
CRISPR/Cas9 Guided Mutagenesis of Grain Size 3 Confers Increased Rice (Oryza sativa L.) Grain Length by Regulating Cysteine Proteinase Inhibitor and Ubiquitin-Related Proteins

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results
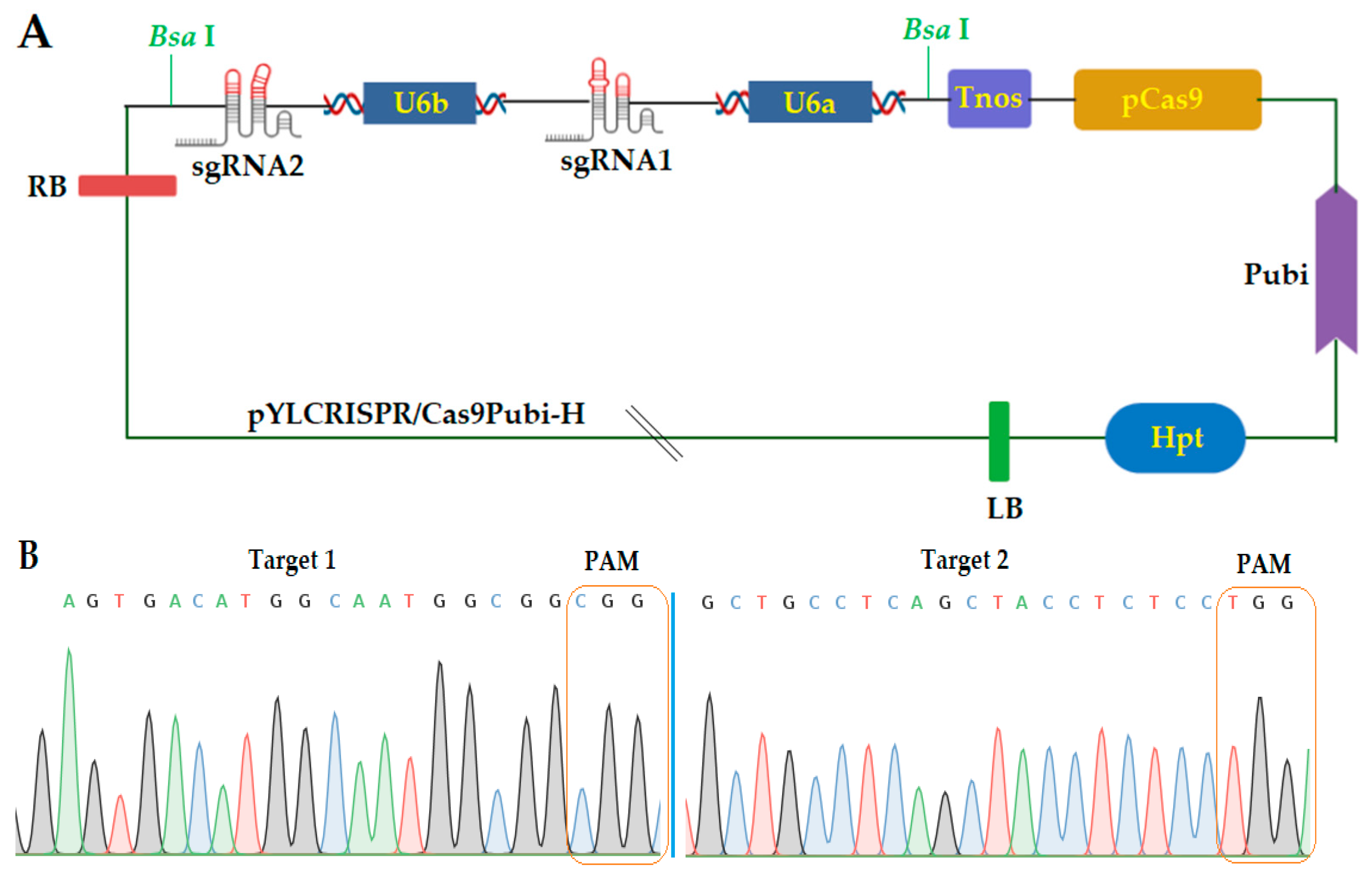
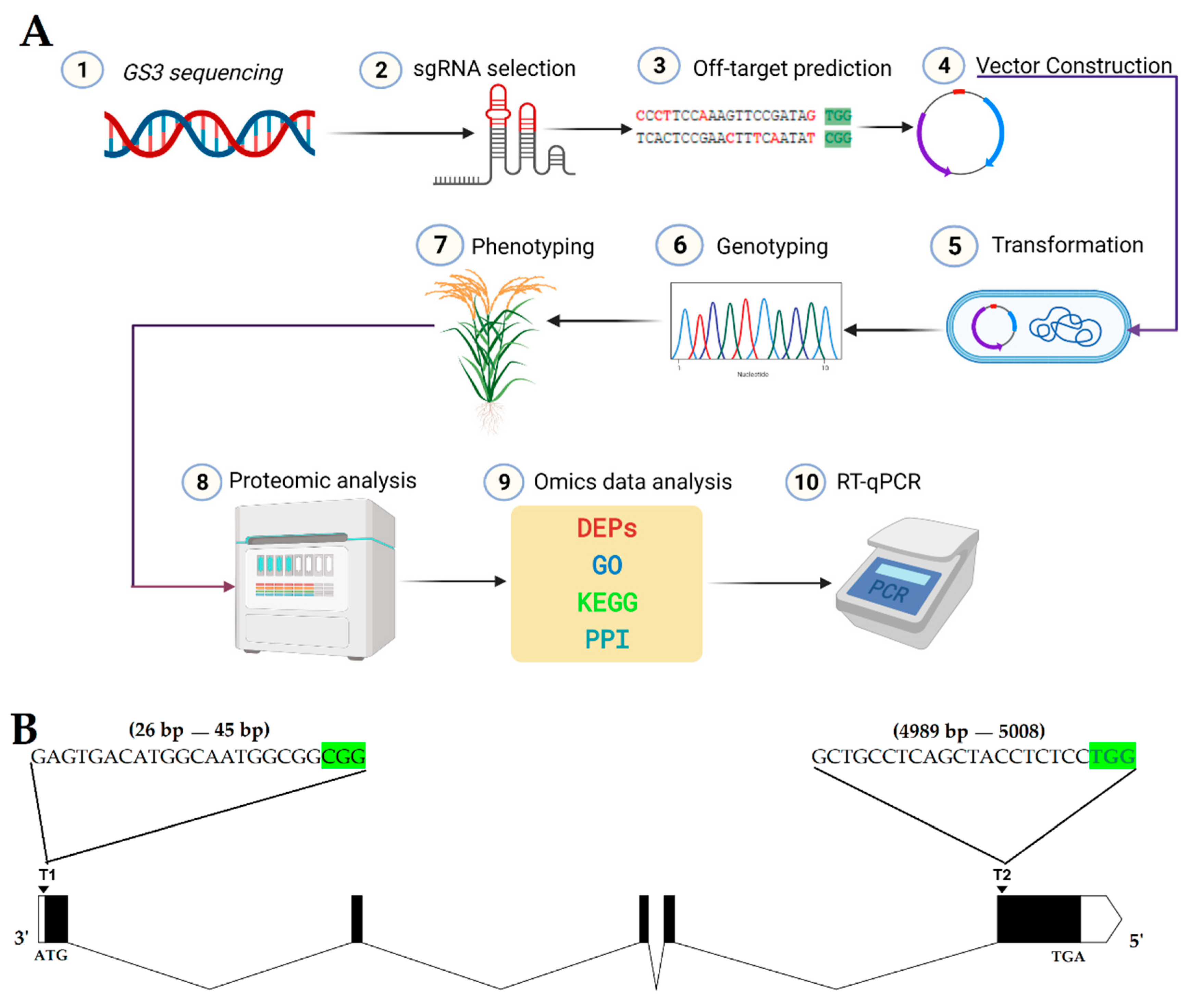
2.1. Construction of CRISPR/Cas9 Expression Vector
2.2. Obtaining Mutant Plants and Genotyping
2.3. Screening of T-DNA-Free Mutant Plants in the T1 Generation and Segregation Analysis
2.4. Investigation of Agronomic Traits
2.5. Proteomic Data Outcome
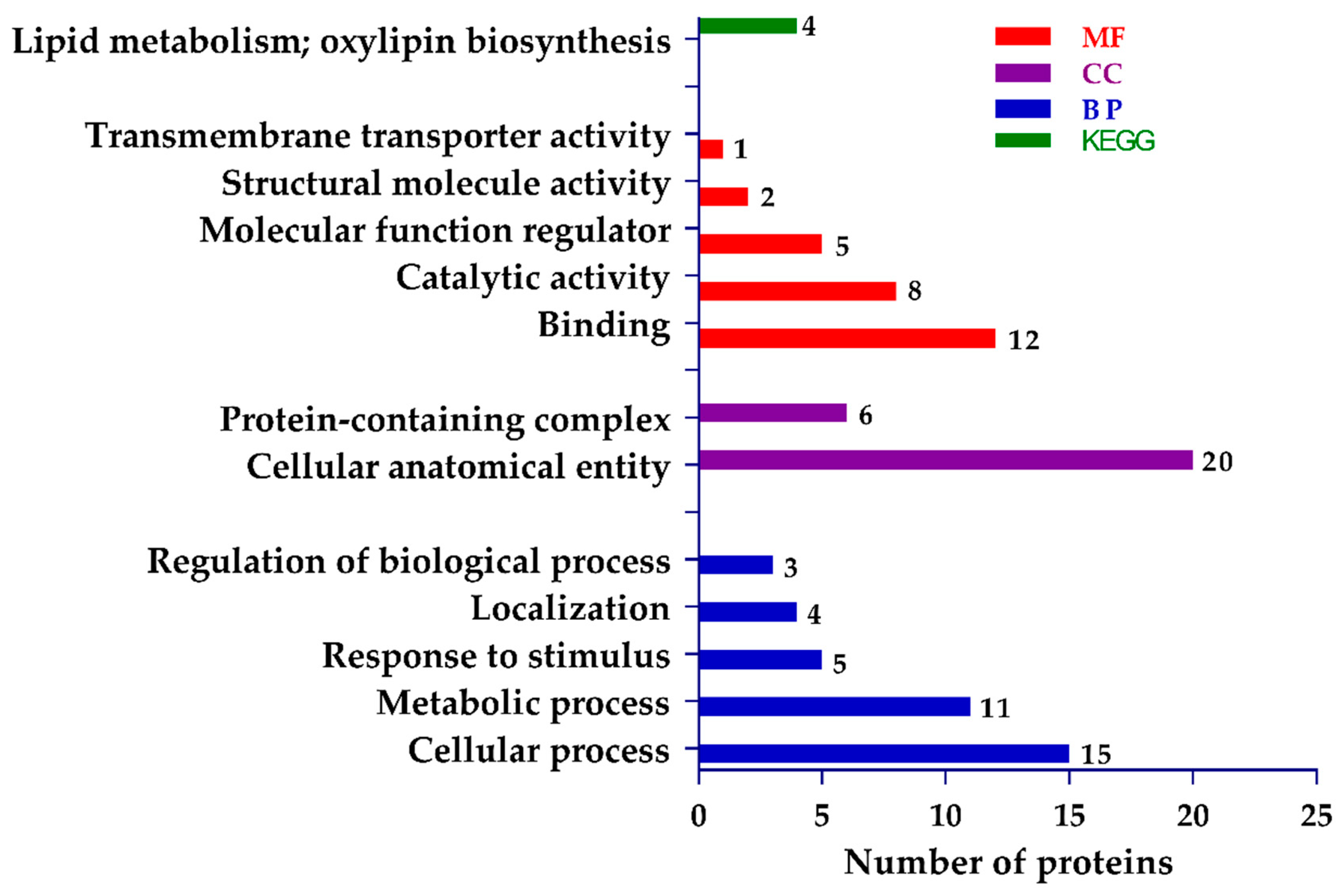
2.6. Functional Assignment and Pathway Analysis
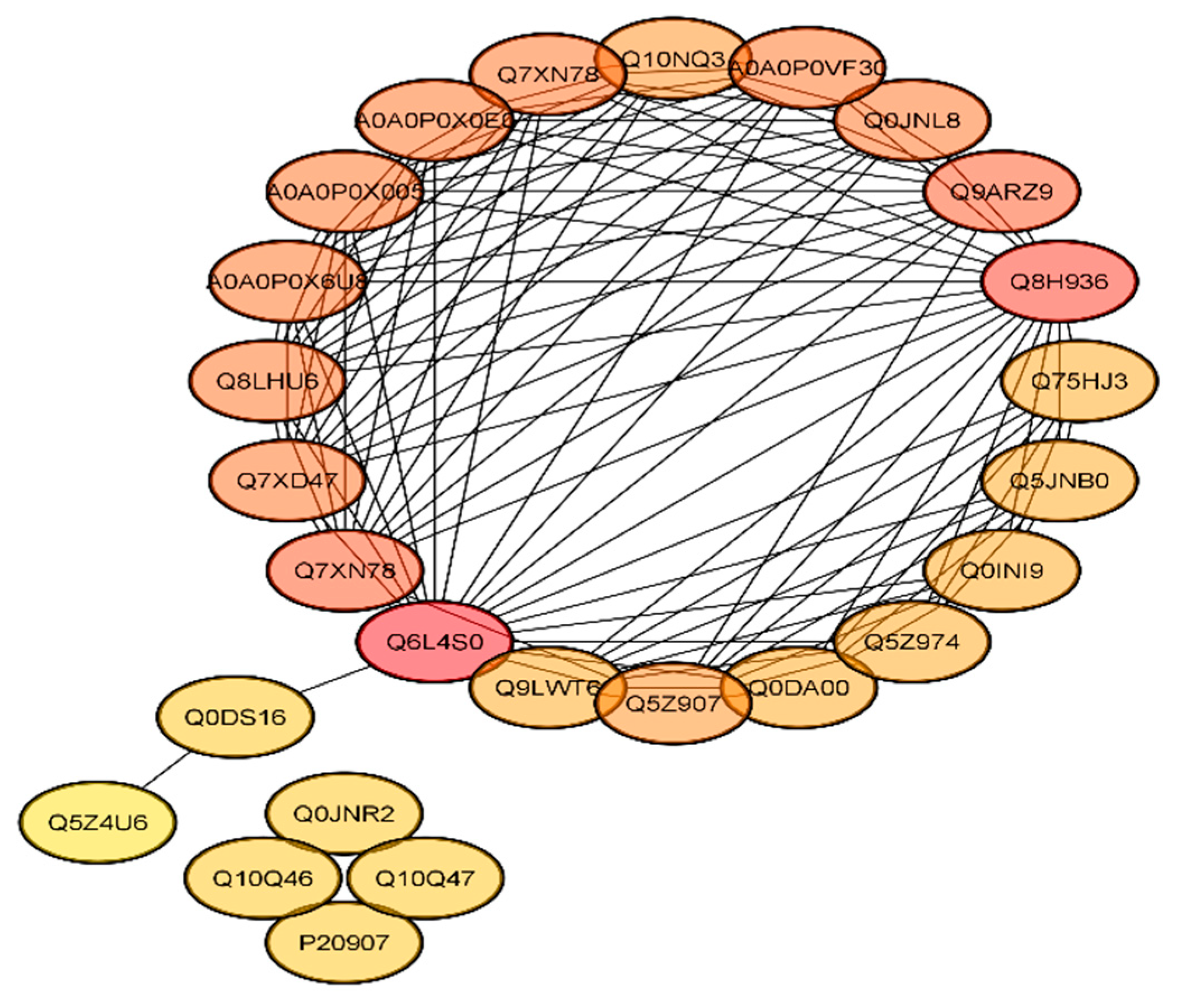
2.7. Functional Interaction Networks of the Differentially Expressed Proteins (DEPs)
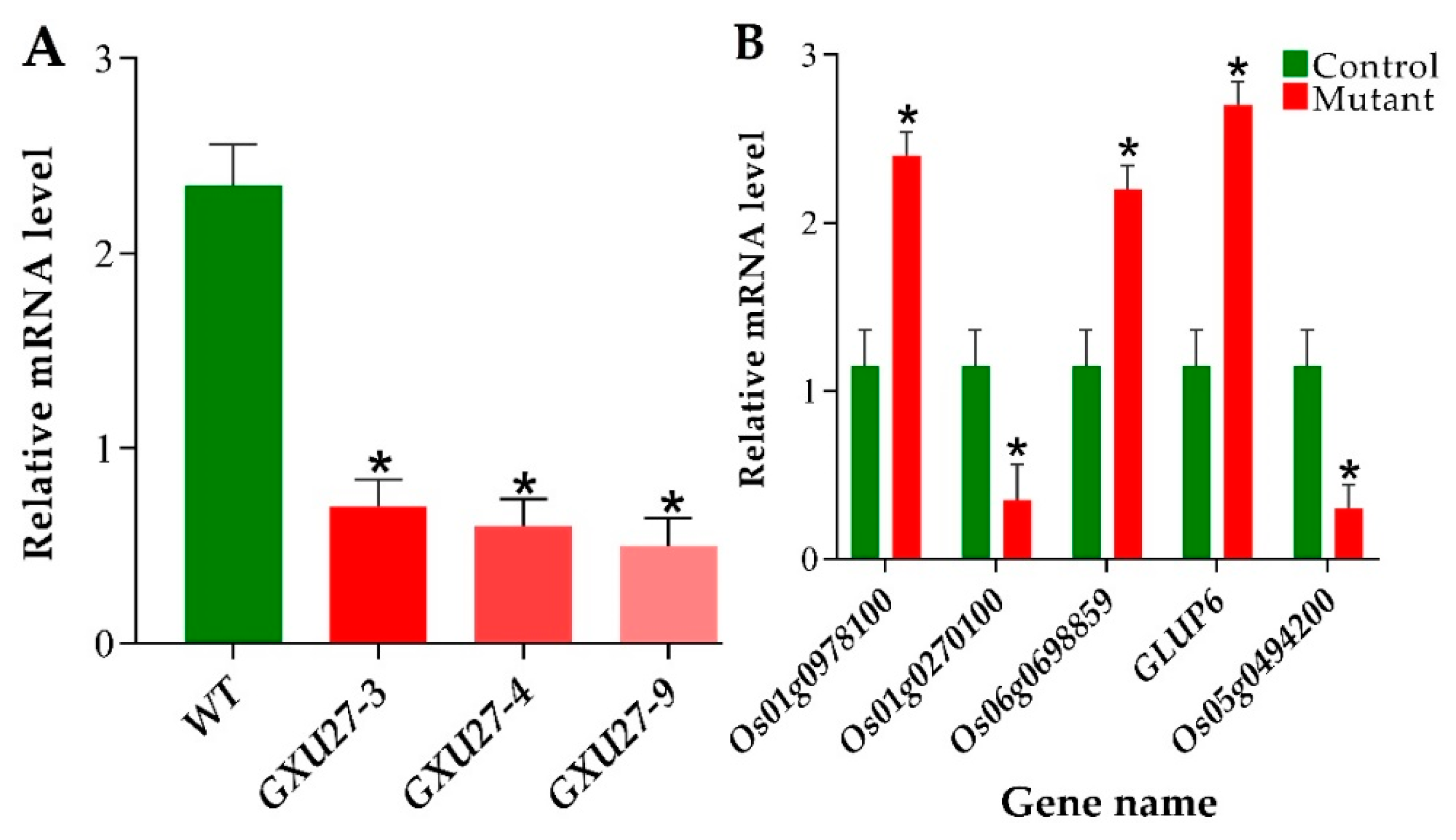
2.8. Verification of mRNA Expression Patterns of GS3 and Differentially Expressed Proteins (DEPs)
3. Discussion
4. Materials and Methods
4.1. Test Materials
4.2. gRNA Design, Vector Construction, and Transformation
4.3. Genotyping, Phenotyping, and Screening of T-DNA-free Plants
4.4. Protein Extraction and Quality Inspection
4.5. Protein Digestion and iTRAQ Labeling
4.6. Strong Cation Exchange Column Classification and Mass Spectrometry Detection of iTRAQ Labeled Samples
4.7. Proteomic Data Analysis and Functional Annotation
4.8. Verification of mRNA Expression of GS3 and Protein-Coding Genes
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| Cas9 | CRISPR-associated protein 9 |
| GWT | 1000-grain weight |
| GL | Grain length |
| iTRAQ | Isobaric tags for relative and absolute quantitation |
| DEPs | Differentially expressed proteins |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO | Gene Ontology |
References
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food security: The challenge of feeding 9 billion people. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Bai, X.; Xing, Y. A rice genetic improvement boom by next generation sequencing. Curr. Issues Mol. Biol. 2018, 27, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Li, Y.; Qian, Q. Recent progress on molecular breeding of rice in China. Plant Cell Rep. 2014, 33, 551–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Mao, Y.; Lu, Y.; Wang, Z.; Tao, X.; Zhu, J.K. Multiplex gene editing in rice with simplified CRISPR-Cpf1 and CRISPR-Cas9 systems. J. Integr. Plant Biol. 2018, 60, 626–631. [Google Scholar] [CrossRef]
- Cai, Y.; Chen, L.; Liu, X.; Guo, C.; Sun, S.; Wu, C.; Jiang, B.; Han, T.; Hou, W. CRISPR/Cas9-mediated targeted mutagenesis of GmFT2a delays flowering time in soya bean. Plant Biotechnol. J. 2018, 16, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.; Zhu, T.; Tian, Z.; Li, C.; Zhang, W.; Song, R. High-efficiency CRISPR/Cas9 multiplex gene editing using the glycine tRNA-processing system-based strategy in maize. BMC Biotechnol. 2016, 16, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhang, R.; Song, G.; Gao, J.; Li, W.; Han, X.; Chen, M.; Li, Y.; Li, G. Targeted mutagenesis using the Agrobacterium tumefaciens-mediated CRISPR-Cas9 system in common wheat. BMC Plant Biol. 2018, 18, 1–12. [Google Scholar] [CrossRef]
