
Novel Ferrocene Derivatives Induce G0/G1 Cell Cycle Arrest and Apoptosis through the Mitochondrial Pathway in Human Hepatocellular Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Compound 1 Was the Most Potent and Selective Compound
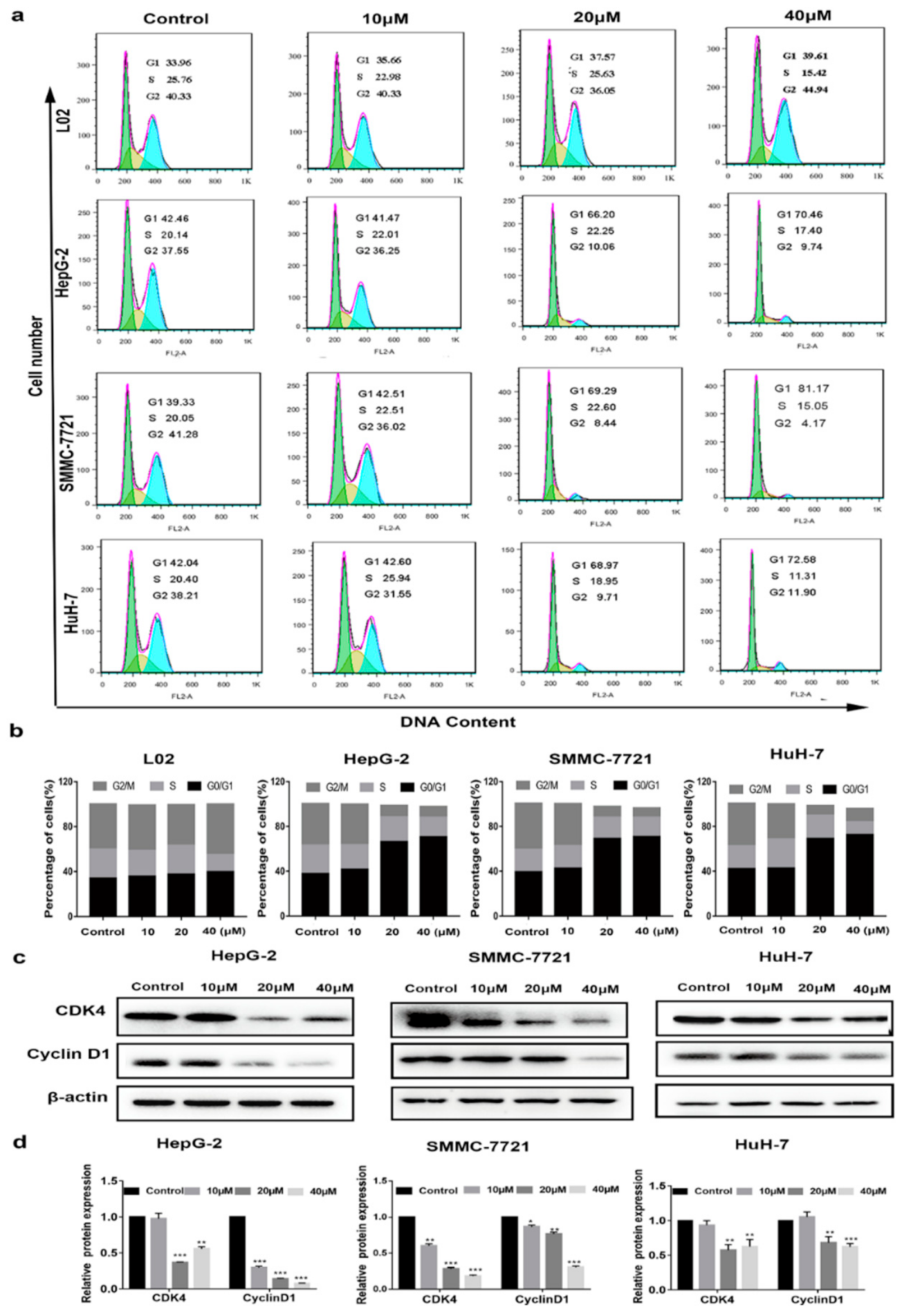
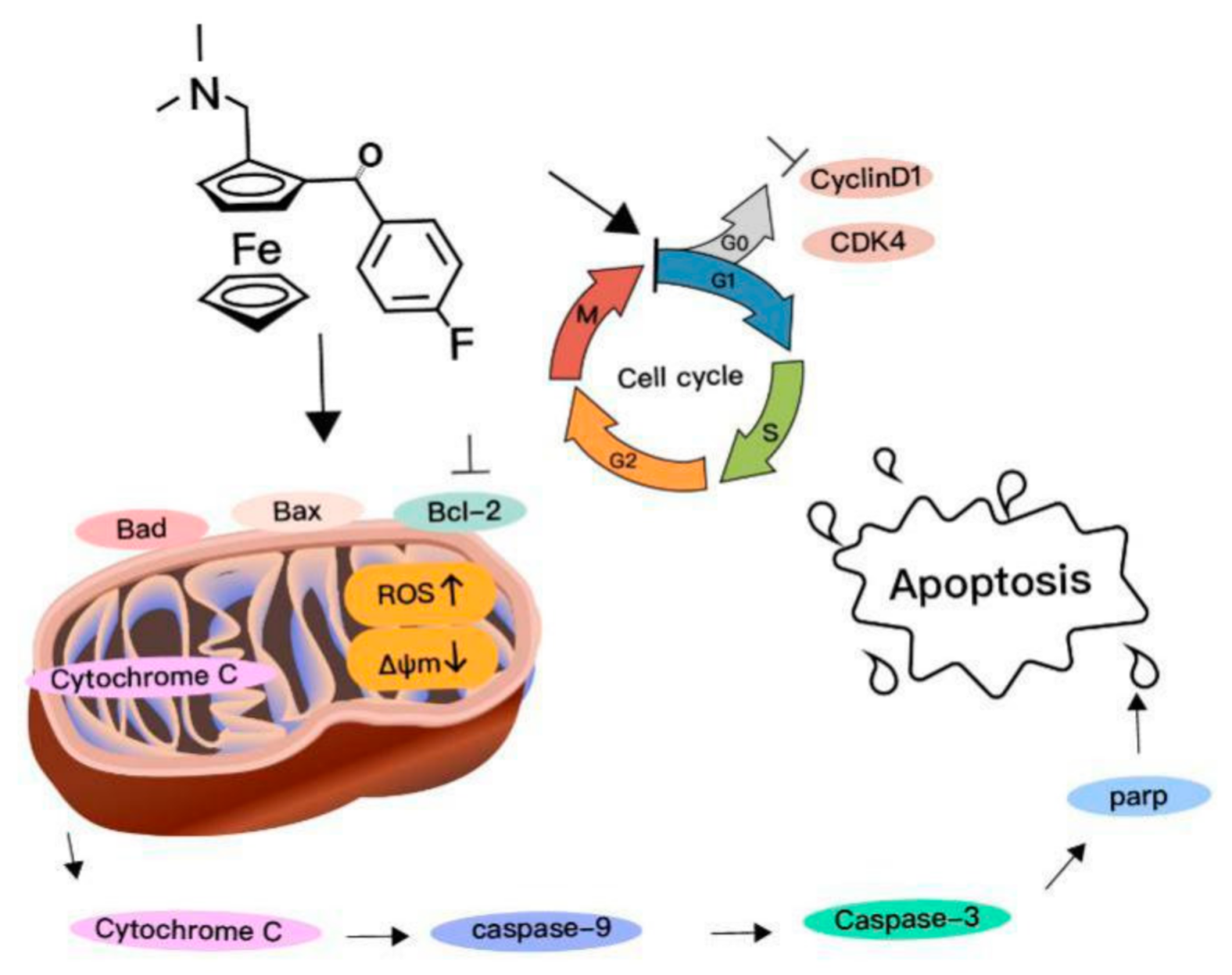
2.2. Cell Cycle Arrest at G0/G1 Phase Treated with Compound 1
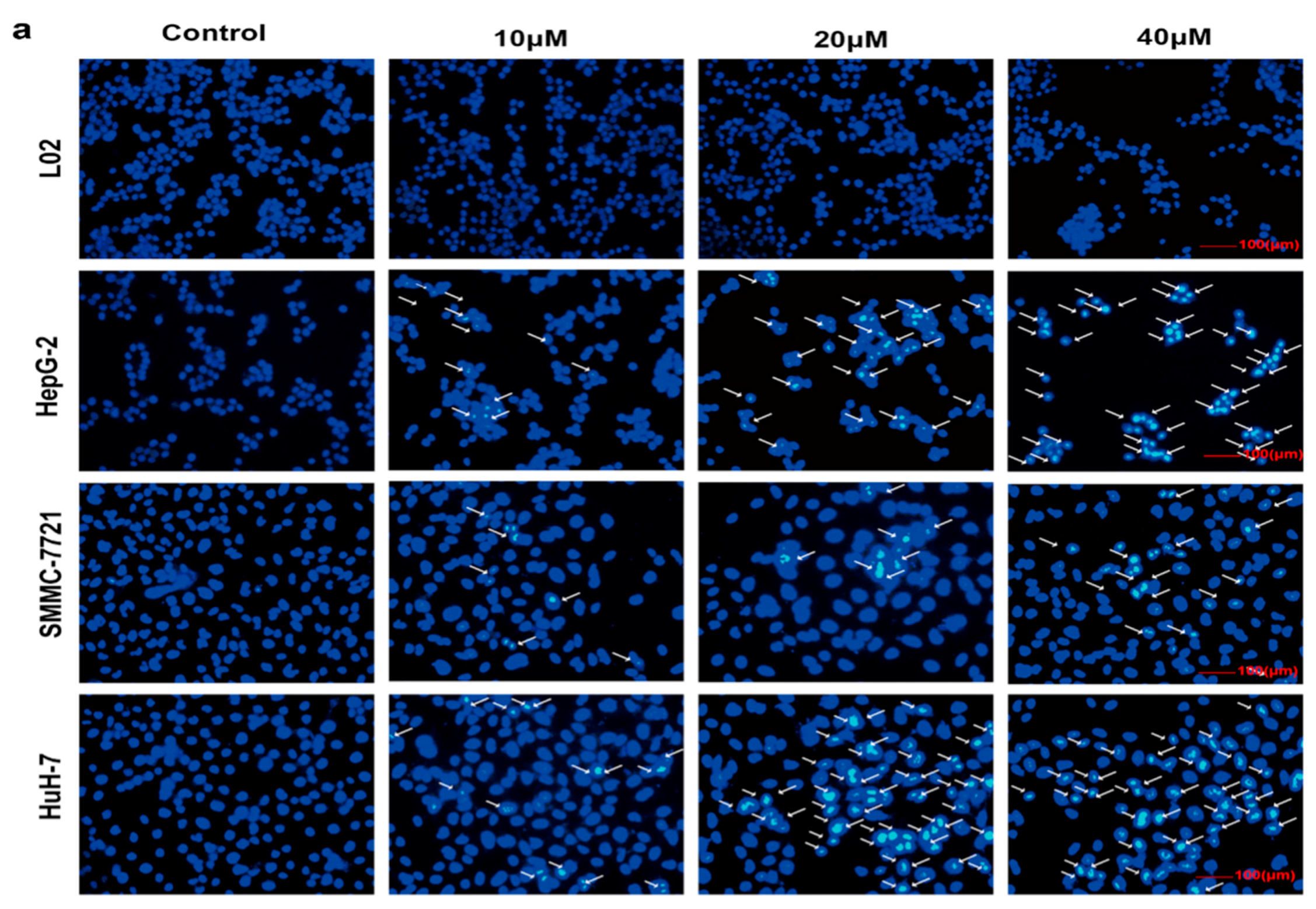
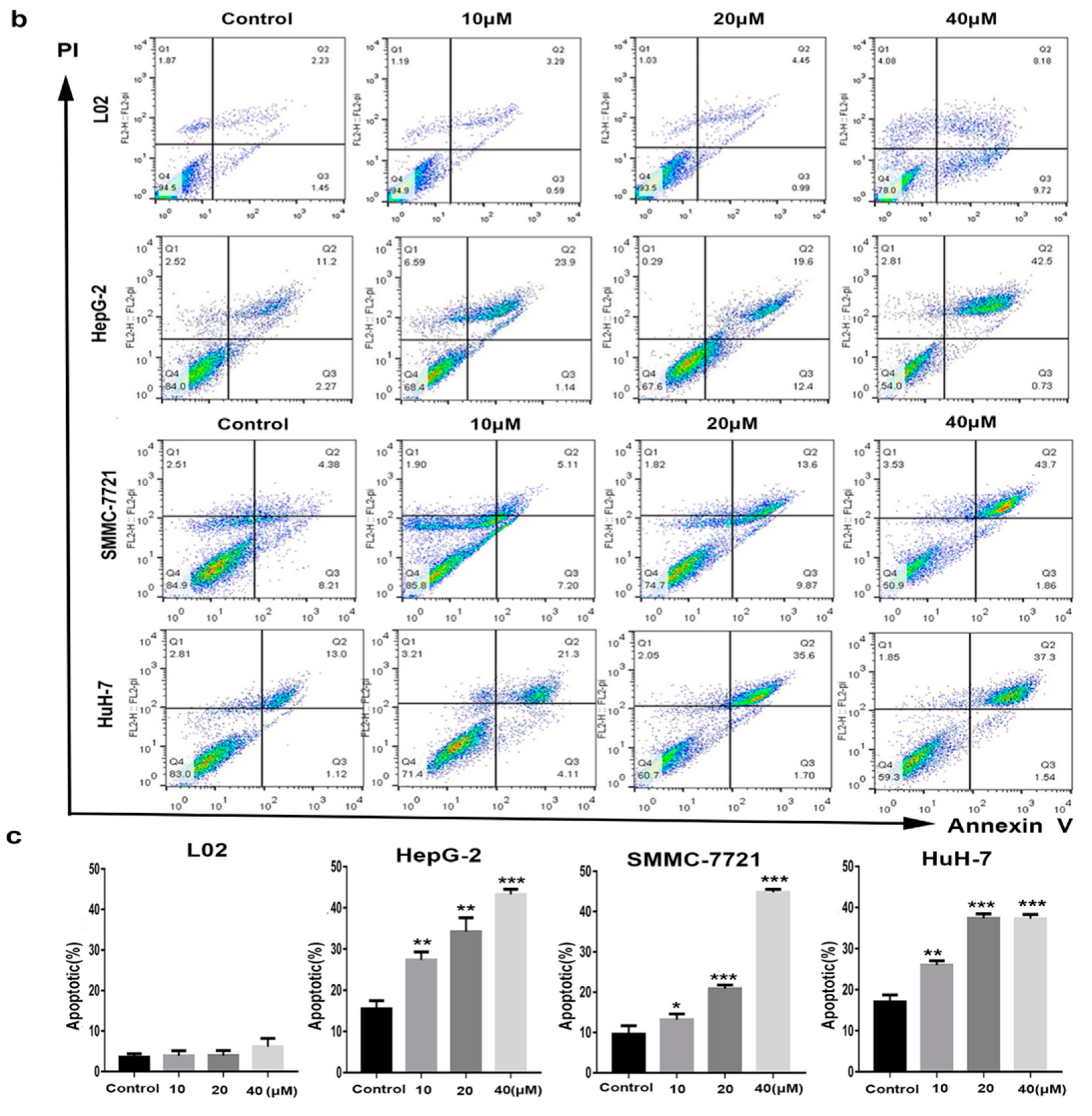
2.3. Selective Apoptosis Induced by Compound 1
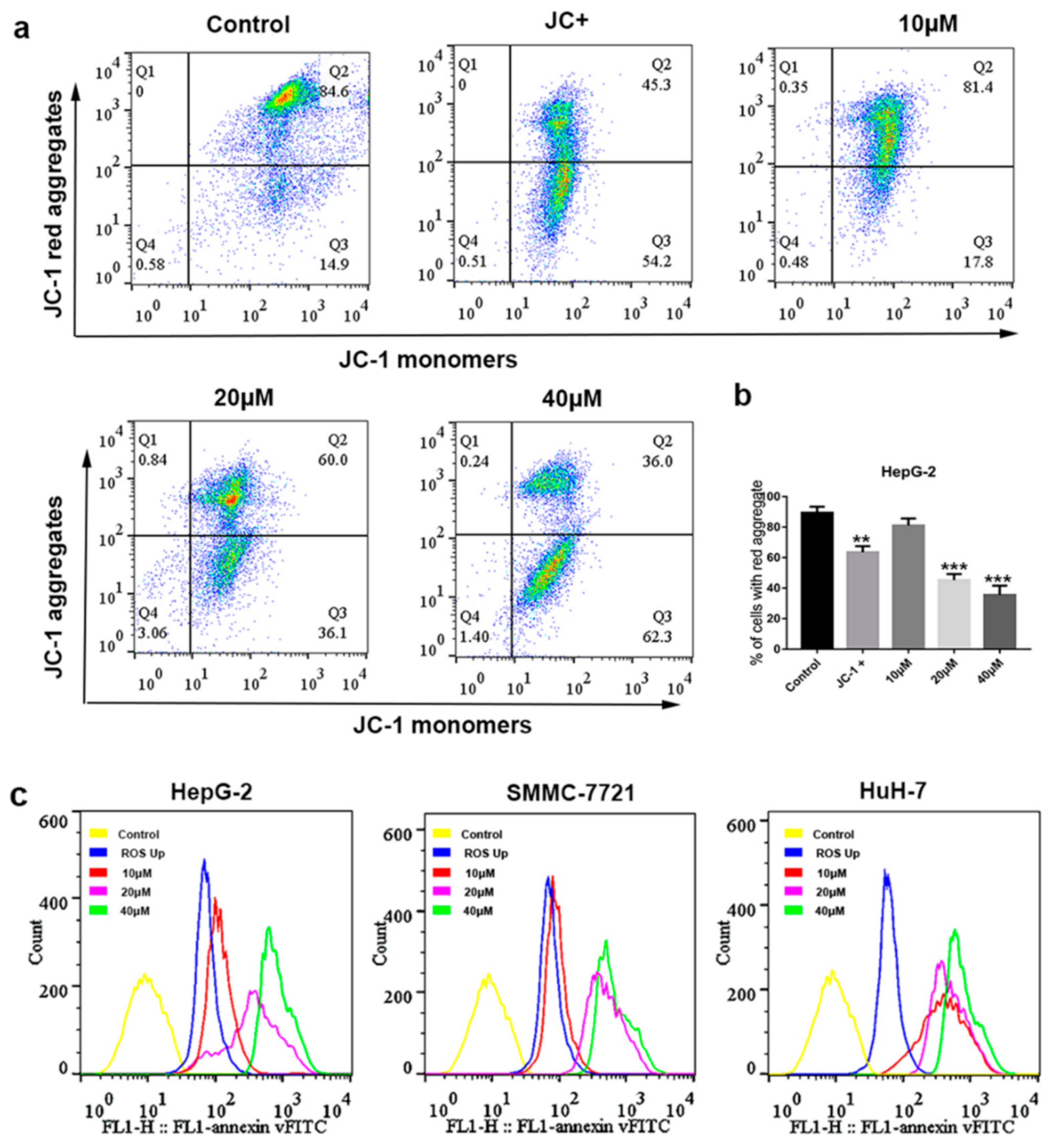
2.4. Mitochondrial Depolarization and ROS Generation Induced by Compound 1
2.5. Mitochondrial Cytochrome c-Dependent Apoptosis Pathway Was Triggered by Compound 1
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cells Culture
4.3. CCK-8 Assay
4.4. Cell Cycle Analysis
4.5. Apoptosis Analysis
4.6. ROS Detection
4.7. The ΔΨm Analysis
4.8. Western Blot Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HCC | Human hepatocellular carcinoma |
| ROS | Reactive oxygen species |
| FBS | Fetal bovine serum |
| DMSO | Dimethyl sulfoxide |
| PBS | Phosphate-buffered saline |
| RIPA | Radio-immunoprecipitation assay |
| ΔΨm (MMP) | Mitochondrial membrane potential |
| DCFH-DA | Dichlorodihydrofluorescein diacetate |
| CDKs | Cyclin-dependent kinases |
