.; Liu, Z.; Balan, K.; Pantazis, P.; Wyche, J.H.; Han, Z. FADD-dependent apoptosis in-duction in Jurkat leukemia T-cells by the resveratrol analogue, 3, 4, 5-trihydroxy-trans-stilbene. Biochem. Pharmacol. 2005, 69, 249–254. [Google Scholar] [CrossRef]
- Reis-Sobreiro, M.; Gajate, C.; Mollinedo, F. Involvement of mitochondria and recruitment of Fas/CD95 signaling in lipid rafts in resveratrol-mediated antimyeloma and antileukemia actions. Oncogene 2009, 28, 3221–3234. [Google Scholar] [CrossRef]
- Li, T.; Wang, W.; Chen, H.; Li, T.; Ye, L. Evaluation of anti-leukemia effect of resveratrol by modulating SATA3 signaling. Int. Immunopharmacol. 2010, 10, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Mertens-Talcott, S.U.; Percival, S.S. Ellagic acid and quercetin interact synergistically with resveratrol in the induction of apoptosis and cause transient cell cycle arrest in human leukemia cells. Cancer Lett. 2005, 218, 141–151. [Google Scholar] [CrossRef]
- Mertens-Talcott, S.U.; Talcott, S.T.; Percival, S.S. Low Concentrations of Quercetin and Ellagic Acid Synergistically Influence Proliferation, Cytotoxicity and Apoptosis in MOLT-4 Human Leukemia Cells. J. Nutr. 2003, 133, 2669–2674. [Google Scholar] [CrossRef] [PubMed]
- Siedlecka-Kroplewska, K.; Wozniak, M.; Kmiec, Z. The wine polyphenol resveratrol modulates autophagy and induces apoptosis in MOLT-4 and HL-60 human leukemia cells. J. Physiol. Pharmacol. 2020, 70, 825–838. [Google Scholar]
- Fernandez, M.V.; Delviks-Frankenberry, K.A.; Scheiblin, D.A.; Happel, C.; Pathak, V.K.; Freed, E.O. Authentication Analysis of MT-4 Cells Distributed by the National Institutes of Health AIDS Reagent Program. J. Virol. 2019, 93, 93. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, C.; Senba, M.; Mori, N. Butein inhibits NF-κB, AP-1 and Akt activation in adult T-cell leukemia/lymphoma. Int. J. Oncol. 2017, 51, 633–643. [Google Scholar] [CrossRef]
- Greil, J.; Gramatzki, M.; Burger, R.; Marschalek, R.; Peltner, M.; Trautmann, U.; Hansen-Hagge, T.E.; Bartram, C.R.; Fey, G.H.; Stehr, K.; et al. The acute lymphoblastic leukaemia cell line SEM with t(4;11) chromosomal rearrangement is biphenotypic and responsive to interleukin-7. Br. J. Haematol. 1994, 86, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Sugamura, K.; Fujii, M.; Kannagi, M.; Sakitani, M.; Takeuchi, M.; Hinuma, Y. Cell surface phenotypes and expression of viral antigens of various human cell lines carrying human T-cell leukemia virus. Int. J. Cancer 1984, 34, 221–228. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Mohamed, A.N.; Hamdan, M.Y.; Vo, T.; Chen, B.; Katato, K.; Abubakr, Y.A.; Dugan, M.C.; Al-Katib, A. Es-tablishment of a human B-CLL xen-ograft model: Utility as a preclinical therapeutic model. Leukemia 1996, 10, 130–137. [Google Scholar] [PubMed]
- Ferry-Dumazet, H.; Garnier, O.; Mamani-Matsuda, M.; Vercauteren, J.; Belloc, F.; Billiard, C.; Dupouy, M.; Thiolat, D.; Kolb, J.P.; Marit, G.; et al. Resveratrol inhibits the growth and induces the apoptosis of both normal and leukemic hematopoietic cells. Carcinogenesis 2002, 23, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Sassi, A.; Bouhlel, I.; Mustapha, N.; Mokdad-Bzeouich, I.; Chaabane, F.; Ghedira, K.; Chekir-Ghedira, L. Assessment in vitro of the genotoxicity, antigenotoxicity and antioxidant of Ceratonia siliqua L. extracts in murine leukaemia cells L1210 by comet assay. Regul. Toxicol. Pharm. 2016, 77, 117–124. [Google Scholar] [CrossRef]
- Orzechowskiinsta, A.; Grzelkowska, K.; Zimowska, W.; Skierski, J.; Ploszaj, T.; Bachanek, K.; Motyl, T.; Karlik, W.; Filipecki, M. Induction of apoptosis and NF-kB by quercetin in growing murine L1210 lymphocytic leukaemic cells potentiated by TNF-α. Reprod. Nutr. Dev. 2000, 40, 441–465. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fan, G.-X.; Wang, W.; Li, T.; Yuan, Y.-K. Resveratrol induces apoptosis, influences IL-6 and exerts immunomodulatory effect on mouse lymphocytic leukemia both in vitro and in vivo. Int. Immunopharmacol. 2007, 7, 1221–1231. [Google Scholar] [CrossRef]
- Kweon, S.H.; Song, J.H.; Kim, T.S. Resveratrol-mediated reversal of doxorubicin resistance in acute myeloid leukemia cells via downregulation of MRP1 expression. Biochem. Biophys. Res. Commun. 2010, 395, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Kim, S.H.; Kweon, S.H.; Lee, T.H.; Kim, H.; Kim, H.; Kim, T.S. Defective expression of deoxycytidine kinase in cy-tarabine-resistant acute myeloid leukemia cells. International J. Oncol. 2009, 34, 1165–1171. [Google Scholar]
- Ruela-de-Sousa, R.R.; Fuhler, G.M.; Blom, N.; Ferreira, C.V.; Aoyama, H.; Peppelenbosch, M.P. Cytotoxicity of apig-enin on leukemia cell lines: Implications for prevention and therapy. Cell Death Dis. 2010, 1, 19. [Google Scholar] [CrossRef] [PubMed]
- Kachadourian, R.; Day, B.J. Flavonoid-induced glutathione depletion: Potential implications for cancer treatment. Free. Radic. Biol. Med. 2006, 41, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.; Kim, M.; Lee, J.; Choi, Y.H.; Kim, G. Butein suppresses c-Myc-dependent transcription and Akt-dependent phos-phorylation of hTERT in human leukemia cells. Cancer Lett. 2009, 286, 172–179. [Google Scholar] [CrossRef]
- Steiner, M.; Priel, I.; Giat, J.; Levy, J.; Sharoni, Y.; Danilenko, M. Carnosic acid inhibits proliferation and augments differentia-tion of human leukemic cells induced by 1, 25-dihydroxyvitamin dsub3 and retinoic acid. Nutr. Cancer 2001, 41, 135–144. [Google Scholar] [CrossRef]
- Pesakhov, S.; Nachliely, M.; Barvish, Z.; Aqaqe, N.; Schwartzman, B.; Voronov, E.; Sharoni, Y.; Studzinski, G.P.; Fishman, D.; Danilenko, M. Cancer-selective cytotoxic Ca2+ overload in acute myeloid leukemia cells and attenuation of disease progression in mice by synergistically acting polyphenols curcumin and carnosic acid. Oncotarget 2016, 7, 31847–31861. [Google Scholar] [CrossRef]
- Pesakhov, S.; Khanin, M.; Studzinski, G.P.; Danilenko, M. Distinct Combinatorial Effects of the Plant Polyphenols Curcumin, Carnosic Acid, and Silibinin on Proliferation and Apoptosis in Acute Myeloid Leukemia Cells. Nutr. Cancer 2010, 62, 811–824. [Google Scholar] [CrossRef]
- Derochette, S.; Franck, T.; Mouithys-Mickalad, A.; Deby-Dupont, G.; Neven, P.; Serteyn, D. Intra- and extracellular antioxidant capacities of the new water-soluble form of curcumin (NDS27) on stimulated neutrophils and HL-60 cells. Chem. Interact. 2013, 201, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Hung, H.; Hour, T.; Hsu, P.; Kao, M.; Tsay, G.J.; Liu, G.Y. Curcumin induces apoptosis through an ornithine de-carboxylase-dependent pathway in human promyelocytic leukemia HL-60 cells. Life Sci. 2008, 82, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Moradzadeh, M.; Roustazadeh, A.; Tabarraei, A.; Erfanian, S.; Sahebkar, A. Epigallocatechin-3-gallate enhances dif-ferentiation of acute promyelocytic leukemia cells via inhibition of PML-RARα and HDAC1. Phytotherapy Res. 2018, 32, 471–479. [Google Scholar] [CrossRef]