- Wang, P.; Zhang, J.; Sun, L.; Ma, Y.; Xu, J.; Liang, S.; Deng, J.; Tan, J.; Zhang, Q.; Tu, L. High efficient multisites genome editing in allotetraploid cotton (Gossypium hirsutum) using CRISPR/Cas9 system. Plant Biotechnol. J. 2018, 16, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Li, J.; Godwin, I.D. Genome editing by CRISPR/Cas9 in sorghum through biolistic bombardment. In Sorghum; Springer: Berlin/Heidelberg, Germany, 2019; pp. 169–183. [Google Scholar] [CrossRef]
- Usman, B.; Nawaz, G.; Zhao, N.; Liu, Y.; Li, R. Generation of high yielding and fragrant rice (Oryza sativa L.) Lines by CRISPR/Cas9 targeted mutagenesis of three homoeologs of cytochrome P450 gene family and OsBADH2 and transcriptome and proteome profiling of revealed changes triggered by mutations. Plants 2020, 9, 788. [Google Scholar] [CrossRef]
- Usman, B.; Nawaz, G.; Zhao, N.; Liao, S.; Liu, Y.; Li, R. Precise Editing of the OsPYL9 Gene by RNA-Guided Cas9 Nuclease Confers Enhanced Drought Tolerance and Grain Yield in Rice (Oryza sativa L.) by Regulating Circadian Rhythm and Abiotic Stress Responsive Proteins. Int. J. Mol. Sci. 2020, 21, 7854. [Google Scholar] [CrossRef]
- Usman, B.; Nawaz, G.; Zhao, N.; Liao, S.; Qin, B.; Liu, F.; Liu, Y.; Li, R. Programmed Editing of Rice (Oryza sativa L.) OsSPL16 Gene Using CRISPR/Cas9 Improves Grain Yield by Modulating the Expression of Pyruvate Enzymes and Cell Cycle Proteins. Int. J. Mol. Sci. 2021, 22, 249. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.Y.; An, H.; Zhang, Y.; Shen, F.C. Study on heredity of morphological characters of rice grain. J. Southwest Agric. Univ. 2000, 22, 102–104. [Google Scholar]
- Song, X.J.; Huang, W.; Shi, M.; Zhu, M.Z.; Lin, H.X. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat. Genet. 2007, 39, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Xing, Y.; Mao, H.; Lu, T.; Han, B.; Xu, C.; Li, X.; Zhang, Q. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor. Appl. Genet. 2006, 112, 1164–1171. [Google Scholar] [CrossRef]
- Zhao, D.S.; Li, Q.F.; Zhang, C.Q.; Zhang, C.; Yang, Q.Q.; Pan, L.X.; Ren, X.Y.; Lu, J.; Gu, M.H.; Liu, Q.Q. GS9 acts as a transcriptional activator to regulate rice grain shape and appearance quality. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, J.; Zheng, X.; Wu, F.; Lin, Q.; Heng, Y.; Tian, P.; Cheng, Z.; Yu, X.; Zhou, K. GW5 acts in the brassinosteroid signalling pathway to regulate grain width and weight in rice. Nat. Plants 2017, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ishimaru, K.; Hirotsu, N.; Madoka, Y.; Murakami, N.; Hara, N.; Onodera, H.; Kashiwagi, T.; Ujiie, K.; Shimizu, B.I.; Onishi, A. Loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield. Nat. Genet. 2013, 45, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Qi, P.; Lin, Y.S.; Song, X.J.; Shen, J.B.; Huang, W.; Shan, J.X.; Zhu, M.Z.; Jiang, L.; Gao, J.P.; Lin, H.X. The novel quantitative trait locus GL3.1 controls rice grain size and yield by regulating Cyclin-T1; 3. Cell Res. 2012, 22, 1666–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Fan, C.; Xing, Y.; Jiang, Y.; Luo, L.; Sun, L.; Shao, D.; Xu, C.; Li, X.; Xiao, J. Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat. Genet. 2011, 43, 1266–1269. [Google Scholar] [CrossRef]
- Wang, Y.; Xiong, G.; Hu, J.; Jiang, L.; Yu, H.; Xu, J.; Fang, Y.; Zeng, L.; Xu, E.; Xu, J. Copy number variation at the GL7 locus contributes to grain size diversity in rice. Nat. Genet. 2015, 47, 944–948. [Google Scholar] [CrossRef]
