
Quantitative Proteomic Approach Reveals Altered Metabolic Pathways in Response to the Inhibition of Lysine Deacetylases in A549 Cells under Normoxia and Hypoxia

 , ,
, , 
 , and
, and 
Abstract
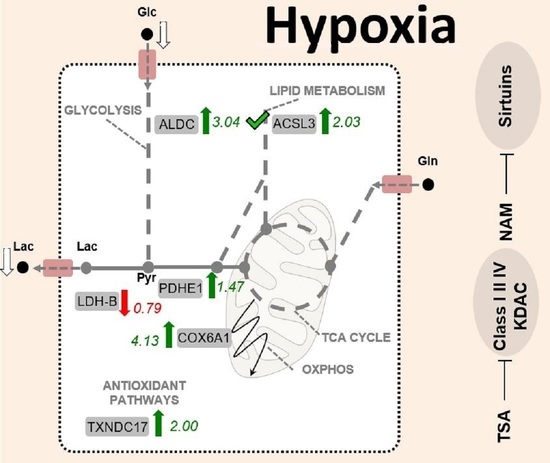
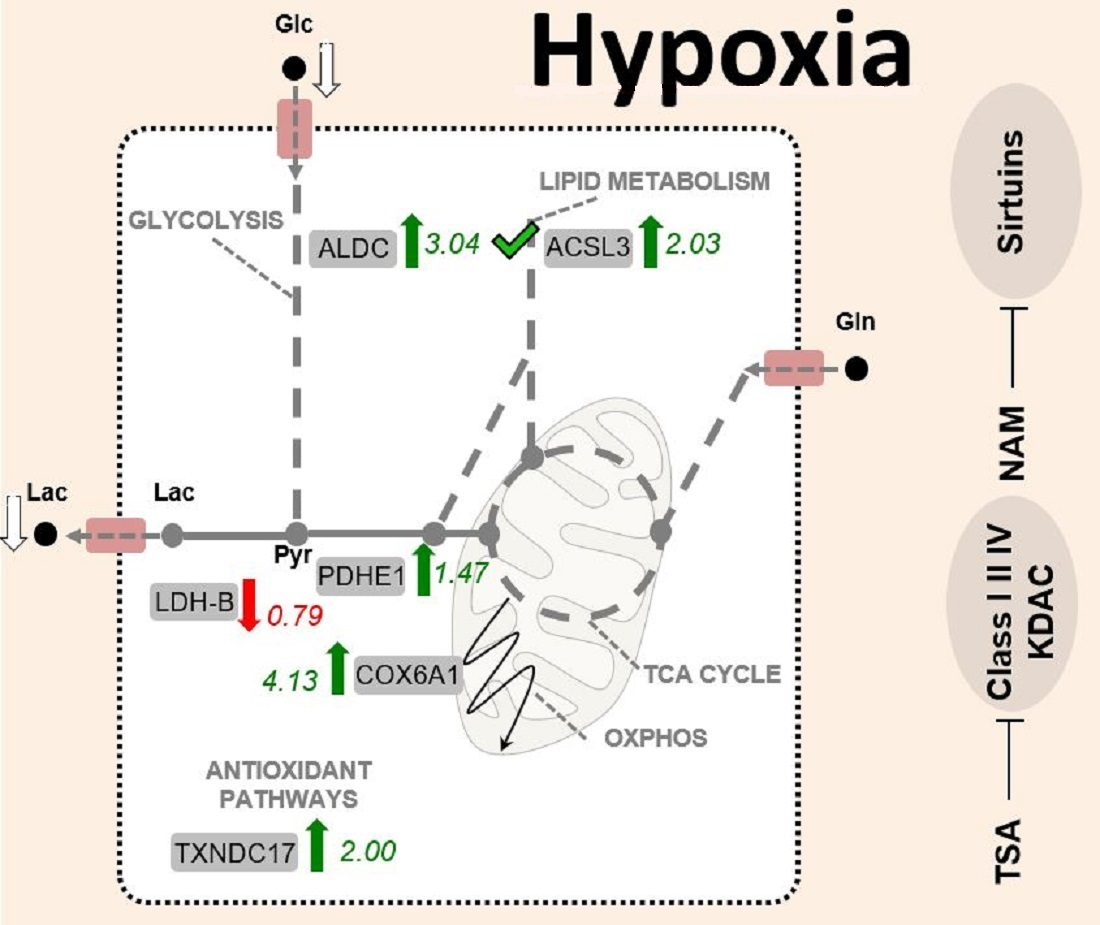
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. KDAC Inhibition Leads to Reduced Cell Proliferation by Inducing Apoptosis, Cell Cycle