- Diab, A.E.K.; Shafik, R.E.; Yasuda, S. In Vitro Antioxidant and Antiproliferative Activities of Novel Orange Peel Extract and It’s Fractions on Leukemia HL-60 Cells. Asian Pac. J. Cancer Prev. 2015, 16, 7053–7060. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.-C.; Huang, T.-C.; Lai, C.-S.; Pan, M.-H. Induction of apoptosis by luteolin through cleavage of Bcl-2 family in human leukemia HL-60 cells. Eur. J. Pharm. 2005, 509, 1–10. [Google Scholar] [CrossRef]
- Alvarez, M.C.; Maso, V.; Torello, C.O.; Ferro, K.P.; Saad, S.T.O. The polyphenol quercetin induces cell death in leukemia by targeting epigenetic regulators of pro-apoptotic genes. Clin. Epigenetics 2018, 10, 1–11. [Google Scholar] [CrossRef]
- De Blas, E.; Estañ, M.C.; de Frutos, M.d.C.G.; Ramos, J.; del Carmen Boyano-Adánez, M.; Aller, P. Selected polyphenols potentiate the apoptotic efficacy of glycolytic inhibitors in human acute myeloid leukemia cell lines. Regulation by protein kinase activities. Cancer Cell Int. 2016, 16, 70. [Google Scholar] [CrossRef]
- Park, E.Y.; Kim, J.-I.; Leem, D.-G.; Shin, J.-S.; Kim, K.-T.; Choi, S.Y.; Lee, M.-H.; Choi, J.-H.; Lee, Y.S.; Lee, K.-T. Resveratrol analogue (E)-8-acetoxy-2-[2-(3,4-diacetoxyphenyl) ethenyl]-quinazoline induces apoptosis via Fas-mediated pathway in HL-60 human leukemia cells. Oncol. Rep. 2016, 36, 3577–3587. [Google Scholar] [CrossRef]
- Fan, Y.; Chen, M.; Meng, J.; Yu, L.; Tu, Y.; Wan, L.; Fang, K.; Zhu, W. Arsenic trioxide and resveratrol show synergistic an-ti-leukemia activity and neutralized cardiotoxicity. PLoS ONE 2014, 9, e105890. [Google Scholar]
- Komina, O.; Węsierska-Gądek, J. Action of resveratrol alone or in combination with roscovitine, a CDK inhibitor, on cell cycle progression in human HL-60 leukemia cells. Biochem. Pharm. 2008, 76, 1554–1562. [Google Scholar] [CrossRef]
- Abubakar, M.B.; Abdullah, W.Z.; Sulaiman, S.A.; Ang, B.S. Polyphenols as Key Players for the Antileukaemic Effects of Propolis. Evid. Based Complement. Altern. Med. 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Li, G.; He, S.; Chang, L.; Lu, H.; Zhang, H.; Zhang, H.; Chiu, J. GADD45α and annexin A1 are involved in the apoptosis of HL-60 induced by resveratrol. Phytomedicine 2011, 18, 704–709. [Google Scholar] [CrossRef]
- Wu, S.-S.; Chen, L.-G.; Lin, R.-J.; Lin, S.-Y.; Lo, Y.-E.; Liang, Y.-C. Cytotoxicity of (-)-vitisin B in human leukemia cells. Drug Chem. Toxicol. 2012, 36, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Mi, X.; Wang, C.; Sun, C.; Chen, X.; Huo, X.; Zhang, Y.; Li, G.; Xu, B.; Zhang, J.; Xie, J.; et al. Xanthohumol induces paraptosis of leukemia cells through p38 mitogen activated protein kinase signaling pathway. Oncotarget 2017, 8, 31297–31304. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, Y.; Kasukabe, T.; Kaneko, Y.; Niitsu, N.; Okabe-Kado, J. Ellagic acid, a natural polyphenolic compound, induces apoptosis and potentiates retinoic acid-induced differentiation of human leukemia HL-60 cells. Int. J. Hematol. 2010, 92, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-J.; Wang, Z.; Li, H.-X.; Wu, R.-C.; Liu, Y.-Z.; Wu, Q.-Y. Mitochondrial dysfunction as an early event in the process of apoptosis induced by woodfordin I in human leukemia K562 cells. Toxicol. Appl. Pharm. 2004, 194, 141–155. [Google Scholar] [CrossRef]
- Han, M.; Lee, W.S.; Nagappan, A.; Kim, H.J.; Park, C.; Kim, G.; Hong, S.H.; Kim, N.D.; Kim, G.; Ryu, C.H.; et al. Polyphenols from Korean prostrate spurge Eu-phorbia supina induce apoptosis through the Fas-associated extrinsic pathway and activation of ERK in human leukemic U937 cells. Oncology Rep. 2016, 36, 99–107. [Google Scholar] [CrossRef][Green Version]
- Blasius, R.; Reuter, S.; Henry, E.; Dicato, M.; Diederich, M. Curcumin regulates signal transducer and activator of transcription (STAT) expression in K562 cells. Biochem. Pharm. 2006, 72, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Kundu, T.; Dey, S.; Bhattacharya, R.K.; Siddiqi, M.; Roy, M. Tea-induced apoptosis in human leuke-mia K562 cells as assessed by comet formation. Asian Pac. J. Cancer Prev. 2006, 7, 201. [Google Scholar]
- Roy, M.; Chakrabarty, S.; Sinha, D.; Bhattacharya, R.K.; Siddiqi, M. Anticlastogenic, antigenotoxic and apoptotic activity of epigallocatechin gallate: A green tea polyphenol. Mutat. Res. Mol. Mech. Mutagen. 2003, 523-524, 33–41. [Google Scholar] [CrossRef]
- Dai, Y.; Rahmani, M.; Pei, X.-Y.; Dent, P.; Grant, S. Bortezomib and flavopiridol interact synergistically to induce apoptosis in chronic myeloid leukemia cells resistant to imatinib mesylate through both Bcr/Abl-dependent and -independent mechanisms. Blood 2004, 104, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Mutlu Altundağ, E.; Yılmaz, A.M.; Koçtürk, S.; Taga, Y.; Yalçın, A.S. Synergistic induction of apoptosis by quercetin and curcumin in chronic myeloid leukemia (K562) cells. Nutr. Cancer 2018, 70, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, C.; Jia, Y.; Liu, Z.; Shu, X.; Liu, K. Resveratrol Increases Anti-Proliferative Activity of Bestatin Through Downregulating P-Glycoprotein Expression Via Inhibiting PI3K/Akt/mTOR Pathway in K562/ADR Cells. J. Cell. Biochem. 2015, 117, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Tsuruo, T.; Iida-Saito, H.; Kawabata, H.; Oh-Hara, T.; Hamada, H.; Utakoji, T. Characteristics of resistance to adriamycin in human myelogenous leukemia K562 resistant to adriamycin and in isolated clones. Jpn. J. Cancer Res. 1986, 77, 682–692. [Google Scholar]
- Grosso, S.; Puissant, A.; Dufies, M.; Colosetti, P.; Jacquel, A.; Lebrigand, K.; Barbry, P.; Deckert, M.; Cassuto, J.P.; Mari, B.; et al. Gene expression profiling of imatinib and PD166326-resistant CML cell lines identifies Fyn as a gene associated with resistance to BCR-ABL inhibitors. Mol. Cancer 2009, 8, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Robert, G.; Fenouille, N.; Luciano, F.; Cassuto, J.-P.; Raynaud, S.; Auberger, P. Resveratrol Promotes Autophagic Cell Death in Chronic Myelogenous Leukemia Cells via JNK-Mediated p62/SQSTM1 Expression and AMPK Activation. Cancer Res. 2010, 70, 1042–1052. [Google Scholar] [CrossRef]
- Hu, L.; Cao, D.; Li, Y.; He, Y.; Guo, K. Resveratrol sensitized leukemia stem cell-like KG-1a cells to cytokine-induced killer cells-mediated cytolysis through NKG2D ligands and TRAIL receptors. Cancer Biol. 2012, 13, 516–526. [Google Scholar] [CrossRef]
- Li, Y.-J.; Xu, H.-J. Relationship between apoptotic effect of Resveratrol on KG-1 cells and expression of bcl-2/bax. Zhongguo shi yan xue ye xue za zhi 2008, 16, 1026–1029. [Google Scholar]
- Lanotte, M.; Martin-Thouvenin, V.; Najman, S.; Balerini, P.; Valensi, F.; Berger, R. NB4, a maturation inducible cell line with t (15; 17) marker isolated from a human acute promyelocytic leukemia (M3). Blood 1991, 77, 1080–1086. [Google Scholar] [CrossRef]
- Quentmeier, H.; Martelli, M.P.; Dirks, W.G.; Bolli, N.; Liso, A.; MacLeod, R.A.F.; Nicoletti, I.; Mannucci, R.; Pucciarini, A.; Bigerna, B.; et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia 2005, 19, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulou, T.; Nakamoto, B.; Kurachi, S.; Tweeddale, M.; Messner, H. Surface Antigenic Profile and Globin Phenotype of Two New Human Erythroleukemia Lines: Characterization and Interpretations. Blood 1988, 72, 1029–1038. [Google Scholar] [CrossRef]