- Si, L.; Chen, J.; Huang, X.; Gong, H.; Luo, J.; Hou, Q.; Zhou, T.; Lu, T.; Zhu, J.; Shangguan, Y. OsSPL13 controls grain size in cultivated rice. Nat. Genet. 2016, 48, 447–456. [Google Scholar] [CrossRef]
- Mao, H.; Sun, S.; Yao, J.; Wang, C.; Yu, S.; Xu, C.; Li, X.; Zhang, Q. Linking differential domain functions of the GS3 protein to natural variation of grain size in rice. Proc. Natl. Acad. Sci. USA 2010, 107, 19579–19584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zhu, Q.; Chen, Y.; Liu, Y.G. CRISPR/Cas9 platforms for genome editing in plants: Developments and applications. Mol. Plant 2016, 9, 961–974. [Google Scholar] [CrossRef] [Green Version]
- Peng, B.; Li, J.; Kong, D.Y.; He, L.L.; Li, M.G.; Nassirou, T.Y.; Song, X.H.; Peng, J.; Jiang, Y.; Sun, Y.F. Genetic Improvement of Grain Quality Promoted by High and New Technology in Rice. J. Agric. Sci. 2019, 11. [Google Scholar] [CrossRef]
- Lowder, L.G.; Zhang, D.; Baltes, N.J.; Paul, J.W.; Tang, X.; Zheng, X.; Voytas, D.F.; Hsieh, T.F.; Zhang, Y.; Qi, Y. A CRISPR/Cas9 toolbox for multiplexed plant genome editing and transcriptional regulation. Plant Physiol. 2015, 169, 971–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, S.; Qin, X.; Luo, L.; Han, Y.; Wang, X.; Usman, B.; Nawaz, G.; Zhao, N.; Liu, Y.; Li, R. CRISPR/Cas9-Induced Mutagenesis of Semi-Rolled Leaf1, 2 Confers Curled Leaf Phenotype and Drought Tolerance by Influencing Protein Expression Patterns and ROS Scavenging in Rice (Oryza sativa L.). Agronomy 2019, 9, 728. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Teng, K.; Nawaz, G.; Feng, X.; Usman, B.; Wang, X.; Luo, L.; Zhao, N.; Liu, Y.; Li, R. Generation of semi-dwarf rice (Oryza sativa L.) lines by CRISPR/Cas9-directed mutagenesis of OsGA20ox2 and proteomic analysis of unveiled changes caused by mutations. 3 Biotech 2019, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, G.; Usman, B.; Peng, H.; Zhao, N.; Yuan, R.; Liu, Y.; Li, R. Knockout of Pi21 by CRISPR/Cas9 and iTRAQ-Based Proteomic Analysis of Mutants Revealed New Insights into M. oryzae Resistance in Elite Rice Line. Genes 2020, 11, 735. [Google Scholar] [CrossRef]
- Nawaz, G.; Han, Y.; Usman, B.; Liu, F.; Qin, B.; Li, R. Knockout of OsPRP1, a gene encoding proline-rich protein, confers enhanced cold sensitivity in rice (Oryza sativa L.) at the seedling stage. 3 Biotech 2019, 9, 1–18. [Google Scholar] [CrossRef]
- Han, Y.; Luo, D.; Usman, B.; Nawaz, G.; Zhao, N.; Liu, F.; Li, R. Development of high yielding glutinous cytoplasmic male sterile rice (Oryza sativa L.) lines through CRISPR/Cas9 based mutagenesis of Wx and TGW6 and proteomic analysis of anther. Agronomy 2018, 8, 290. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A. The ubiquitin system for protein degradation and some of its roles in the control of the cell division cycle. Cell Death Diff. 2005, 12, 1191–1197. [Google Scholar] [CrossRef] [Green Version]
- Dielen, A.S.; Badaoui, S.; Candresse, T.; German-Retana, S. The ubiquitin/26S proteasome system in plant–pathogen interactions: A never-ending hide-and-seek game. Mol. Plant Pathol. 2010, 11, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Stone, S.L. The role of ubiquitin and the 26S proteasome in plant abiotic stress signaling. Front. Plant Sci. 2014, 5, 135. [Google Scholar] [CrossRef]