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Sueangoen, N.; Tantiwetrueangdet, A.; Panvichian, R. HCC-derived EGFR mutants are functioning, EGF-dependent, and erlotinib-resistant. Cell Biosci. 2020, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, Y. Promising new strategies for hepatocellular carcinoma. Hepatol. Res. 2017, 47, 251–265. [Google Scholar] [CrossRef]
- Reig, M.; Torres, F.; Rodriguez-Lope, C.; Forner, A.; LLarch, N.; Rimola, J.; Darnell, A.; Rios, J.; Ayuso, C.; Bruix, J. Early dermatologic adverse events predict better outcome in HCC patients treated with sorafenib. J. Hepatol. 2014, 61, 318–324. [Google Scholar] [CrossRef]
- Wang, R.; Chen, H.; Yan, W.; Zheng, M.; Zhang, T.; Zhang, Y. Ferrocene-containing hybrids as potential anticancer agents: Current developments, mechanisms of action and structure-activity relationships. Eur. J. Med. Chem. 2020, 190, 112109. [Google Scholar] [CrossRef]
- Sansook, S.; Hassell-Hart, S.; Ocasio, C.; Spencer, J. Ferrocenes in medicinal chemistry: A personal perspective. J. Organomet. Chem. 2019, 905, 121017. [Google Scholar] [CrossRef]
- Xiao, J.; Sun, Z.; Kong, F.; Gao, F. Current scenario of ferrocene-containing hybrids for antimalarial activity. Eur. J. Med. Chem. 2019, 185, 111791. [Google Scholar] [CrossRef]