- Chen, S.; Xue, Y.; Zhang, X.; Wu, Y.; Pan, J.; Wang, Y.; Ceng, J. A new human acute monocytic leukemia cell line SHI-1 with t(6;11)(q27;q23), p53 gene alterations and high tumorigenicity in nude mice. Haematology 2005, 90, 766–775. [Google Scholar]
- Zhu, G.; Shen, Q.; Jiang, H.; Ji, O.; Zhu, L.; Zhang, L. Curcumin inhibited the growth and invasion of human monocytic leu-kaemia SHI-1 cells in vivo by altering MAPK and MMP signalling. Pharm. Biol. 2020, 58, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Han, M.H.; Lee, W.S.; Jung, J.H.; Jeong, J.; Park, C.; Kim, H.J.; Kim, G.; Jung, J.M.; Kwon, T.K.; Kim, G.Y.; et al. Polyphenols isolated from Allium cepa, L. induces apoptosis by suppressing IAP-1 through inhibiting PI3K/Akt signaling pathways in human leu-kemic cells. Food Chem. Toxicol. 2013, 62, 382–389. [Google Scholar] [CrossRef]
- Song, J.; Seo, Y.; Park, H. Pinosylvin enhances leukemia cell death via down-regulation of AMPKα expression. Phytother. Res. 2018, 32, 2097–2104. [Google Scholar] [CrossRef]
- Díaz-Rodríguez, E.; Álvarez-Fernández, S.; Chen, X.; Paiva, B.D.L.; López-Pérez, R.; García-Hernández, J.L.; Miguel, J.F.S.; Pandiella, A. Deficient Spindle Assembly Checkpoint in Multiple Myeloma. PloS ONE 2011, 6, e27583. [Google Scholar] [CrossRef] [PubMed]
- Osanai, K.; Landis-Piwowar, K.R.; Dou, Q.P.; Chan, T.H. A para-amino substituent on the D-ring of green tea poly-phenol epigallocatechin-3-gallate as a novel proteasome inhibitor and cancer cell apoptosis inducer. Bioorganic Med. Chem. 2007, 15, 5076–5082. [Google Scholar] [CrossRef]
- Zhamanbayeva, G.T.; Aralbayeva, A.N.; Murzakhmetova, M.K.; Tuleukhanov, S.T.; Danilenko, M. Cooperative an-ti-proliferative and differentiation-enhancing activity of medicinal plant extracts in acute myeloid leukemia cells. Biomed. Pharmacother. 2016, 82, 80–89. [Google Scholar] [CrossRef]
- Shishodia, S.; Sethi, G.; Ahn, K.S.; Aggarwal, B.B. Guggulsterone inhibits tumor cell proliferation, induces S-phase arrest, and promotes apoptosis through activation of c-Jun N-terminal kinase, suppression of Akt pathway, and downregulation of antiapoptotic gene products. Biochem. Pharm. 2007, 74, 118–130. [Google Scholar] [CrossRef]
- Kang, S.; Jeong, S.; Kim, S.; Kim, J.; Jung, J.H.; Koh, W.; Kim, J.H.; Kim, D.K.; Chen, C.Y.; Kim, S.H. Icariside II induces apoptosis in U937 acute myeloid leukemia cells: Role of inactivation of STAT3-related signaling. PLoS ONE 2012, 7, e28706. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.-H.; Chang, L.-S. Piceatannol induces Fas and FasL up-regulation in human leukemia U937 cells via Ca2+/p38α MAPK-mediated activation of c-Jun and ATF-2 pathways. Int. J. Biochem. Cell Biol. 2010, 42, 1498–1506. [Google Scholar] [CrossRef]
- Guha, P.; Dey, A.; Sen, R.; Chatterjee, M.; Chattopadhyay, S.; Bandyopadhyay, S.K. Intracellular GSH depletion trig-gered mitochondrial Bax translocation to accomplish resveratrol-induced apoptosis in the U937 cell line. J. Pharmacol. Exp. Ther. 2011, 336, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Lee, W.S.; Han, M.; Song, K.S.; Hong, S.; Nagappan, A.; Kim, G.Y.; Kim, G.S.; Jung, J.M.; Ryu, C.H.; et al. Lonicera japonica Thunb. Induces caspa-se-dependent apoptosis through death receptors and suppression of AKT in U937 human leukemic cells. Phytother. Res. 2018, 32, 504–513. [Google Scholar] [CrossRef]
- Udhayakumar, V.; Brodeur, P.H.; Rajagopalan, M.; Zimmer, S.; Pollok, K.E.; Subbarao, B. Isolation and immunological characterization of a group of new B lymphomas from CBA mice. Clin. Immunol. Immunopathol. 1989, 51, 240–251. [Google Scholar] [CrossRef]
- Bange, E.; Timlin, C.; Kabel, C.; Svoboda, J.; Roeker, L.; Mato, A.R. Evidence for and Against Green Tea and Turmeric in the Management of Chronic Lymphocytic Leukemia. Clin. Lymphoma Myeloma Leuk. 2018, 18, e421–e426. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Zhang, W.; Sanderson, B.J.S. Selective Growth Inhibition of Human Leukemia and Human Lymphoblastoid Cells by Resveratrol via Cell Cycle Arrest and Apoptosis Induction. J. Agric. Food Chem. 2008, 56, 7572–7577. [Google Scholar] [CrossRef]
- Turrini, E.; Ferruzzi, L.; Fimognari, C. Potential Effects of Pomegranate Polyphenols in Cancer Prevention and Therapy. Oxidative Med. Cell. Longev. 2015, 2015, 1–19. [Google Scholar] [CrossRef]
- Mohan, A.; Narayanan, S.; Sethuraman, S.; Maheswari Krishnan, U. Combinations of plant polyphenols & anti-cancer mol-ecules: A novel treatment strategy for cancer chemotherapy. Anti-Cancer Agents Med. Chem. 2013, 13, 281–295. [Google Scholar]
- Todoric, J.; Antonucci, L.; Karin, M. Targeting Inflammation in Cancer Prevention and Therapy. Cancer Prev. Res. 2016, 9, 895–905. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Sethi, G.; Ahn, K.S.; Sandur, S.K.; Pandey, M.K.; Kunnumakkara, A.B.; Sung, B.; Ichikawa, H. Targeting sig-nal-transducer-and-activator-of-transcription-3 for prevention and therapy of cancer: Modern target but ancient solution. Ann. N. Y. Acad. Sci. 2006, 1091, 151–169. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Kordes, U.; Krappmann, D.; Heissmeyer, V.; Ludwig, W.D.; Scheidereit, C. Transcription factor NF-κB is constitutively acti-vated in acute lymphoblastic leukemia cells. Leukemia 2000, 14, 399–402. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, Z.; Qu, X.; Zhu, X.; Zhao, L.; Wei, R.; Guo, Q.; Sun, L.; Yin, X.; Zhang, Y.; et al. Roles of STAT3 in leukemia. Inter-Natl. J. Oncol. 2018, 53, 7–20. [Google Scholar]
- Baud, V.; Karin, M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef]
- Basseres, D.S.; Baldwin, A.S. Nuclear factor-κ B and inhibitor of κ B kinase pathways in oncogenic initiation and pro-gression. Oncogene 2006, 25, 6817–6830. [Google Scholar] [CrossRef]
- Aggarwal, B.B. Nuclear factor-κB: The enemy within. Cancer Cell 2004, 6, 203–208. [Google Scholar] [CrossRef]
- Dutta, J.; Fan, Y.; Gupta, N.; Fan, G.; Gelinas, C. Current insights into the regulation of programmed cell death by NF-κB. Oncogene 2006, 25, 6800–6816. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Kamata, H.; Karin, M. IKK/NF-κB signaling: Balancing life and death–a new approach to cancer therapy. J. Clin. Investig. 2005, 115, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z. NF-κB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Hazan-Halevy, I.; Harris, D.M.; Li, P.; Ferrajoli, A.; Faderl, S.; Keating, M.J.; Estrov, Z. STAT-3 activates NF-κB in chronic lym-phocytic leukemia cells. Mol. Cancer Res. 2011, 9, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Karin, M. The IκB kinase–a bridge between inflammation and cancer. Cell Res. 2008, 18, 334–342. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- McFarland, B.C.; Gray, G.K.; Nozell, S.E.; Hong, S.W.; Benveniste, E.N. Activation of the NF-κB pathway by the STAT3 in-hibitor JSI-124 in human glioblastoma cells. Mol. Cancer Res. 2013, 11, 494–505. [Google Scholar] [CrossRef]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-κB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Bollrath, J.; Greten, F.R. IKK/NF-κB and STAT3 pathways: Central signalling hubs in inflammation-mediated tumour promo-tion and metastasis. EMBO Rep. 2009, 10, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Joyce, D.; Albanese, C.; Steer, J.; Fu, M.; Bouzahzah, B.; Pestell, R.G. NF-κB and cell-cycle regulation: The cyclin con-nection. Cytokine Growth Factor Rev. 2001, 12, 73–90. [Google Scholar] [CrossRef]