- Marino, D.; Peeters, N.; Rivas, S. Ubiquitination during plant immune signaling. Plant Physiol. 2012, 160, 15–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandman, O.; Stewart-Ornstein, J.; Wong, D.; Larson, A.; Williams, C.C.; Li, G.W.; Zhou, S.; King, D.; Shen, P.S.; Weibezahn, J. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 2012, 151, 1042–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershko, A.; Ciechanover, A.; Heller, H.; Haas, A.L.; Rose, I.A. Proposed role of ATP in protein breakdown: Conjugation of protein with multiple chains of the polypeptide of ATP-dependent proteolysis. Proc. Natl. Acad. Sci. USA 1980, 77, 1783–1786. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar] [CrossRef]
- Johnson, E.S.; Bartel, B.; Seufert, W.; Varshavsky, A. Ubiquitin as a degradation signal. EMBO J. 1992, 11, 497–505. [Google Scholar] [CrossRef]
- Hicke, L.; Riezman, H. Ubiquitination of a yeast plasma membrane receptor signals its ligand-stimulated endocytosis. Cell 1996, 84, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Hurst, J.H.; Dohlman, H.G. Dynamic ubiquitination of the mitogen-activated protein kinase kinase (MAPKK) Ste7 determines mitogen-activated protein kinase (MAPK) specificity. J. Biol. Chem. 2013, 288, 18660–18671. [Google Scholar] [CrossRef] [Green Version]
- Jentsch, S.; McGrath, J.P.; Varshavsky, A. The yeast DNA repair gene RAD6 encodes a ubiquitin-conjugating enzyme. Nature 1987, 329, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.; Layfield, R.; Dhananjayan, S.C.; Ismail, A.; Nawaz, Z. Ubiquitin and control of transcription. Essays Biochem. 2005, 41, 69–80. [Google Scholar] [CrossRef]
- An, H.; Harper, J.W. Ribosome abundance control via the ubiquitin–proteasome system and autophagy. J. Mol. Biol. 2020, 432, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Back, S.; Gorman, A.W.; Vogel, C.; Silva, G.M. Site-specific K63 ubiquitinomics provides insights into translation regulation under stress. J. Prot. Res. 2018, 18, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.K.; Porras-Yakushi, T.R.; Reitsma, J.M.; Huber, F.M.; Sweredoski, M.J.; Hoelz, A.; Hess, S.; Deshaies, R.J. A conserved quality-control pathway that mediates degradation of unassembled ribosomal proteins. eLife 2016, 5, e19105. [Google Scholar] [CrossRef]
- Wei, J.; Qiu, X.; Chen, L.; Hu, W.; Hu, R.; Chen, J.; Sun, L.; Li, L.; Zhang, H.; Lv, Z. The E3 ligase AtCHIP positively regulates Clp proteolytic subunit homeostasis. J. Exp. Bot. 2015, 66, 5809–5820. [Google Scholar] [CrossRef] [Green Version]
- Tsai, B.; Ye, Y.; Rapoport, T.A. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 2002, 3, 246–255. [Google Scholar] [CrossRef]
- Ye, Y.; Meyer, H.H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 2001, 414, 652–656. [Google Scholar] [CrossRef]
- Lee, S.; Lee, D.W.; Lee, Y.; Mayer, U.; Stierhof, Y.D.; Lee, S.; Jürgens, G.; Hwang, I. Heat shock protein cognate 70-4 and an E3 ubiquitin ligase, CHIP, mediate plastid-destined precursor degradation through the ubiquitin-26S proteasome system in Arabidopsis. Plant Cell 2009, 21, 3984–4001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shomura, A.; Izawa, T.; Ebana, K.; Ebitani, T.; Kanegae, H.; Konishi, S.; Yano, M. Deletion in a gene associated with grain size increased yields during rice domestication. Nat. Genet. 2008, 40, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Weng, J.; Gu, S.; Wan, X.; Gao, H.; Guo, T.; Su, N.; Lei, C.; Zhang, X.; Cheng, Z.; Guo, X. Isolation and initial characterization of GW5, a major QTL associated with rice grain width and weight. Cell Res. 2008, 18, 1199–1209. [Google Scholar] [CrossRef]