- Swetha, Y.; Reddy, E.R.; Kumar, J.R.; Trivedi, R.; Giribabu, L.; Sridhar, B.; Rathod, B.; Prakasham, R.S. Synthesis, characterization and antimicrobial evaluation of ferrocene–oxime ether benzyl 1H-1,2,3-triazole hybrids. New J. Chem. 2019, 43, 8341–8351. [Google Scholar]
- Monserrat, J.P.; Al-Safi, R.I.; Tiwari, K.N.; Quentin, L.; Chabot, G.G.; Vessières, A.; Jaouen, G.; Neamati, N.; Hillard, E.A. Ferrocenyl chalcone difluoridoborates inhibit HIV-1 integrase and display low activity towards cancer and endothelial cells. Bioorg. Med. Chem. Lett. 2011, 21, 6195–6197. [Google Scholar] [CrossRef] [PubMed]
- Peter, S.; Aderibigbe, B.A. Ferrocene-Based Compounds with Antimalaria/Anticancer Activity. Molecules 2019, 24, 3604. [Google Scholar] [CrossRef] [Green Version]
- Pi, C.; Cui, X.; Liu, X.; Guo, M.; Zhang, H.; Wu, Y. Synthesis of ferrocene derivatives with planar chirality via palladium-catalyzed enantioselective C-H bond activation. Org. Lett. 2014, 16, 5164–5167. [Google Scholar] [CrossRef]
- Sun, A.; Lin, J.; Pi, C.; Xu, R.; Cui, X. Biological Evaluation of Ferrocenyl Olefins: Cancer Cell Growth Inhibition, ROS Production, and Apoptosis Activity. Arch. Pharm. Chem. Life Sci. 2016, 349, 186–192. [Google Scholar] [CrossRef]
- Dy, G.K.; Adjei, A.A. Understanding, recognizing, and managing toxicities of targeted anticancer therapies. CA-Cancer J. Clin. 2013, 63, 249–279. [Google Scholar] [CrossRef]