- Imbert, V.; Peyron, J. NF-κB in hematological malignancies. Biomedicines 2017, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Carvalho, G.; Fabre, C.; Grosjean, J.; Fenaux, P.; Kroemer, G. Targeting NF-κB in hematologic malignancies. Cell Death Differ. 2006, 13, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Hewamana, S.; Alghazal, S.; Lin, T.T.; Clement, M.; Jenkins, C.; Guzman, M.L.; Jordan, C.T.; Neelakantan, S.; Crooks, P.A.; Burnett, A.K.; et al. The NF-κB subunit Rel A is asso-ciated with in vitro survival and clinical disease progression in chronic lymphocytic leukemia and represents a promising therapeutic target. J. Am. Soc. Hematol. 2008, 111, 4681–4689. [Google Scholar]
- Cuni, S.; Perez-Aciego, P.; Perez-Chacon, G.; Vargas, J.A.; Sanchez, A.; Martin-Saavedra, F.M.; Ballester, S.; Garcia-Marco, J.; Jorda, J.; Durantez, A. A sustained acti-vation of PI3K/NF-κ B pathway is critical for the survival of chronic lymphocytic leukemia B cells. Leukemia 2004, 18, 1391–1400. [Google Scholar] [CrossRef]
- Schuh, K.; Avots, A.; Tony, H.-P.; Serfling, E.; Kneitz, C. Nuclear NF-ATp is a Hallmark of Unstimulated B Cells from B-CLL Patients. Leuk. Lymphoma 1996, 23, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Delaval, B.; Lelièvre, H.; Birnbaum, D. Myeloproliferative disorders: The centrosome connection. Leukemia 2005, 19, 1739–1744. [Google Scholar] [CrossRef]
- Reuther, J.Y.; Reuther, G.W.; Cortez, D.; Pendergast, A.M.; Baldwin, A.S. A requirement for NF-κB activation in Bcr–Abl-mediated transformation. Genes Dev. 1998, 12, 968–981. [Google Scholar] [CrossRef]
- Hamdane, M.; David-Cordonnier, M.; D’Halluin, J.C. Activation of p65 NF-κB protein by p210 BCR–ABL in a myeloid cell line (P210 BCR–ABL activates p65 NF-κB). Oncogene 1997, 15, 2267–2275. [Google Scholar] [CrossRef]
- Davoudi, Z.; Akbarzadeh, A.; Rahmatiyamchi, M.; Movassaghpour, A.A.; Alipour, M.; Nejati-Koshki, K.; Sadeghi, Z.; Da-riushnejad, H.; Zarghami, N. Molecular target therapy of AKT and NF-kB signaling pathways and multidrug resistance by specific cell penetrating inhibitor peptides in HL-60 cells. Asian Pac. J. Cancer Prev. 2014, 15, 4353–4358. [Google Scholar] [CrossRef] [PubMed]
- Notarbartolo, M.; Cervello, M.; Dusonchet, L.; Cusimano, A.; D’Alessandro, N. Resistance to diverse apoptotic triggers in multidrug resistant HL60 cells and its possible relationship to the expression of P-glycoprotein, Fas and of the novel anti-apoptosis factors IAP (inhibitory of apoptosis proteins). Cancer Lett. 2002, 180, 91–101. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, M.M.; Sung, B.; Yadav, V.R.; Kannappan, R.; Aggarwal, B.B. NF-κB addiction and its role in cancer:‘one size does not fit all’. Oncogene 2011, 30, 1615–1630. [Google Scholar] [CrossRef]
- Mankan, A.K.; Greten, F.R. Inhibiting signal transducer and activator of transcription 3: Rationality and rationale design of inhibitors. Expert Opin. Investig. Drugs 2011, 20, 1263–1275. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar]
- Darnell, J.E.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extra-cellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Redell, M.S.; Ruiz, M.J.; Alonzo, T.A.; Gerbing, R.B.; Tweardy, D.J. Stat3 signaling in acute myeloid leukemia: Lig-and-dependent and-independent activation and induction of apoptosis by a novel small-molecule Stat3 inhibitor. Blood J. Am. Soc. Hematol. 2011, 117, 5701–5709. [Google Scholar] [CrossRef]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in re-sponse to IL-6 and activates transcription by binding to NFκB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef]
- Braunstein, J.; Brutsaert, S.; Olson, R.; Schindler, C. STATs Dimerize in the Absence of Phosphorylation. J. Biol. Chem. 2003, 278, 34133–34140. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Harris, D.; Liu, Z.; Liu, J.; Li, P.; Chen, X.; Shanker, S.; Ferrajoli, A.; Keating, M.J.; Estrov, Z. STAT3 is constitu-tively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 2010, 115, 2852–2863. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zhong, Z.; Darnell, J.E. Maximal activation of transcription by statl and stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Groner, B.; Lucks, P.; Borghouts, C. The function of Stat3 in tumor cells and their microenvironment. In Seminars in Cell and Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 19, pp. 4341–4350. [Google Scholar]
- Kubo, M.; Hanada, T.; Yoshimura, A. Suppressors of cytokine signaling and immunity. Nat. Immunol. 2003, 4, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 Balance by Stat3 Signaling in the Tumor Microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.Y. STAT3 in immune responses and inflammatory bowel diseases. Cell Res. 2006, 16, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef] [PubMed]
- Haura, E.B.; Turkson, J.; Jove, R. Mechanisms of Disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat. Clin. Pr. Oncol. 2005, 2, 315–324. [Google Scholar] [CrossRef]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of un-phosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar] [PubMed]
- Darnell, J.E. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar]
- Mohassab, A.M.; Hassan, H.A.; Abdelhamid, D.; Abdel-Aziz, M. STAT3 transcription factor as target for anti-cancer therapy. Pharm. Rep. 2020, 72, 1101–1124. [Google Scholar] [CrossRef]
- Boudný, M.; Trbušek, M. The Important Role of STAT3 in Chronic Lymphocytic Leukaemia Biology. Klin. Onkol. 2020, 33, 32–38. [Google Scholar] [CrossRef]
- Liu, F.; Jia, L.; Wang, P.; Wang, H.; Farren, T.W.; Agrawal, S.G. STAT3 and NF-κB cooperatively control in vitro spon-taneous apoptosis and poor chemo-responsiveness in patients with chronic lymphocytic leukemia. Oncotarget 2016, 7, 32031. [Google Scholar] [CrossRef] [PubMed]
- Spiekermann, K.; Biethahn, S.; Wilde, S.; Hiddemann, W.; Alves, F. Constitutive activation of STAT transcription factors in acute myelogenous leukemia. Eur. J. Haematol. 2001, 67, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Bi, C.; Janakakumara, J.V.; Liu, S.; Chng, W.; Tay, K.; Poon, L.F.; Xie, Z.; Palaniyandi, S.; Yu, H.; et al. Enhanced activation of STAT pathways and over-expression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood 2009, 113, 4052–4062. [Google Scholar] [CrossRef]
- Bewry, N.N.; Nair, R.R.; Emmons, M.F.; Boulware, D.; Pinilla-Ibarz, J.; Hazlehurst, L.A. Stat3 contributes to resistance toward BCR-ABL inhibitors in a bone marrow microenvironment model of drug resistance. Mol. Cancer 2008, 7, 3169–3175. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Frelin, C.; Imbert, V.; Griessinger, E.; Peyron, A.; Rochet, N.; Philip, P.; Dageville, C.; Sirvent, A.; Hummelsberger, M.; Bérard, E.; et al. Targeting NF-κB activation via pharma-cologic inhibition of IKK2-induced apoptosis of human acute myeloid leukemia cells. Blood 2005, 105, 804–811. [Google Scholar] [CrossRef]