- Todd, I.; Radford, P.M.; Draper-Morgan, K.A.; McIntosh, R.; Bainbridge, S.; Dickinson, P.; Jamhawi, L.; Sansaridis, M.; Huggins, M.L.; Tighe, P.J. Mutant forms of tumour necrosis factor receptor I that occur in TNF-receptor-associated periodic syndrome retain signalling functions but show abnormal behaviour. Immunology 2004, 113, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Bode, W. The cystatins: Protein inhibitors of cysteine proteinases. FEBS Lett. 1991, 285, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Van Wyk, S.G.; Kunert, K.J.; Cullis, C.A.; Pillay, P.; Makgopa, M.E.; Schlüter, U.; Vorster, B.J. The future of cystatin engineering. Plant Sci. 2016, 246, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, M.; Abraham, Z.; Carbonero, P.; Díaz, I. Comparative phylogenetic analysis of cystatin gene families from arabidopsis, rice and barley. Mol. Genet. Genom. 2005, 273, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Belenghi, B.; Acconcia, F.; Trovato, M.; Perazzolli, M.; Bocedi, A.; Polticelli, F.; Ascenzi, P.; Delledonne, M. AtCYS1, a cystatin from Arabidopsis thaliana, suppresses hypersensitive cell death. Eur. J. Biochem. 2003, 270, 2593–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, J.J.; Yang, W.J.; Yuan, S.H.; Ying, F.Y.; Qiong, L.C. Study on transformation of cysteine proteinase inhibitor gene into cabbage (Brassica oleracea var. capitata L.). In Proceedings of the IV International Symposium on Brassicas and XIV Crucifer Genetics Workshop 706, Daejeon, Korea, 1 April 2006. [Google Scholar] [CrossRef]
- Bouchard, E.; Michaud, D.; Cloutier, C. Molecular interactions between an insect predator and its herbivore prey on transgenic potato expressing a cysteine proteinase inhibitor from rice. Mol. Ecol. 2003, 12, 2429–2437. [Google Scholar] [CrossRef]
- Gholizadeh, A. The possible involvement of d-amino acids or their metabolites in Arabidopsis cysteine proteinase/cystatin-dependent proteolytic pathway. Cytol. Genet. 2015, 49, 73–79. [Google Scholar] [CrossRef]
- Irie, K.; Hosoyama, H.; Takeuchi, T.; Iwabuchi, K.; Watanabe, H.; Abe, M.; Abe, K.; Arai, S. Transgenic rice established to express corn cystatin exhibits strong inhibitory activity against insect gut proteinases. Plant Mol. Biol. 1996, 30, 149–157. [Google Scholar] [CrossRef]
- Rassam, M.; Laing, W.A. Purification and characterization of phytocystatins from kiwifruit cortex and seeds. Phytochemistry 2004, 65, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Wang, S.; Liang, D.; Li, M.; Ma, F. Genome-wide identification and expression profiling of the cystatin gene family in apple (Malus × domestica Borkh.). Plant Physiol. Biochem. 2014, 79, 88–97. [Google Scholar] [CrossRef]
- Zhao, P.; Zhou, X.M.; Zou, J.; Wang, W.; Wang, L.; Peng, X.B.; Sun, M.X. Comprehensive analysis of cystatin family genes suggests their putative functions in sexual reproduction, embryogenesis, and seed formation. J. Exp. Bot. 2014, 65, 5093–5107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.E.; Hong, J.K.; Je, J.H.; Lee, K.O.; Kim, D.Y.; Lee, S.Y.; Lim, C.O. Regulation of seed germination and seedling growth by an Arabidopsis phytocystatin isoform, AtCYS6. Plant Cell Rep. 2009, 28, 1623–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernas, M.; Sánchez-Monge, R.; Salcedo, G. Biotic and abiotic stress can induce cystatin expression in chestnut. FEBS Lett. 2000, 467, 206–210. [Google Scholar] [CrossRef]