- Mattia, E.D.; Cecchin, E.; Guardascione, M.; Foltran, L.; Toffoli, G. Pharmacogenetics of the systemic treatment in advanced hepatocellular carcinoma. World J. Gastroenterol. 2019, 25, 3870–3896. [Google Scholar] [CrossRef]
- Hsieh, F.S.; Chen, Y.L.; Hung, M.H.; Chu, P.Y.; Tsai, M.H.; Chen, L.J.; Hsiao, Y.J.; Shih, C.T.; Chang, M.J.; Chao, T.I. Palbociclib induces activation of AMPK and inhibits hepatocellular carcinoma in a CDK4/6 independent manner. Mol. Oncol. 2017, 11, 1035–1049. [Google Scholar] [CrossRef]
- Villa, E.; Piscaglia, F.; Geva, R.; Dalecos, G.; Sangiovanni, A. Phase IIa safety and efficacy of milciclib, a pan-cyclin dependent kinase inhibitor, in unresectable, sorafenib-refractory or -intolerant hepatocellular carcinoma patients. J. Clin. Oncol. 2020, 38, 16711. [Google Scholar] [CrossRef]
- Fesik, S.W. Promoting apoptosis as a strategy for cancer drug discovery. Nat. Rev. Cancer 2005, 5, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Doonan, F.; Cotter, T.G. Morphological assessment of apoptosis. Methods 2008, 44, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Mita, M.M.; Mita, A.C.; Tolcher, A.W. Apoptosis: Mechanisms and implications for cancer therapeutics. Target. Oncol. 2006, 1, 197–214. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Qu, X.; Sheng, J.; Shen, L.; Su, J.; Xu, Y.; Xie, Q.; Wu, Y.; Zhang, X.; Sun, L. Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PLoS ONE 2017, 12, e0173712. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.L.; Li, D.; Sun, K.; Wang, J. Inhibition of autophagy enhances anticancer effects of bevacizumab in hepatocarcinoma. J. Biol. Chem. 2013, 91, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Moldoveanu, T.; Follis, A.V.; Kriwacki, R.W.; Green, D.R. Many players in BCL-2 family affairs. Trends Biochem. Sci. 2014, 39, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.M.; MacFarlane, M.; Zhuang, J.; Wolf, B.B.; Green, D.R.; Cohen, G.M. Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J. Biol. Chem. 1999, 274, 5053–5060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, K.; Mestres-Arenas, A.; Fajas, L.; Leal-Esteban, L.C. The multifaceted role of cell cycle regulators in the coordination of growth and metabolism. FEBS J. 2020. [Google Scholar] [CrossRef]
- Marzec, M.; Kasprzycka, M.; Lai, R.; Gladden, A.B.; Wlodarski, P.; Tomczak, E.; Nowell, P.; Deprimo, S.E.; Sadis, S.; Eck, S.; et al. Mantle cell lymphoma cells express predominantly cyclin D1a isoform and are highly sensitive to selective inhibition of CDK4 kinase activity. Blood 2006, 108, 1744–1750. [Google Scholar] [CrossRef] [Green Version]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Chang, H.Y.; Deng, J.S.; Chen, J.J.; Huang, G.J. Hispolon from Phellinus linteus induces G0/G1 cell cycle arrest and apoptosis in NB4 human leukaemia cells. Am. J. Chin. Med. 2013, 41, 1439–1457. [Google Scholar] [CrossRef]