- Guzman, M.L.; Swiderski, C.F.; Howard, D.S.; Grimes, B.A.; Rossi, R.M.; Szilvassy, S.J.; Jordan, C.T. Preferential induction of apoptosis for primary human leukemic stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16220–16225. [Google Scholar] [CrossRef]
- Xiao, X.; Jiang, K.; Xu, Y.; Peng, H.; Wang, Z.; Liu, S.; Zhang, G. (−)-Epigallocatechin-3-gallate induces cell apoptosis in chronic myeloid leukaemia by regulating Bcr/Abl-mediated p38-MAPK/JNK and JAK 2/STAT 3/AKT signalling pathways. Clin. Exp. Pharmacol. Physiol. 2019, 46, 126–136. [Google Scholar] [CrossRef]
- Udensi, U.K.; Tchounwou, P.B. Dual effect of oxidative stress on leukemia cancer induction and treatment. J. Exp. Clin. Cancer Res. 2014, 33, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Moskaug, J.Ø.; Carlsen, H.; Myhrstad, M.C.W.; Blomhoff, R. Polyphenols and glutathione synthesis regulation. Am. J. Clin. Nutr. 2005, 81, 277S–283S. [Google Scholar] [CrossRef]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of Glutathione in Cancer Progression and Chemoresistance. Oxidative Med. Cell. Longev. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Irwin, M.E.; Rivera-Del Valle, N.; Chandra, J. Redox control of leukemia: From molecular mechanisms to therapeutic oppor-tunities. Antioxid. Redox Signal. 2013, 18, 1349–1383. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Maraldi, T.; Prata, C.; Sega, F.V.D.; Caliceti, C.; Zambonin, L.; Fiorentini, D.; Hakim, G. NAD(P)H oxidase isoform Nox2 plays a prosurvival role in human leukaemia cells. Free. Radic. Res. 2009, 43, 1111–1121. [Google Scholar] [CrossRef]
- Kwon, D.H.; Cha, H.; Lee, H.; Hong, S.; Park, C.; Park, S.; Kim, G.Y.; Kim, S.; Kim, H.S.; Hwang, H.J.; et al. Protective Effect of Glutathione against Oxidative Stress-induced Cytotoxicity in RAW 264.7 Macrophages through Activating the Nuclear Factor Erythroid 2-Related Fac-tor-2/Heme Oxygenase-1 Pathway. Antioxidants 2019, 8, 82. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: Signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-González, J.; Wilkins-Rodríguez, A.A.; Gutiérrez-Kobeh, L. Role of glutathione, ROS, and Bcl-xL in the inhibition of apoptosis of monocyte-derived dendritic cells by Leishmania mexicana promastigotes. Parasitol. Res. 2018, 117, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharm. 2014, 5, 196. [Google Scholar] [CrossRef]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef]
- Kearns, P.; Pieters, R.; Rottier, M.; Veerman, A.; Schmiegalow, K.; Pearson, A. Hall AG. Glutathione in childhood acute leu-kaemias. In Drug Resistance in Leukemia and Lymphoma III. Advances in Experi-Mental Medicine and Biology; Kaspers, G.J.L., Pieters, R., Veerman, A.J.P., Eds.; Springer: Boston, MA, USA, 1999; Volume 457, pp. 211–216. [Google Scholar]
- Ortega, A.L.; Mena, S.; Estrela, J.M. Glutathione in Cancer Cell Death. Cancers 2011, 3, 1285–1310. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 2016, 21, 648–657. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferrop-totic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Chen, H.H.W.; Kuo, M.T. Role of Glutathione in the Regulation of Cisplatin Resistance in Cancer Chemotherapy. Met. Drugs 2010, 2010, 1–7. [Google Scholar] [CrossRef]
- Zhang, K.; Mack, P.; Wong, K.P. Glutathione-related mechanisms in cellular resistance to anticancer drugs. Int. J. Oncol. 1998, 12, 871–953. [Google Scholar] [CrossRef]
- Baguley, B.C. Multidrug resistance in cancer. Mol. Biol. 2010, 596, 1–14. [Google Scholar]
- Cole, S.P.; Deeley, R.G. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharm. Sci. 2006, 27, 438–446. [Google Scholar] [CrossRef]
- Keppler, D. Multidrug resistance proteins (MRPs, ABCCs): Importance for pathophysiology and drug therapy. In Drug Transporters. Handbook of Experimental Pharmacology; Fromm, M., Kim, R., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 201, pp. 299–323. [Google Scholar]
- McLellan, L.I.; Wolf, C.R. Glutathione and glutathione-dependent enzymes in cancer drug resistance. Drug Resist. Updat. 1999, 2, 153–164. [Google Scholar] [CrossRef]
- Dethmers, J.K.; Meister, A. Glutathione export by human lymphoid cells: Depletion of glutathione by inhibition of its synthesis decreases export and increases sensitivity to irradiation. Proc. Natl. Acad. Sci. USA 1981, 78, 7492–7496. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Coppola, S.; Ghibelli, L.; Cho, S.H.; Kagawa, S.; Spurgers, K.B.; Brisbay, S.M.; Roth, J.A.; Meyn, R.E.; Fang, B.; et al. GSH depletion enhances adenoviral bax-induced apoptosis in lung cancer cells. Cancer Gene 2004, 11, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Babich, H.; Schuck, A.G.; Weisburg, J.H.; Zuckerbraun, H.L. Research Strategies in the Study of the Pro-Oxidant Nature of Polyphenol Nutraceuticals. J. Toxicol. 2011, 2011, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hsuuw, Y.-D.; Chan, W.-H. Epigallocatechin Gallate Dose-Dependently Induces Apoptosis or Necrosis in Human MCF-7 Cells. Ann. N. Y. Acad. Sci. 2007, 1095, 428–440. [Google Scholar] [CrossRef]
- Raza, H.; John, A. Green tea polyphenol epigallocatechin-3-gallate differentially modulates oxidative stress in PC12 cell compartments. Toxicol. Appl. Pharm. 2005, 207, 212–220. [Google Scholar] [CrossRef]
- Myhrstad, M.C.; Carlsen, H.; Nordström, O.; Blomhoff, R.; Moskaug, J.Ø. Flavonoids increase the intracellular gluta-thione level by transactivation of the γ- glutamylcysteine synthetase catalytical subunit promoter. Free Radic. Biol. Med. 2002, 32, 386–393. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assays. Assay Guidance Manual; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2016. [Google Scholar]
- Otto, W. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Intracellular control of cell-cycle events. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Vermeulen, K.; Van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and thera-peutic targets in cancer. Cell Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; De Bruin, R.A.M. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995, 9, 1149–1163. [Google Scholar] [CrossRef]
- Carnero, A.; Hannon, G.J. The INK4 family of CDK inhibitors. In Cyclin Dependent Kinase (CDK) Inhibitors. Current Topics in Microbiology and Immunology; Vogt, P.K., Reed, S.I., Eds.; Springer: Berlin/Heidelberg, Germany, 1998; Volume 227, pp. 43–55. [Google Scholar]
- Harper, J.W.; Elledge, S.J.; Keyomarsi, K.; Dynlacht, B.; Tsai, L.; Zhang, P.; Dobrowolski, S.; Bai, C.; Connell-Crowley, L.; Swindell, E. Inhibition of cyclin-dependent kinases by p21. Mol. Biol. Cell 1995, 6, 387–400. [Google Scholar] [CrossRef]
- Lee, M.H.; Reynisdottir, I.; Massague, J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev. 1995, 9, 639–649. [Google Scholar] [CrossRef]
- Goodger, N.; Gannon, J.; Hunt, T.; Morgan, P. Cell cycle regulatory proteins—An overview with relevance to oral cancer. Oral Oncol. 1997, 33, 61–73. [Google Scholar] [CrossRef]
- Hajleh, M.A.; Al-Dujaili, A. Anti-cancer activity of pomegranate and its biophenols; general review. EC Nutr. 2016, 6, 28–52. [Google Scholar]
- Dahlawi, H.; Jordan-Mahy, N.; Clench, M.R.; Le Maitre, C.L. Bioactive Actions of Pomegranate Fruit Extracts on Leukemia Cell Lines In Vitro Hold Promise for New Therapeutic Agents for Leukemia. Nutr. Cancer 2012, 64, 100–110. [Google Scholar] [CrossRef]