- Van der Vyver, C.; Schneidereit, J.; Driscoll, S.; Turner, J.; Kunert, K.; Foyer, C.H. Oryzacystatin I expression in transformed tobacco produces a conditional growth phenotype and enhances chilling tolerance. Plant Biotechnol. J. 2003, 1, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papolu, P.K.; Dutta, T.K.; Tyagi, N.; Urwin, P.E.; Lilley, C.J.; Rao, U. Expression of a cystatin transgene in eggplant provides resistance to root-knot nematode, Meloidogyne incognita. Front. Plant Sci. 2016, 7, 1122. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhao, P.; Zhou, X.; Xiong, H.; Sun, M. Genome-wide identification and characterization of cystatin family genes in rice (Oryza sativa L.). Plant Cell Rep. 2015, 34, 1579–1592. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Li, S.; Chen, M.; Cai, Y.; Xie, Y.; Li, B.; Sun, Q.; Jiang, H.; Pan, Z.; Gao, Y.L. Molecular cloning and expression of cDNA encoding the cysteine proteinase inhibitor from upland cotton. J. Plant Biol. 2009, 52, 426–432. [Google Scholar] [CrossRef]
- Miyaji, T.; Murayama, S.; Kouzuma, Y.; Kimura, N.; Kanost, M.R.; Kramer, K.J.; Yonekura, M. Molecular cloning of a multidomain cysteine protease and protease inhibitor precursor gene from the tobacco hornworm (Manduca sexta) and functional expression of the cathepsin F-like cysteine protease domain. Insect Biochem. Mol. Biol. 2010, 40, 835–846. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Hou, P.; Zhu, H.T.; Li, G.D.; Liu, X.G.; Xie, X.M. Knockout of the VPS22 component of the ESCRT-II complex in rice (Oryza sativa L.) causes chalky endosperm and early seedling lethality. Mol. Biol. Rep. 2013, 40, 3475–3481. [Google Scholar] [CrossRef]
- Ariizumi, T.; Toriyama, K. Genetic regulation of sporopollenin synthesis and pollen exine development. Ann. Rev. Plant Biol. 2011, 62, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Cui, M.; Yang, L.; Kim, Y.J.; Zhang, D. Genetic and biochemical mechanisms of pollen wall development. Trends Plant Sci. 2015, 20, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Shang, B.; Xu, C.; Zhang, X.; Cao, H.; Xin, W.; Hu, Y. Very-long-chain fatty acids restrict regeneration capacity by confining pericycle competence for callus formation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2016, 113, 5101–5106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acosta, I.F.; Laparra, H.; Romero, S.P.; Schmelz, E.; Hamberg, M.; Mottinger, J.P.; Moreno, M.A.; Dellaporta, S.L. tasselseed1 is a lipoxygenase affecting jasmonic acid signaling in sex determination of maize. Science 2009, 323, 262–265. [Google Scholar] [CrossRef] [Green Version]
- Cai, Q.; Yuan, Z.; Chen, M.; Yin, C.; Luo, Z.; Zhao, X.; Liang, W.; Hu, J.; Zhang, D. Jasmonic acid regulates spikelet development in rice. Nat. Comm. 2014, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Yuan, Z.; Zhao, Z.; Li, C.; Zhang, X.; Liang, H.; Liu, Y.; Xu, Q.; Liu, H. Tasselseed5 encodes a cytochrome C oxidase that functions in sex determination by affecting jasmonate catabolism in maize. J. Integr. Plant Biol. 2020, 62, 247–255. [Google Scholar] [CrossRef]