- Li, M.; Zhang, N.; Li, M. Capecitabine treatment of HCT-15 colon cancer cells induces apoptosis via mitochondrial pathway. Trop. J. Pharm. Res. 2017, 16, 1529–1536. [Google Scholar] [CrossRef] [Green Version]
- Marinelli, S.; Granito, A.; Piscaglia, F.; Renzulli, M.; Stagni, A.; Bolondi, L. Metronomic capecitabine in patients with hepatocellular carcinoma unresponsive to or ineligible for sorafenib treatment: Report of two cases. Hepat. Mon. 2013, 13, e11721. [Google Scholar] [CrossRef] [Green Version]
- Granito, A.; Marinelli, S.; Terzi, E.; Piscaglia, F.; Renzulli, M.; Venerandi, L.; Benevento, F.; Bolondi, L. Metronomic capecitabine as second-line treatment in hepatocellular carcinoma after sorafenib failure. Dig. Liver. Dis. 2015, 47, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Wandee, J.; Prawan, A.; Senggunprai, L.; Kongpetch, S.; Kukongviriyapan, V. Metformin sensitizes cholangiocarcinoma cell to cisplatin-induced cytotoxicity through oxidative stress mediated mitochondrial pathway. Life Sci. 2019, 217, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Takaki, A.; Yamamoto, K. Control of oxidative stress in hepatocellular carcinoma: Helpful or harmful? World J. Hepatol. 