- Senderowicz, A.M. Flavopiridol: The First Cyclin-Dependent Kinase Inhibitor in Human Clinical Trials. Investig. New Drugs 1999, 17, 313–320. [Google Scholar] [CrossRef]
- Sedlacek, H. Mechanisms of action of flavopiridol. Crit. Rev. Oncol. 2001, 38, 139–170. [Google Scholar] [CrossRef]
- Shapiro, G.I. Preclinical and Clinical Development of the Cyclin-Dependent Kinase Inhibitor Flavopiridol. Clin. Cancer Res. 2004, 10, 4270–4275. [Google Scholar] [CrossRef]
- Zaman, S.; Wang, R.; Gandhi, V. Targeting the apoptosis pathway in hematologic malignancies. Leuk. Lymphoma 2014, 55, 1980–1992. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Bhardwaj, A.; Aggarwal, R.S.; Seeram, N.P.; Shishodia, S.; Takada, Y. Role of resveratrol in prevention and therapy of cancer: Preclinical and clinical studies. Anticancer. Res. 2004, 24, 2783–2840. [Google Scholar]
- Goldar, S.; Khaniani, M.S.; Derakhshan, S.M.; Baradaran, B. Molecular mechanisms of apoptosis and roles in cancer devel-opment and treatment. Asian Pac. J. Cancer Prev. 2015, 16, 2129–2144. [Google Scholar] [CrossRef]
- Yamada, A.; Arakaki, R.; Saito, M.; Kudo, Y.; Ishimaru, N. Dual role of Fas/FasL-mediated signal in peripheral im-mune tol-erance. Front. Immunol. 2017, 8, 403. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Liu, H.; Su, D.; Zhang, J.; Ge, S.; Li, Y.; Wang, F.; Gravel, M.; Roulston, A.; Song, Q.; Xu, W.; et al. Improvement of pharmacokinetic profile of TRAIL via Tri-mer-Tag enhances its antitumor activity in vivo. Sci. Rep. 2017, 7, 8953. [Google Scholar] [CrossRef]
- Athar, M.; Back, J.H.; Kopelovich, L.; Bickers, D.R.; Kim, A.L. Multiple molecular targets of resveratrol: An-ti-carcinogenic mechanisms. Arch. Biochem. Biophys. 2009, 486, 95–102. [Google Scholar] [CrossRef]
- Torello, C.O.; Shiraishi, R.N.; Della Via, F.I.; De Castro, T.C.L.; Longhini, A.L.; Santos, I.; Bombeiro, A.L.; Silva, C.L.A.; Queiroz, M.L.D.S.; Rego, E.M.; et al. Reactive oxygen species production triggers green tea-induced anti-leukaemic effects on acute promyelocytic leukaemia model. Cancer Lett. 2018, 414, 116–126. [Google Scholar] [CrossRef]
- Maruszewska, A.; Tarasiuk, J. Antitumour effects of selected plant polyphenols, gallic acid and ellagic acid, on sensi-tive and multidrug-resistant leukaemia HL60 cells. Phytother. Res. 2019, 33, 1208–1221. [Google Scholar] [CrossRef] [PubMed]
- Kundu, T.; Dey, S.; Roy, M.; Siddiqi, M.; Bhattacharya, R. Induction of apoptosis in human leukemia cells by black tea and its polyphenol theaflavin. Cancer Lett. 2005, 230, 111–121. [Google Scholar] [CrossRef]
- Xu, W.; Jing, L.; Wang, Q.; Lin, C.; Chen, X.; Diao, J.; Liu, Y.; Sun, X. Bax-PGAM5L-Drp1 complex is required for intrinsic apop-tosis execution. Oncotarget 2015, 6, 30017. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2011, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Papaliagkas, V.; Anogianaki, A.; Anogianakis, G.; Ilonidis, G. The proteins and the mechanisms of apoptosis: A mini-review of the fundamentals. Hippokratia 2007, 11, 108–113. [Google Scholar]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Lopez, J.; Tait, S. Mitochondrial apoptosis: Killing cancer using the enemy within. Br. J. Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef]
- Hassan, M.; Watari, H.; AbuAlmaaty, A.; Ohba, Y.; Sakuragi, N. Apoptosis and Molecular Targeting Therapy in Cancer. Biomed Res. Int. 2014, 2014, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef]
- Shahrabi, S.; Paridar, M.; Zeinvand-Lorestani, M.; Jalili, A.; Zibara, K.; Abdollahi, M.; Khosravi, A. Autophagy regulation and its role in normal and malignant hematopoiesis. J. Cell. Physiol. 2019, 234, 21746–21757. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.B.; Dhamija, S. Beclin 1 phosphorylation–at the center of autophagy regulation. Frontiers Cell Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy—Unanswered questions. J. Cell Sci. 2010, 124, 161–170. [Google Scholar] [CrossRef]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and Function of Autophagy during Cell Survival and Cell Death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef]
- Lorin, S.; Hamaï, A.; Mehrpour, M.; Codogno, P. Autophagy regulation and its role in cancer. Semin. Cancer Biol. 2013, 23, 361–379. [Google Scholar] [CrossRef]
- Rubinstein, A.D.; Kimchi, A. Life in the balance–a mechanistic view of the crosstalk between autophagy and apoptosis. J. Cell Sci. 2012, 125, 5259–5268. [Google Scholar] [CrossRef]
- Oral, O.; Oz-Arslan, D.; Itah, Z.; Naghavi, A.; Deveci, R.; Karacali, S.; Gozuacik, D. Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis 2012, 17, 810–820. [Google Scholar] [CrossRef]
- Betin, V.M.S.; Lane, J.D. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci. 2009, 122, 2554–2566. [Google Scholar] [CrossRef]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef]
- Zhu, H.; He, L. Beclin 1 Biology and its Role in Heart Disease. Curr. Cardiol. Rev. 2015, 11, 229–237. [Google Scholar] [CrossRef]
- Pagliarini, V.; Wirawan, E.; Romagnoli, A.; Ciccosanti, F.; Lisi, G.; Lippens, S.; Cecconi, F.; Fimia, G.M.; Vandenabeele, P.; Corazzari, M.; et al. Proteolysis of Ambra1 during apoptosis has a role in the inhibition of the autophagic pro-survival response. Cell Death Differ. 2012, 19, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 regulates autophagy and development of the nervous system. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The Role of Autophagy in Cancer: Therapeutic Implications. Mol. Cancer 2011, 10, 1533–1541. [Google Scholar] [CrossRef]
- Schmeisser, K.; Parker, J.A. Pleiotropic Effects of mTOR and Autophagy During Development and Aging. Front. Cell Dev. Biol. 2019, 7, 192. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef]
- Pattingre, S.; Espert, L.; Biard-Piechaczyk, M.; Codogno, P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie 2008, 90, 313–323. [Google Scholar] [CrossRef]
- Matsushita, M.; Suzuki, N.N.; Obara, K.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. Structure of Atg5· Atg16, a complex es-sential for autophagy. J. Biol. Chem. 2007, 282, 6763–6772. [Google Scholar] [CrossRef]
- Satoo, K.; Noda, N.N.; Kumeta, H.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. The structure of Atg4B–LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009, 28, 1341–1350. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Alegre, F.; Moragrega, Á.B.; Polo, M.; Marti-Rodrigo, A.; Esplugues, J.V.; Blas-Garcia, A.; Apostolova, N. Role of p62/SQSTM1 beyond autophagy: A lesson learned from drug-induced toxicity in vitro. Br. J. Pharm. 2018, 175, 440–455. [Google Scholar] [CrossRef]
- Zhang, Y.; Mun, S.R.; Linares, J.F.; Ahn, J.; Towers, C.G.; Ji, C.H.; Fitzwalter, B.E.; Holden, M.R.; Mi, W.; Shi, X.; et al. ZZ-dependent regulation of p62/SQSTM1 in autophagy. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Fuchs, Y.; Steller, H. Live to die another way: Modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apop-tosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef]
- Tohru, Y.; Andrew, T. Autophagy and cell death. Essays Biochem. 2013, 55, 105–117. [Google Scholar]