- Yan, Y.; Christensen, S.; Isakeit, T.; Engelberth, J.; Meeley, R.; Hayward, A.; Emery, R.N.; Kolomiets, M.V. Disruption of OPR7 and OPR8 reveals the versatile functions of jasmonic acid in maize development and defense. Plant Cell 2012, 24, 1420–1436. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Li, J.; Garcia-Ruiz, H.; Bates, P.D.; Mirkov, T.E.; Wang, X. A stearoyl-acyl carrier protein desaturase, N b SACPD-C, is critical for ovule development in N icotiana benthamiana. Plant J. 2014, 80, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.U.; Huang, A.H. Plastid lysophosphatidyl acyltransferase is essential for embryo development in Arabidopsis. Plant Physiol. 2004, 134, 1206–1216. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.F.; Xiao, S.; Qi, W.; Mishra, G.; Ma, J.; Wang, M.; Chye, M.L. The Arabidopsis acbp1acbp2 double mutant lacking acyl-CoA-binding proteins ACBP1 and ACBP2 is embryo lethal. New Phytol. 2010, 186, 843–855. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, S.; Kawai-Oda, A.; Ueda, J.; Nishida, I.; Okada, K. The Defective in Anther Dehiscence1 gene encodes a novel phospholipase A1 catalyzing the initial step of jasmonic acid biosynthesis, which synchronizes pollen maturation, anther dehiscence, and flower opening in Arabidopsis. Plant Cell 2001, 13, 2191–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConn, M. The critical requirement for linolenic acid is pollen development, not photosynthesis, in an Arabidopsis mutant. Plant Cell 1996, 8, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Stintzi, A. The Arabidopsis male-sterile mutant, opr3, lacks the 12-oxophytodienoic acid reductase required for jasmonate synthesis. Proc. Natl. Acad. Sci. USA 2000, 97, 10625–10630. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Halitschke, R.; Kim, H.B.; Baldwin, I.T.; Feldmann, K.A.; Feyereisen, R. A knock-out mutation in allene oxide synthase results in male sterility and defective wound signal transduction in Arabidopsis due to a block in jasmonic acid biosynthesis. Plant J. 2002, 31, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, Y.; Charnikhova, T.; Mulder, P.P.; Heijmans, J.; Hoogenboom, A.; Agalou, A.; Michel, C.; Morel, J.B.; Dreni, L. OsJAR1 is required for JA-regulated floret opening and anther dehiscence in rice. Plant Mol. Biol. 2014, 86, 19–33. [Google Scholar] [CrossRef]
- Riemann, M.; Haga, K.; Shimizu, T.; Okada, K.; Ando, S.; Mochizuki, S.; Nishizawa, Y.; Yamanouchi, U.; Nick, P.; Yano, M. Identification of rice Allene Oxide Cyclase mutants and the function of jasmonate for defence against Magnaporthe oryzae. Plant J. 2013, 74, 226–238. [Google Scholar] [CrossRef]
- Kim, E.H.; Kim, Y.S.; Park, S.H.; Koo, Y.J.; Do Choi, Y.; Chung, Y.Y.; Lee, I.J.; Kim, J.K. Methyl jasmonate reduces grain yield by mediating stress signals to alter spikelet development in rice. Plant Physiol. 2009, 149, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, Y.; Duan, E.; Qi, Q.; Zhou, K.; Lin, Q.; Wang, D.; Wang, Y.; Long, W.; Zhao, Z. OPEN GLUME1: A key enzyme reducing the precursor of JA, participates in carbohydrate transport of lodicules during anthesis in rice. Plant Cell Rep. 2018, 37, 329–346. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. 1988. Available online: http://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 14 August 2018).