2015, 7, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 16, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zong, M.; Xu, W.; Zhang, Y.; Wang, B.; Yang, M.; Tao, L. Natural pyrethrins induces apoptosis in human hepatocyte cells via Bax- and Bcl-2-mediated mitochondrial pathway. Chem. Biol. Interact. 2017, 262, 38–45. [Google Scholar] [CrossRef]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a Mitochondrial Protein that Promotes Cytochrome c Dependent Caspase Activation by Eliminating IAP Inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.S.; Hsu, S.C.; Weng, S.W.; Yu, C.C.; Yang, J.S.; Lai, K.C.; Lin, J.P.; Lin, J.G.; Chung, J.G. Crude extract of Rheum palmatum L induced cell death in LS1034 human colon cancer cells acts through the caspase-dependent and -independent pathways. Environ. Toxicol. 2014, 29, 969–980. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef]
- Olsson, M.; Zhivotovsky, B. Caspases and cancer. Cell Death Differ. 2011, 18, 1441–1449. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.Y.; Li, Z.X.; Li, H.Y.; Wang, H.J.; Zhu, X.W.; Dou, Z.H. Effects of 14 single herbs on the induction of caspase-3 in tumor cells: A brief review. Chin. J. Integr. Med. 2013, 19, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lee, C. Porcine deltacoronavirus induces caspase-dependent apoptosis through activation of the cytochrome c-mediated intrinsic mitochondrial pathway. Virus Res. 2018, 253, 112–123. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, J.; Zeng, L.; Tang, M.; Lin, H.; Pi, C.; Xu, R.; Cui, X. Novel Ferrocene Derivatives Induce G0/G1 Cell Cycle Arrest and Apoptosis through the Mitochondrial Pathway in Human Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 3097. https://doi.org/10.3390/ijms22063097
Zheng J, Zeng L, Tang M, Lin H, Pi C, Xu R, Cui X. Novel Ferrocene Derivatives Induce G0/G1 Cell Cycle Arrest and Apoptosis through the Mitochondrial Pathway in Human Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2021; 22(6):3097. https://doi.org/10.3390/ijms22063097
Chicago/Turabian StyleZheng, Jianrong, Liao Zeng, Mingqing Tang, Hongjun Lin, Chao Pi, Ruian Xu, and Xiuling Cui. 2021. "Novel Ferrocene Derivatives Induce G0/G1 Cell Cycle Arrest and Apoptosis through the Mitochondrial Pathway in Human Hepatocellular Carcinoma" International Journal of Molecular Sciences 22, no. 6: 3097. https://doi.org/10.3390/ijms22063097
APA StyleZheng, J., Zeng, L., Tang, M., Lin, H., Pi, C., Xu, R., & Cui, X. (2021). Novel Ferrocene Derivatives Induce G0/G1 Cell Cycle Arrest and Apoptosis through the Mitochondrial Pathway in Human Hepatocellular Carcinoma. International Journal of Molecular Sciences, 22(6), 3097. https://doi.org/10.3390/ijms22063097