- Gump, J.M.; Thorburn, A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011, 21, 387–392. [Google Scholar] [CrossRef]
- Fitzwalter, B.E.; Thorburn, A. Recent insights into cell death and autophagy. FEBS J. 2015, 282, 4279–4288. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, Y.M.; Kaushik, V.; Azad, N.; Wright, C.; Rojanasakul, Y.; Odoherty, G.A.; Iyer, A.K.V. Autophagy-Induced Apoptosis in Lung Cancer Cells by a Novel Digitoxin Analog. J. Cell. Physiol. 2016, 231, 817–828. [Google Scholar] [CrossRef]
- Shen, S.; Kepp, O.; Kroemer, G. The end of autophagic cell death? Autophagy 2012, 8, 1–3. [Google Scholar] [CrossRef]
- Shen, H.-M.; Codogno, P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy 2011, 7, 457–465. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Cooper, K.F. Till Death Do Us Part: The Marriage of Autophagy and Apoptosis. Oxidative Med. Cell. Longev. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Delou, J.M.; Biasoli, D.; Borges, H.L. The Complex Link between Apoptosis and Autophagy: A Promising New Role for RB. An. Da Acad. Bras. De Ciências 2016, 88, 2257–2275. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, K.; Lei, Y.; Li, Q.; Nice, E.C.; Huang, C. Redox signaling: Potential arbitrator of autophagy and apoptosis in therapeutic response. Free. Radic. Biol. Med. 2015, 89, 452–465. [Google Scholar] [CrossRef]
- Su, M.; Mei, Y.; Sinha, S. Role of the Crosstalk between Autophagy and Apoptosis in Cancer. J. Oncol. 2013, 2013, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Radoshevich, L.; Murrow, L.; Chen, N.; Fernandez, E.; Roy, S.; Fung, C.; Debnath, J. ATG12 Conjugation to ATG3 Regulates Mitochondrial Homeostasis and Cell Death. Cell 2010, 142, 590–600. [Google Scholar] [CrossRef]
- Fu, L.-L.; Cheng, Y.; Liu, B. Beclin-1: Autophagic regulator and therapeutic target in cancer. Int. J. Biochem. Cell Biol. 2013, 45, 921–924. [Google Scholar] [CrossRef]
- Furuya, D.; Tsuji, N.; Yagihashi, A.; Watanabe, N. Beclin 1 augmented cis-diamminedichloroplatinum induced apop-tosis via enhancing caspase-9 activity. Exp. Cell Res. 2005, 307, 26–40. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-Induced Modulator of Autophagy, Is Critical for Apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef]
- Wirawan, E.; Walle, L.V.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- Luo, S.; Rubinsztein, D.C. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010, 17, 268–277. [Google Scholar] [CrossRef]
- Larsen, A.K.; Escargueil, A.E.; Skladanowski, A. Resistance mechanisms associated with altered intracellular distribution of anticancer agents. Pharmacol. Ther. 2000, 85, 217–229. [Google Scholar] [CrossRef]
- Gillet, J.-P.; Gottesman, M.M. Mechanisms of multidrug resistance in cancer. In Methods in Molecular Biology; Metzler, J.B., Ed.; Humana Press: New York, NY, USA, 2010; Volume 596, pp. 47–76. [Google Scholar]
- Leith, C. Multidrug resistance in leukemia. Curr. Opin. Hematol. 1998, 5, 287–291. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Pastan, I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem. 1993, 62, 385–427. [Google Scholar] [CrossRef]
- Consoli, U.; Santonocito, A.; Stagno, F.; Fiumara, P.; Privitera, A.; Parisi, G.; Giustolisi, G.M.; Pavone, B.; Palumbo, G.A.; Di Raimondo, F.; et al. Multidrug resistance mechanisms in chronic lymphocytic leukaemia. Br. J. Haematol. 2002, 116, 774–780. [Google Scholar] [CrossRef]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Hyde, S.C.; Emsley, P.; Hartshorn, M.J.; Mimmack, M.M.; Gileadi, U.; Pearce, S.R.; Gallagher, M.P.; Gill, D.R.; Hubbard, R.E.; Higgins, C.F. Structural model of ATP-binding proteing associated with cystic fibrosis, multidrug resistance and bacterial transport. Nature 1990, 346, 362–365. [Google Scholar] [CrossRef]
- Van Der Deen, M.; De Vries, E.G.; Timens, W.; Scheper, R.J.; Timmer-Bosscha, H.; Postma, D.S. ATP-binding cassette (ABC) transporters in normal and pathological lung. Respir. Res. 2005, 6, 59. [Google Scholar] [CrossRef]
- Izquierdo, M.A.; Shoemaker, R.H.; Flens, M.J.; Scheffer, G.L.; Wu, L.; Prather, T.R.; Scheper, R.J. Overlapping phenotypes of multidrug resistance among panels of human cancer-cell lines. Int. J. Cancer 1996, 65, 230–237. [Google Scholar] [CrossRef]
- Cao, H.; Ou, J.; Chen, L.; Zhang, Y.; Szkudelski, T.; Delmas, D.; Daglia, M.; Xiao, J. Dietary polyphenols and type 2 diabetes: Human Study and Clinical Trial. Crit. Rev. Food Sci. Nutr. 2019, 59, 3371–3379. [Google Scholar] [CrossRef] [PubMed]
- Alam, N.; Almoyad, M.; Huq, F. Polyphenols in Colorectal Cancer: Current State of Knowledge including Clinical Trials and Molecular Mechanism of Action. Biomed Res. Int. 2018, 2018, 1–29. [Google Scholar] [CrossRef]
- Cory, H.; Passarelli, S.; Szeto, J.; Tamez, M.; Mattei, J. The Role of Polyphenols in Human Health and Food Systems: A Mini-Review. Front. Nutr. 2018, 5, 87. [Google Scholar] [CrossRef]
- Piotrowska, H.; Kucinska, M.; Murias, M. Biological activity of piceatannol: Leaving the shadow of resveratrol. Mutat. Res. Mutat. Res. 2012, 750, 60–82. [Google Scholar] [CrossRef]
- Kouhpeikar, H.; Butler, A.E.; Bamian, F.; Barreto, G.E.; Majeed, M.; Sahebkar, A. Curcumin as a therapeutic agent in leukemia. J. Cell. Physiol. 2019, 234, 12404–12414. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
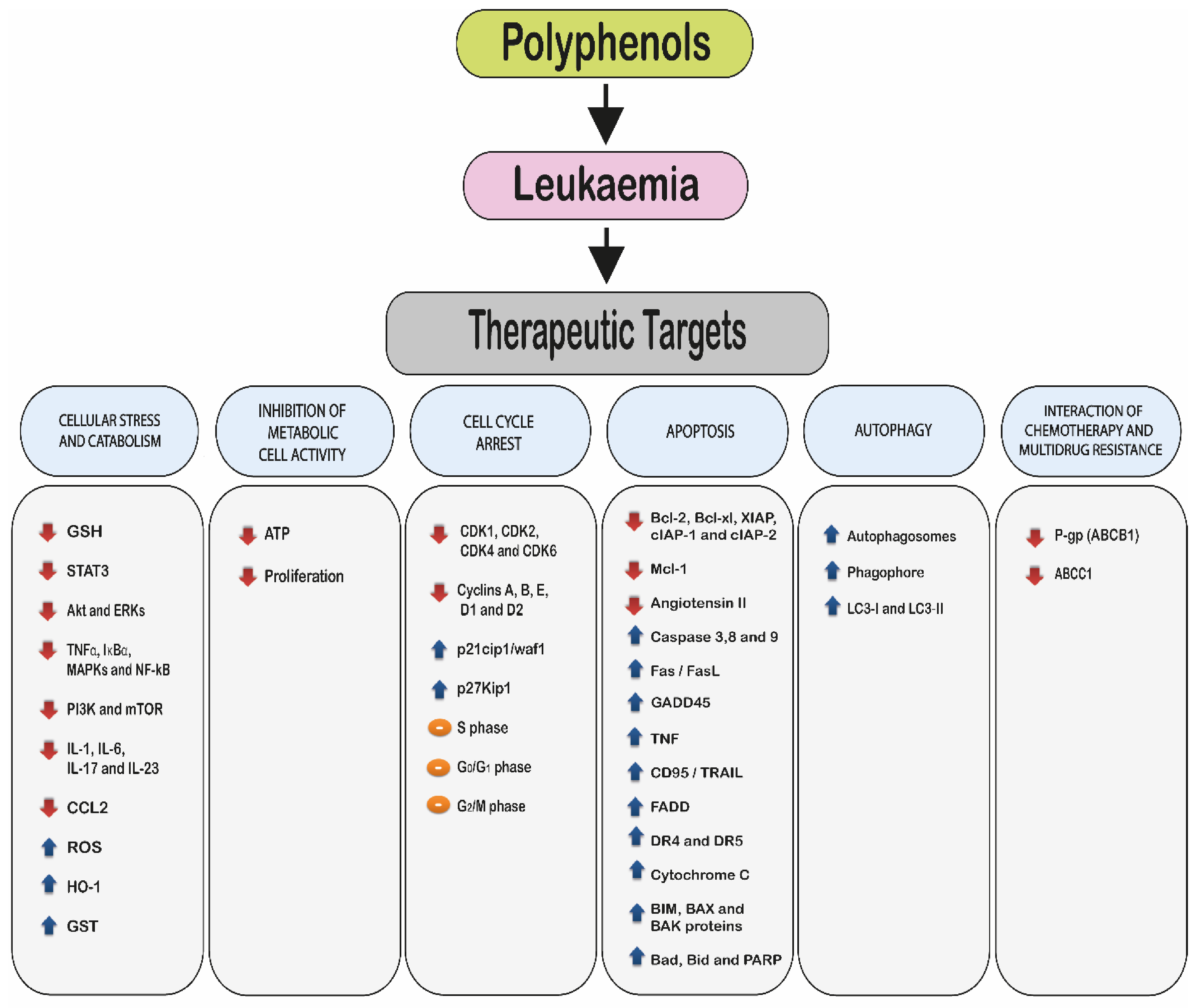{kind=link}
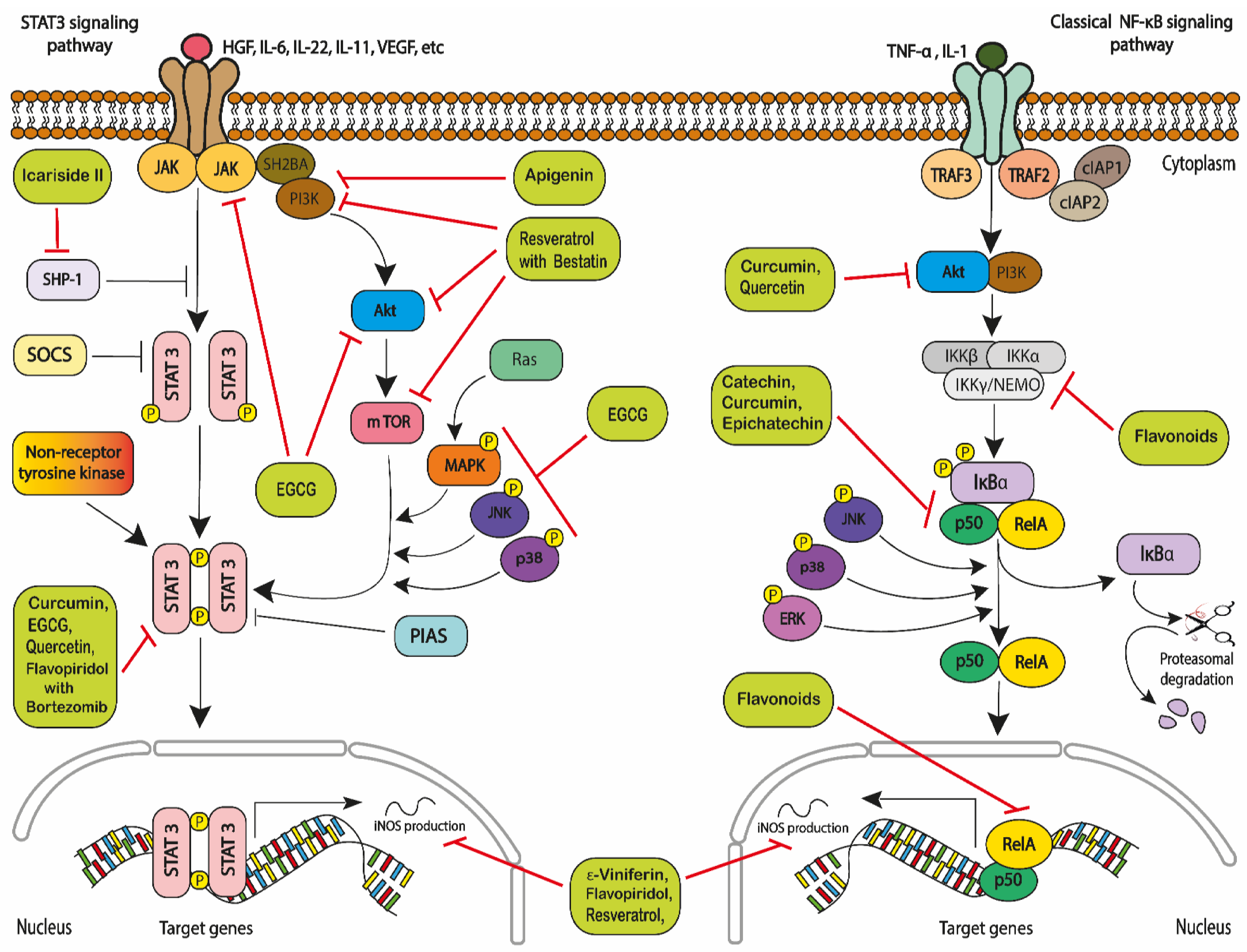{kind=link}
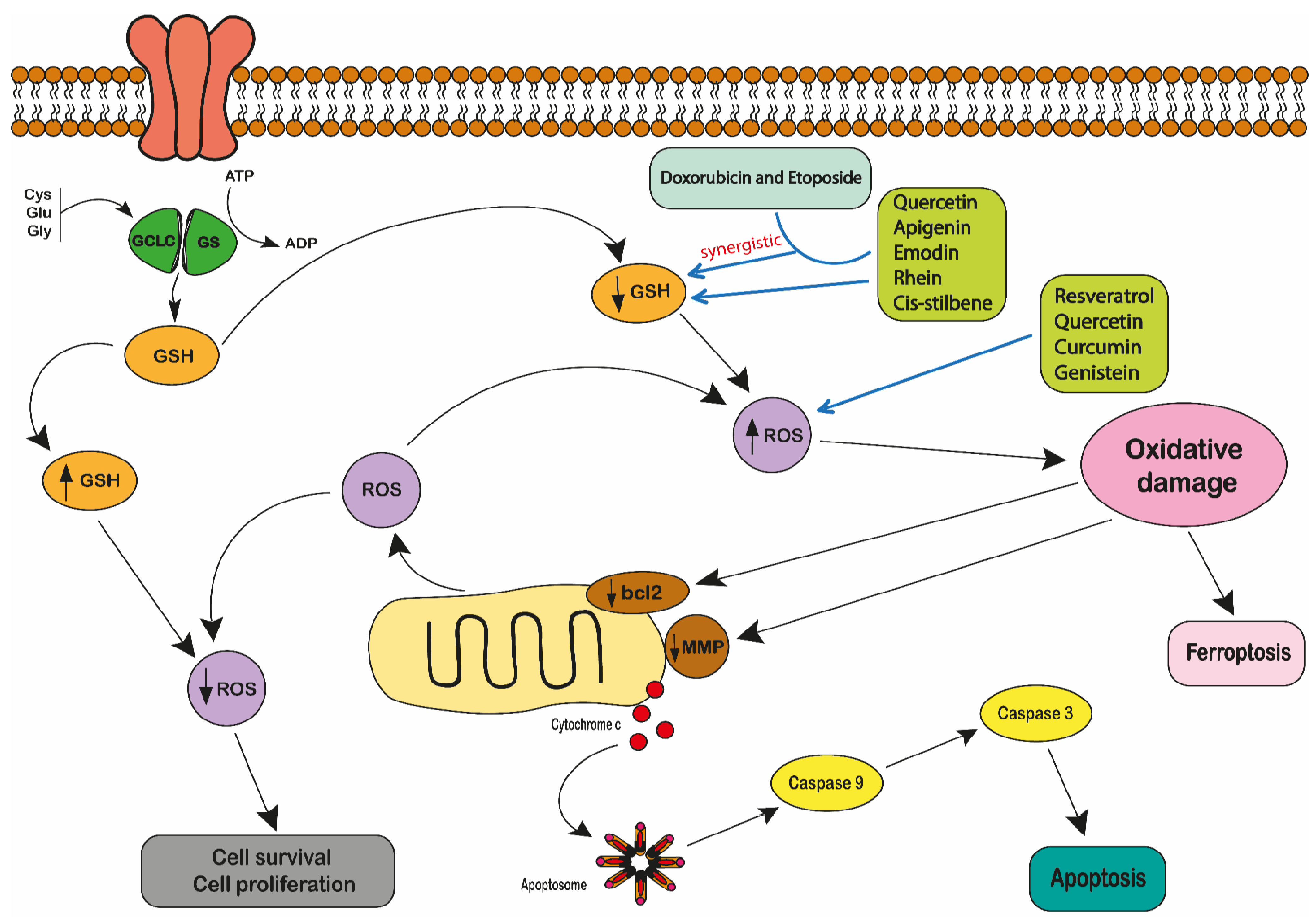{kind=link}
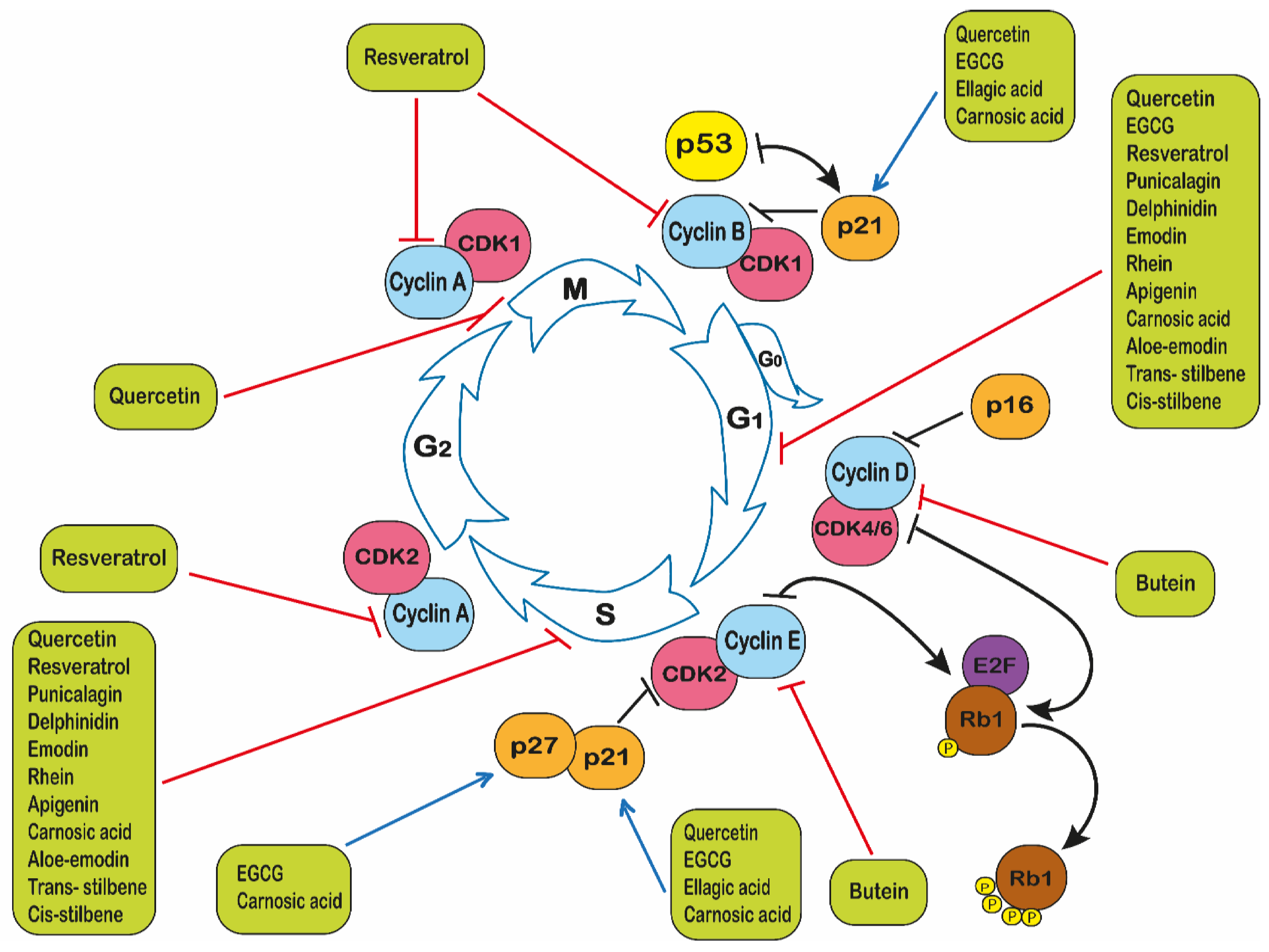{kind=link}
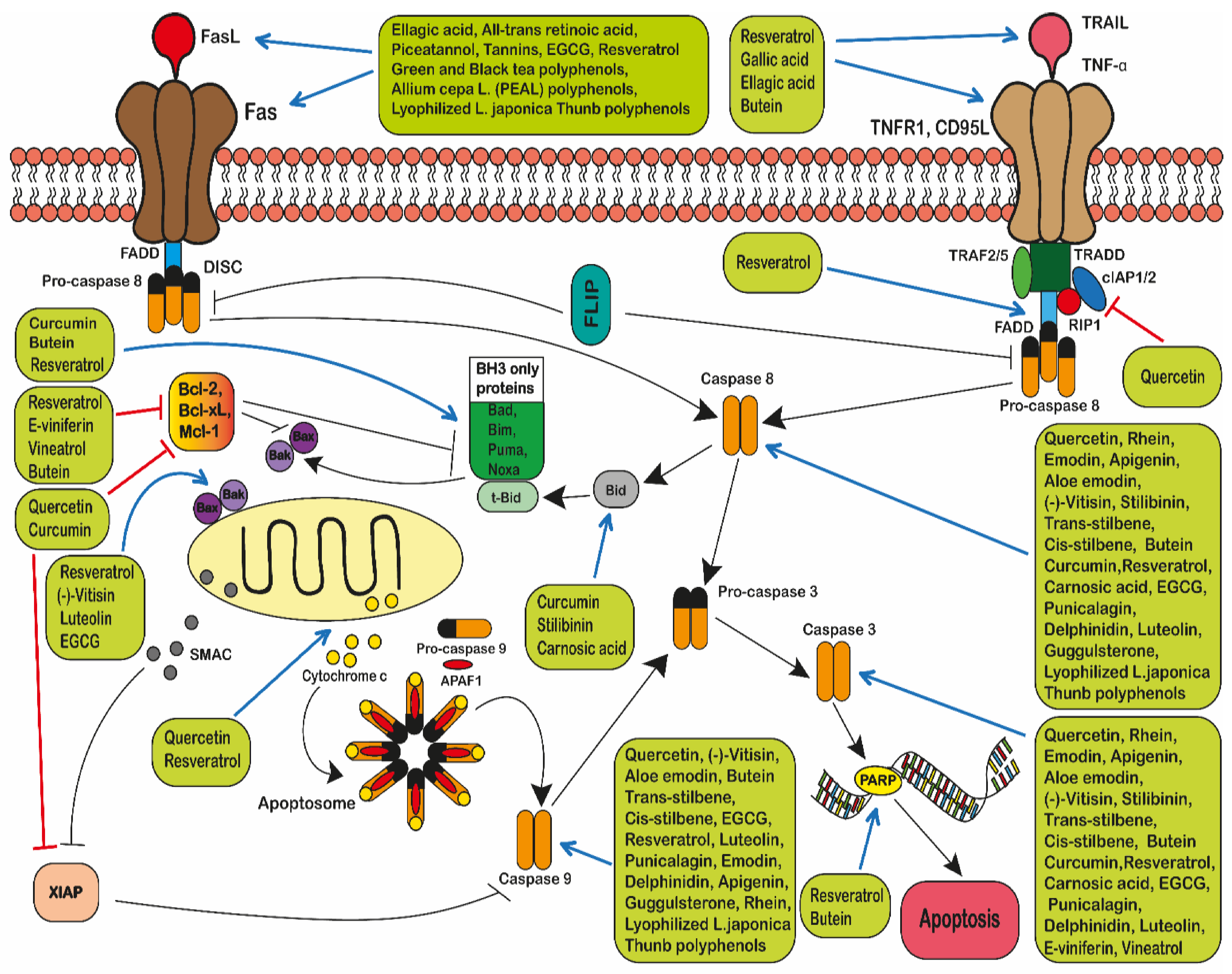{kind=link}
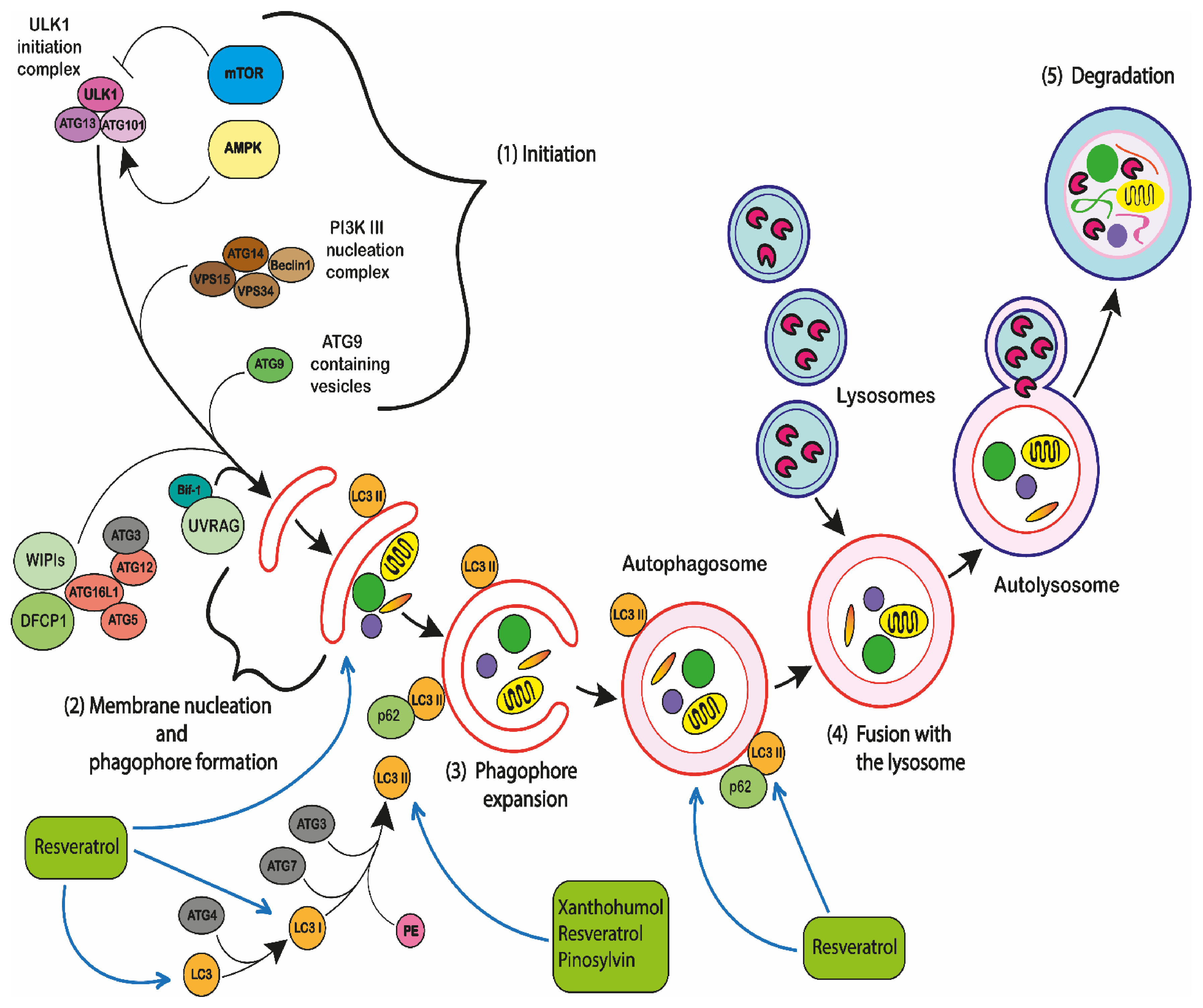{kind=link}
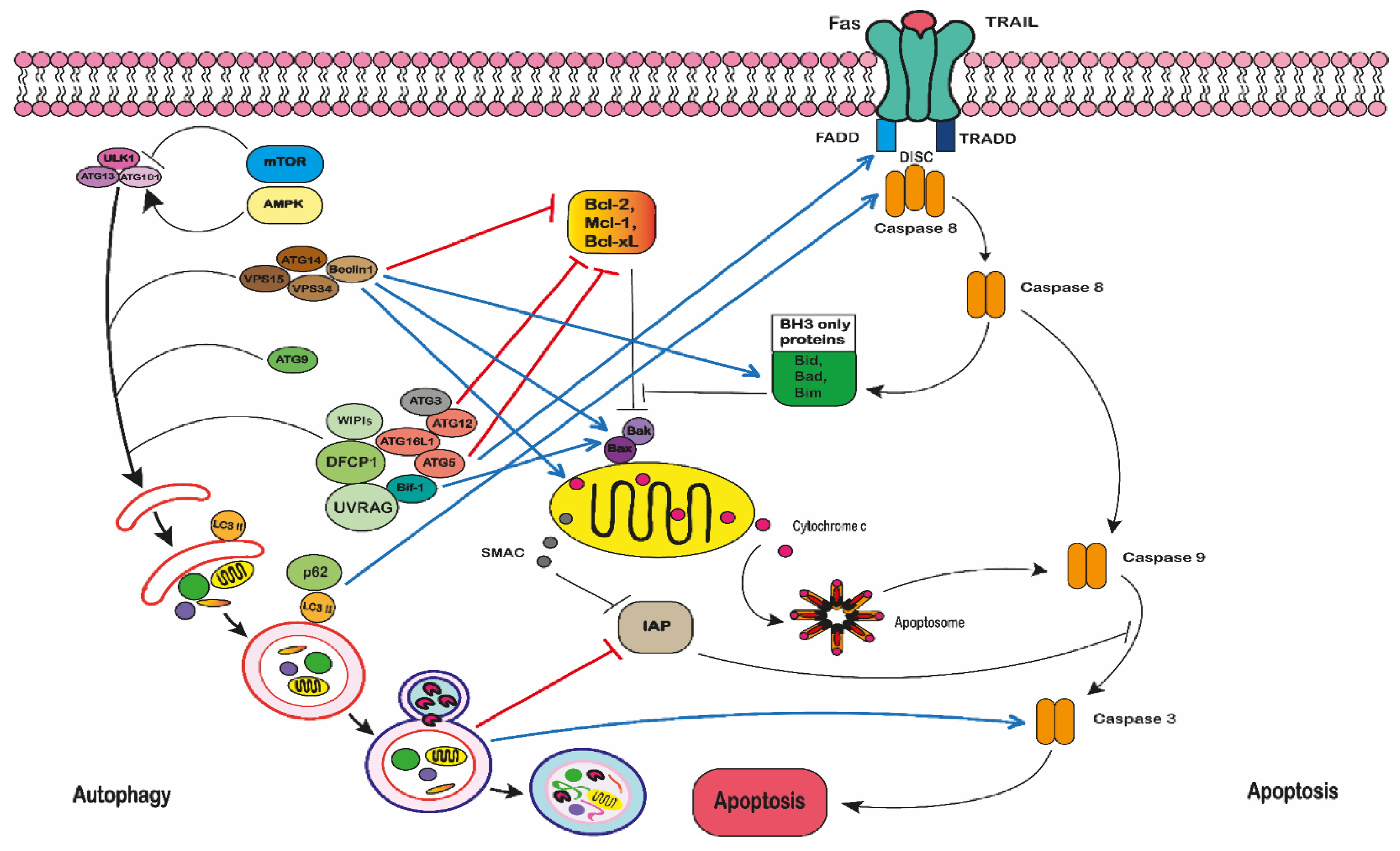{kind=link}
| CANCER TYPE | CELL LINE | DESCRIPTION | SPECIES | SOURCE OR REF | POLYPHENOLS |
|---|---|---|---|---|---|
| LYMPHOID LEUKAEMIA | 232B4 | Chronic lymphocytic leukaemia | Human | Wendel-Hansen et al. 1994 [60] | Quercetin [61] Resveratrol [61] |
| B-CLL | Chronic lymphocytic leukaemia | Human | Hoogendoorn et al. 2004 [62] | Curcumin [63,64] Epsilon-viniferin (ε-viniferin) [65] Flavopiridol [65] Quercetin [19] Resveratrol [66] | |
| CCRF-CEM | Acute lymphocytic leukaemia | Human | ATCC | Aloe-emodin [67] Apigenin [67] Delphinidin [68] Emodin [67] Flavonoids [69] Punicalagin [68] Quercetin [67,68] Rhein [67] Resveratrol [70] Trans-Stilbene [67] cis-Stilbene [67] Quercetin, apigenin, emodin, rhein or cis-stilbene plus doxorubicin or etoposide [24] Quercetin, apigenin, emodin, rhein, or cis-Stilbene plus chlorambucil, cisplatin or cyclophosphamide [71] Quercetin, apigenin, emodin, rhein, or cis-stilbene plus methotrexate, 6-mercaptopurine or 5-fluorouracil [23] EGCG, tannic acid or curcumin plus doxorubicin [72] | |
| CCRF-CEM-C7H2 | Acute lymphoblastic leukaemia | Human | Strasser-Wozak et al. 1995 [73] | Resveratrol [74] | |
| CEM | Acute lymphoblastic leukaemia | Human | ATCC | Resveratrol [75,76] | |
| CEM-C1-15 | Acute lymphoblastic leukaemia | Human | ATCC | Pinosylvin [77] Resveratrol [77] | |
| CEM-C7-14 | Acute lymphocytic leukaemia | Human | Gu et al. 2019 [78] | Pinosylvin [77] Resveratrol [77] | |
| ESKOL | B-lymphoblastoid hairy cell leukaemia cell lines | Human | Harvey et al. 1991 [79] | Resveratrol [66] e-Viniferin plus Vineatrol [80] | |