- Xie, X.; Ma, X.; Zhu, Q.; Zeng, D.; Li, G.; Liu, Y.G. CRISPR-GE: A convenient software toolkit for CRISPR-based genome editing. Mol. Plant 2017, 10, 1246–1249. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Ding, Y.; Zhou, Y.; Jin, W.; Xie, K.; Chen, L.L. CRISPR-P 2.0: An improved CRISPR-Cas9 tool for genome editing in plants. Mol. Plant 2017, 10, 530–532. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Zhang, Q.; Zhu, Q.; Liu, W.; Chen, Y.; Qiu, R.; Wang, B.; Yang, Z.; Li, H.; Lin, Y. A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol. Plant 2015, 8, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Hiei, Y.; Ohta, S.; Komari, T.; Kumashiro, T. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J. 1994, 6, 271–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Xie, X.; Ma, X.; Li, J.; Chen, J.; Liu, Y.G. DSDecode: A web-based tool for decoding of sequencing chromatograms for genotyping of targeted mutations. Mol. Plant 2015, 8, 1431–1433. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Xu, X.Y.; Gong, Q.Q.; Xie, C.; Fan, W.; Yang, J.L.; Lin, Q.S.; Zheng, S.J. Root proteome of rice studied by iTRAQ provides integrated insight into aluminum stress tolerance mechanisms in plants. J. Proteom. 2014, 98, 189–205. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Generation | Genotypes | PH | PN | FLL | FLW | GNPP | GL | GWD | GWT |
|---|---|---|---|---|---|---|---|---|---|
| T0 | WT | 145.3 ± 3.6 | 9.5 ± 1.6 | 60.2 ± 2.3 | 2.6 ± 0.3 | 148.6 ± 0.9 | 8.7 ± 0.2 | 2.9 ± 0.1 | 31.3 ± 1.3 |
| GXU27-3 | 144.6 ± 4.2 ns | 9.7 ± 2.2 ns | 59.9 ± 3.1 ns | 2.5 ± 0.5 ns | 147.4 ± 0.8 ns | 11.1 ± 0.3 * | 3.0 ± 0.3 ns | 39.8 ± 1.2 * | |
| GXU27-4 | 146.4 ± 5.6 ns | 9.6 ± 1.4 ns | 60.3 ± 2.2 ns | 2.6 ± 0.4 ns | 146.5 ± 0.7 ns | 10.8 ± 0.5 * | 2.9 ± 0.2 ns | 39.1 ± 1.6 * | |
| GXU27-9 | 145.9 ± 4.8 ns | 9.7 ± 1.8 ns | 59.8 ± 2.5 ns | 2.7 ± 0.3 ns | 149.2 ± 0.8 ns | 10.9 ± 0.2 * | 2.9 ± 0.1 ns | 39.0 ± 1.4 * | |
| T1 | WT | 146.5 ± 2.8 | 9.6 ± 1.5 | 61.3 ± 2.6 | 2.5 ± 0.2 | 149.4 ± 0.6 | 8.8 ± 0.3 | 3.0 ± 0.1 | 32.1 ± 1.1 |
| GXU27-3 | 145.4 ± 3.4 ns | 9.8 ± 1.9 ns | 60.2 ± 2.4 ns | 2.7 ± 0.2 ns | 148.5 ± 0.9 ns | 11.3 ± 0.2 * | 3.1 ± 0.1 ns | 39.7 ± 1.4 * | |
| GXU27-4 | 147.3 ± 4.2 ns | 9.8 ± 1.2 ns | 59.9 ± 2.5 ns | 2.4 ± 0.5 ns | 148.2 ± 0.5 ns | 10.9 ± 0.3 * | 3.0 ± 0.2 ns | 39.3 ± 1.0 * | |
| GXU27-9 | 145.8 ± 3.5 ns | 9.6 ± 1.4 ns | 60.1 ± 2.2 ns | 2.6 ± 0.4 ns | 149.6 ± 0.4 ns | 10.6 ± 0.4 * | 2.9 ± 0.2 ns | 39.2 ± 1.2 * | |
| T2 | WT | 144.6 ± 2.8 | 9.8 ± 1.3 | 59.9 ± 2.4 | 2.7 ± 0.2 | 148.2 ± 0.6 | 8.6 ± 0.3 | 2.9 ± 0.2 | 31.6 ± 1.2 |
| GXU27-3 | 144.8 ± 3.7 ns | 9.6 ± 2.0 ns | 60.1 ± 3.2 ns | 2.6 ± 0.3 ns | 145.4 ± 0.6 ns | 11.2 ± 0.4 * | 3.0 ± 0.1 ns | 39.6 ± 1.5 * | |
| GXU27-4 | 145.9 ± 3.5 ns | 9.8 ± 1.6 ns | 60.0 ± 2.3 ns | 2.6 ± 0.4 ns | 147.5 ± 0.5 ns | 10.9 ± 0.3 * | 3.0 ± 0.1 ns | 39.3 ± 1.2 * | |
| GXU27-9 | 145.3 ± 4.2 ns | 9.9 ± 1.7 ns | 59.9 ± 2.4 ns | 2.5 ± 0.2 ns | 149.3 ± 0.7 ns | 10.7 ± 0.2 * | 2.9 ± 0.3 ns | 39.1 ± 1.3 * |
| Protein ID | Protein Names | log2 FC | Regulation |
|---|---|---|---|
| A2ZMY2 | Cysteine synthase | 1.66 | Up |
| Q5JNB0 | Cysteine synthase | 1.55 | Up |
| B8AJV7 | Cysteine synthase | 1.46 | Up |
| Q0JNR2 | Cysteine proteinase inhibitor 12 | −2.38 | Down |
| A0A0A7EQF3 | Cysteine proteinase inhibitor | −2.66 | Down |
| P20907 | Cysteine proteinase inhibitor 2 | −2.30 | Down |
| A2XEA1 | Ubiquitin | −1.50 | Down |
| Q10NQ3 | Vacuolar protein sorting-associated protein 9A | 2.87 | Up |
| A2X377 | Vacuolar protein sorting-associated protein 29 | 3.12 | Up |
| A0A0E0GXY5 | Vacuolar protein sorting-associated protein 41 homolog | 3.09 | Up |
| Q8H8K1 | Putative vacuolar sorting receptor protein | 2.13 | Up |
| Q6L4S0 | DNA damage-binding protein 1 | 1.86 | Up |
| Q7XD67 | DNA ligase | 3.83 | Up |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usman, B.; Zhao, N.; Nawaz, G.; Qin, B.; Liu, F.; Liu, Y.; Li, R. CRISPR/Cas9 Guided Mutagenesis of Grain Size 3 Confers Increased Rice (Oryza sativa L.) Grain Length by Regulating Cysteine Proteinase Inhibitor and Ubiquitin-Related Proteins. Int. J. Mol. Sci. 2021, 22, 3225. https://doi.org/10.3390/ijms22063225
Usman B, Zhao N, Nawaz G, Qin B, Liu F, Liu Y, Li R. CRISPR/Cas9 Guided Mutagenesis of Grain Size 3 Confers Increased Rice (Oryza sativa L.) Grain Length by Regulating Cysteine Proteinase Inhibitor and Ubiquitin-Related Proteins. International Journal of Molecular Sciences. 2021; 22(6):3225. https://doi.org/10.3390/ijms22063225
Chicago/Turabian StyleUsman, Babar, Neng Zhao, Gul Nawaz, Baoxiang Qin, Fang Liu, Yaoguang Liu, and Rongbai Li. 2021. "CRISPR/Cas9 Guided Mutagenesis of Grain Size 3 Confers Increased Rice (Oryza sativa L.) Grain Length by Regulating Cysteine Proteinase Inhibitor and Ubiquitin-Related Proteins" International Journal of Molecular Sciences 22, no. 6: 3225. https://doi.org/10.3390/ijms22063225
APA StyleUsman, B., Zhao, N., Nawaz, G., Qin, B., Liu, F., Liu, Y., & Li, R. (2021). CRISPR/Cas9 Guided Mutagenesis of Grain Size 3 Confers Increased Rice (Oryza sativa L.) Grain Length by Regulating Cysteine Proteinase Inhibitor and Ubiquitin-Related Proteins. International Journal of Molecular Sciences, 22(6), 3225. https://doi.org/10.3390/ijms22063225