| HSB-2 | Acute lymphoblastic leukaemia | Human | ATCC | Resveratrol [81] | |
| Jurkat | Acute lymphocytic leukaemia | Human | ATCC | Aloe-emodin [67] Apigenin [67] Butein [82] Catechin [83] Emodin [67] Epichatechin [83] Epigallocatechin gallate [84] Flavonoids [83] Pinosylvin [77] Quercetin [67] Resveratrol [75,76,77,85,86,87] Rhein [67] trans- stilbene cis-Stilbene [24,67] Quercetin, apigenin, emodin, rhein, or cis-stilbene plus chlorambucil, cisplatin or cyclophosphamide [71] Quercetin, apigenin, emodin, rhein, or cis-Stilbene plus methotrexate, 6-mercaptopurine or 5-fluorouracil [23] Quercetin, apigenin, emodin, rhein, or cis-Stilbene plus doxorubicin or etoposide [24,67] | |
| MOLT-3 | Acute lymphocytic leukaemia | Human | ATCC | Delphinidin [68] Quercetin [68] | |
| MOLT-4 | Acute lymphocytic leukaemia | Human | ATCC | Pinosylvin [77] Quercetin [88,89] Resveratrol [76,77,90] Quercetin plus ellagic acid [88] | |
| MT-4 | Acute lymphoblastic leukaemia | Human | Fernandez et al. 2019 [91] | Butein [92] | |
| Nalm-6 | Acute lymphoblastic leukaemia | Human | ATCC | Resveratrol [66,75,76] | |
| REH | Acute lymphoblastic leukaemia | Human | ATCC | Resveratrol [75,76] | |
| RS4;11 | Acute lymphoblastic leukaemia | Human | ATCC | Resveratrol [75,76] | |
| SEM | Acute lymphoblastic leukaemia | Human | Greil et al. 1994 [93] | Resveratrol [75,76] | |
| SUP-B15 | Acute lymphocytic leukaemia | Human | ATCC | Resveratrol [87] | |
| TL-Oml | Acute lymphoblastic leukaemia | Human | Sugamura et al. 1984 [94] | Butein [92] | |
| WSU-CLL | Chronic lymphocytic leukaemia | Human | Mohammad et al. 1996 [95] | Resveratrol [66,96] e-Viniferin plus Vineatrol [80] | |
| L1210 | Lymphocytic leukaemia | Mouse | ATCC | Curcumin [97] Gallic acid [97] Quercetin [97,98] Tannic acid [97] Resveratrol [99] | |
| MYELOID LEUKAEMIA | AML-2/DX30, AML-2/DX100 AML-2/DX300 | Doxorubicin resistant acute myeloid leukaemia cell lines | Human | Kweon et al. 2010 [100] | Resveratrol [100] |
| AML-2/WT | Ara-C resistant acute myeloid leukaemia | Human | Song et al. 2009 [101] | Resveratrol [100] | |
| HL-60 | Acute promyelocytic leukaemia | Human | ATCC | Apigenin [102,103] Butein [104] Chrysin [103] Carnosic acid [105,106] Curcumin [106,107,108,109] Delphinidin [68] Ellagic acid [66] Epigallocatechin gallate [110] Flavonoids [111] Gallic acid [66] Luteolin [112] Punicalagin [68] Quercetin [68,108,109,113,114] Resveratrol [76,90,96,115,116,117,118,119] Silibinin [107] (-)-Vitisin [120] Xanthohumol [121] Curcumin plus silibinin or carnosic acid [107] Ellagic acid plus all-trans-retinoic acid [122] Gallic acid, ellagic acid plus all-trans retinoic acid [122] Green or black tea [123] | |
| K562 | Chronic myelogenous leukaemia | Human | ATCC | Allium cepa L. (PEAL) polyphenols [124] Butein [82,104] Curcumin [125] Epigallocatechin gallate [126,127] Flavopiridol [128] Quercetin [129] Resveratrol [76,81,96,130] Green or black tea [123] Resveratrol plus bestatin [130] Woodfordin I extract (high in tannins) [123] | |
| K562/ADR | Adriamycin-resistant chronic myeloid leukaemia cell line | Human | Tsuruo et al. 1986 [131] | Resveratrol plus bestatin [130] | |
| IM-S and IM-R K562 | Imatinib-sensitive and resistant chronic myelogenous leukaemia | Human | Grosso et al. 2009 [132] | Resveratrol [133] | |
| Kasumi-1 | Acute myeloblastic leukaemia | Human | ATCC | Resveratrol [87] | |
| KCL22 | Chronic myeloid leukaemia | Human | ATCC | Resveratrol [96] | |
| KG-1a | Acute myelogenous leukaemia | Human | ATCC | Aloe-emodin [67] Apigenin [23,67] Carnosic acid [106,107] Curcumin [106,107] Quercetin [23,67] Emodin [23,67] Rhein [23,67] Resveratrol [134,135] Silibinin [107] cis-Stilbene [23,67] trans-Stilbene [67] Curcumin plus silibinin or carnosic acid [107] Quercetin, apigenin plus doxorubicin or etoposide [24] Quercetin, apigenin plus cis-platin, cyclophosphamide or chlorambucil [71] | |
| LAMA84 | Chronic myeloid leukaemia | Human | ATCC | Flavopiridol [128] | |
| MV4:11 | Biphenotypic B myelomonocytic myeloid leukaemia | Human | ATCC | Resveratrol [75,76] | |
| NB4 | Acute promyelocytic leukaemia | Human | Lanotte et al. 1991 [136] | Carnosic acid [106] Curcumin [106] Epigallocatechin gallate [110] Genistein [114,115] Quercetin [114] Resveratrol [115] | |
| OCI/AML3 | Acute myeloid leukaemia | Human | Quentmeier et al. 2005 [137] | Resveratrol [76] | |
| OCIM2 | Acute myeloid leukaemia | Human | Papayannopoulou et al. 1988 [138] | Resveratrol [76] | |
| SHI-1 | Acute monocytic leukaemia | Human | Chen et al. 2005 [139] | Curcumin [140] | |
| THP-1 | Acute monocytic leukaemia | Human | ATCC | Allium cepa L. (PEAL) polyphenols [141] Aloe-emodin [67] Apigenin [67] Butein (104] Delphinidin [68] Emodin [67] Flavonoids [69] Genistein [114] Pinosylvin [142] Punicalagin [68] Quercetin [67,68,114] Resveratrol [96,142] Rhein [67] cis-stilbene [67] trans-Stilbene [67] Quercetin, apigenin, emodin, rhein, or cis-stilbene plus chlorambucil, cisplatin or cyclophosphamide [71] Quercetin, apigenin, emodin, rhein, or cis-Stilbene plus methotrexate, 6-mercaptopurine or 5-fluorouracil [23] Quercetin, apigenin, emodin, rhein, or cis-Stilbene plus doxorubicin or etoposide [24] | |
| C1498 (TIB-49) | Acute myeloid leukaemia | Mouse | ATCC | Carnosic acid [106] Curcumin [106] | |
| MYELOMA | MM144 | Plasma cell myeloma | Human | Díaz-Rodríguez et al. 2011 [143] | Resveratrol [86] |
| MM1S | Immunoglobulin A lambda myeloma | Human | ATCC | Resveratrol [86] | |
| U266 | Myeloma; plasmacytoma | Human | ATCC | Resveratrol [86] | |
| LYMPHOMA | Raji | Burkitt’s lymphoma cell line | Human | ATCC | Epigallocatechin-gallate [144] |
| U-937 | Histiocytic lymphoma | Human | ATCC | Butein [82,104] Carnosic acid [105,106] Curcumin [106] Flavonoids [145] Guggulsterone [146] Icariside II [147] Piceatannol [148] Pinosylvin [142] Quercetin [113,124] Resveratrol [96,115,142,149] Polyphenols extracted from lyophilized Lonicera japonica (PELJ) [150] | |
| BKS-2 | B lymphoma cell line | Mouse | Udhayakumar et al. 1989 [151] | Curcumin [152] | |
| WEHI-231 | B lymphoma cell line | Mouse | ATCC | Curcumin [152] | |
| HEREDITARY SPHEROCYTSIS | WIL2-NS | B lymphocyte | Human | ATCC | Resveratrol [153] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alaswad, H.A.; Mahbub, A.A.; Le Maitre, C.L.; Jordan-Mahy, N. Molecular Action of Polyphenols in Leukaemia and Their Therapeutic Potential. Int. J. Mol. Sci. 2021, 22, 3085. https://doi.org/10.3390/ijms22063085
Alaswad HA, Mahbub AA, Le Maitre CL, Jordan-Mahy N. Molecular Action of Polyphenols in Leukaemia and Their Therapeutic Potential. International Journal of Molecular Sciences. 2021; 22(6):3085. https://doi.org/10.3390/ijms22063085
Chicago/Turabian StyleAlaswad, Hamza A., Amani A. Mahbub, Christine L. Le Maitre, and Nicola Jordan-Mahy. 2021. "Molecular Action of Polyphenols in Leukaemia and Their Therapeutic Potential" International Journal of Molecular Sciences 22, no. 6: 3085. https://doi.org/10.3390/ijms22063085
APA StyleAlaswad, H. A., Mahbub, A. A., Le Maitre, C. L., & Jordan-Mahy, N. (2021). Molecular Action of Polyphenols in Leukaemia and Their Therapeutic Potential. International Journal of Molecular Sciences, 22(6), 3085. https://doi.org/10.3390/ijms22063085