Arrest, and Oxidative Stress in A549 Cells
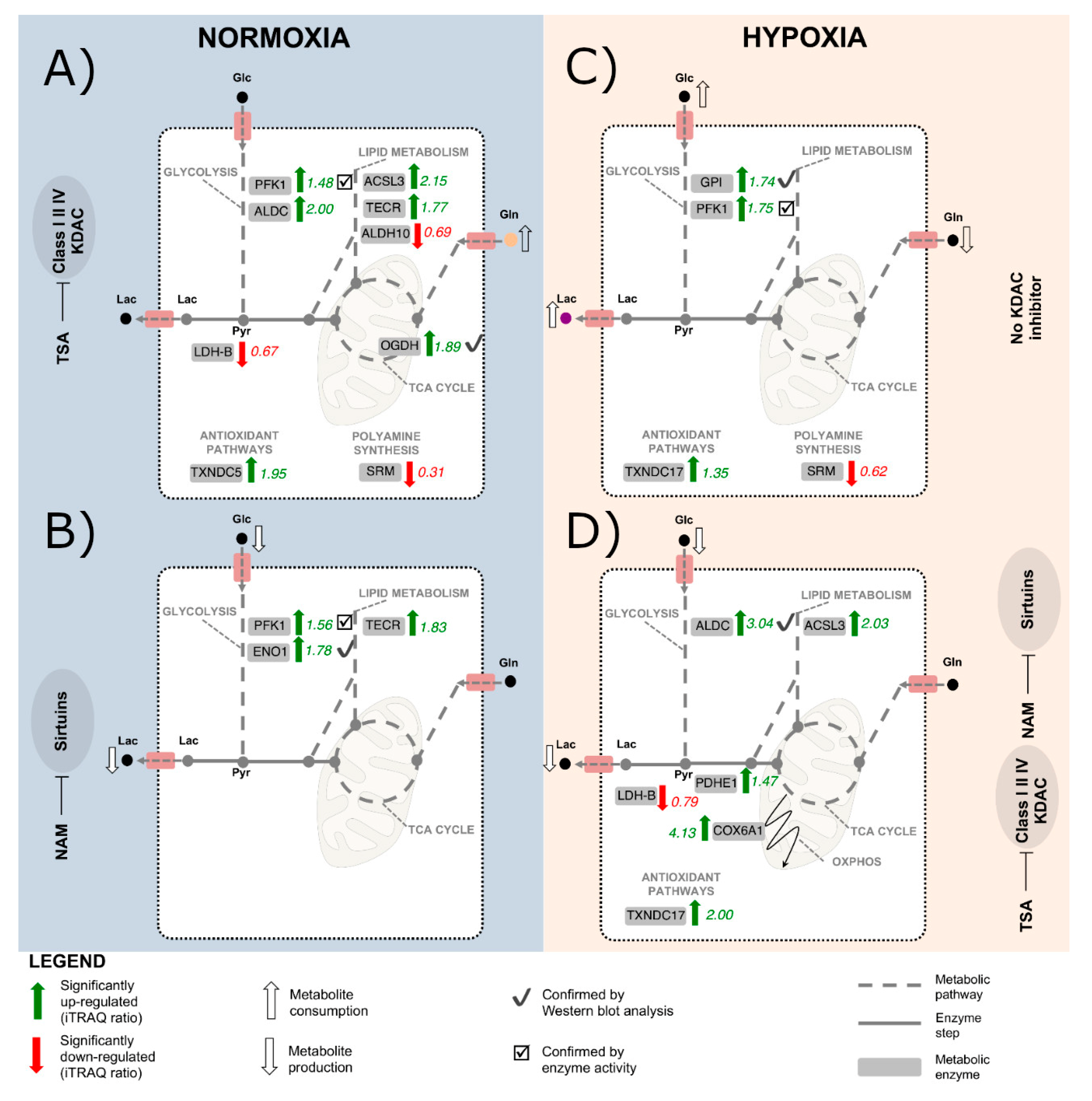
2.2. KDAC Inhibition Modulates the Tumor Phenotype through Changes in the Metabolic Profile
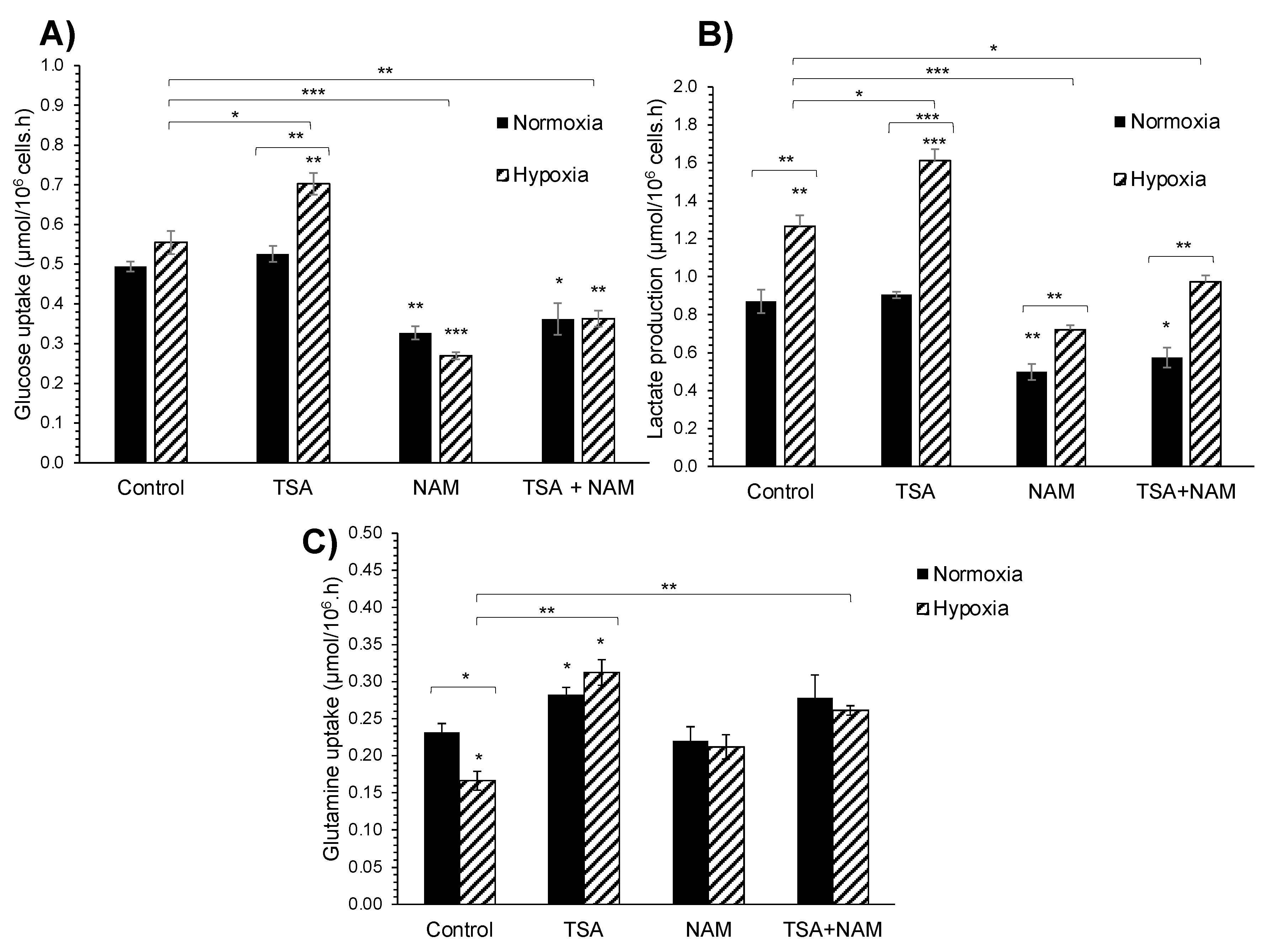
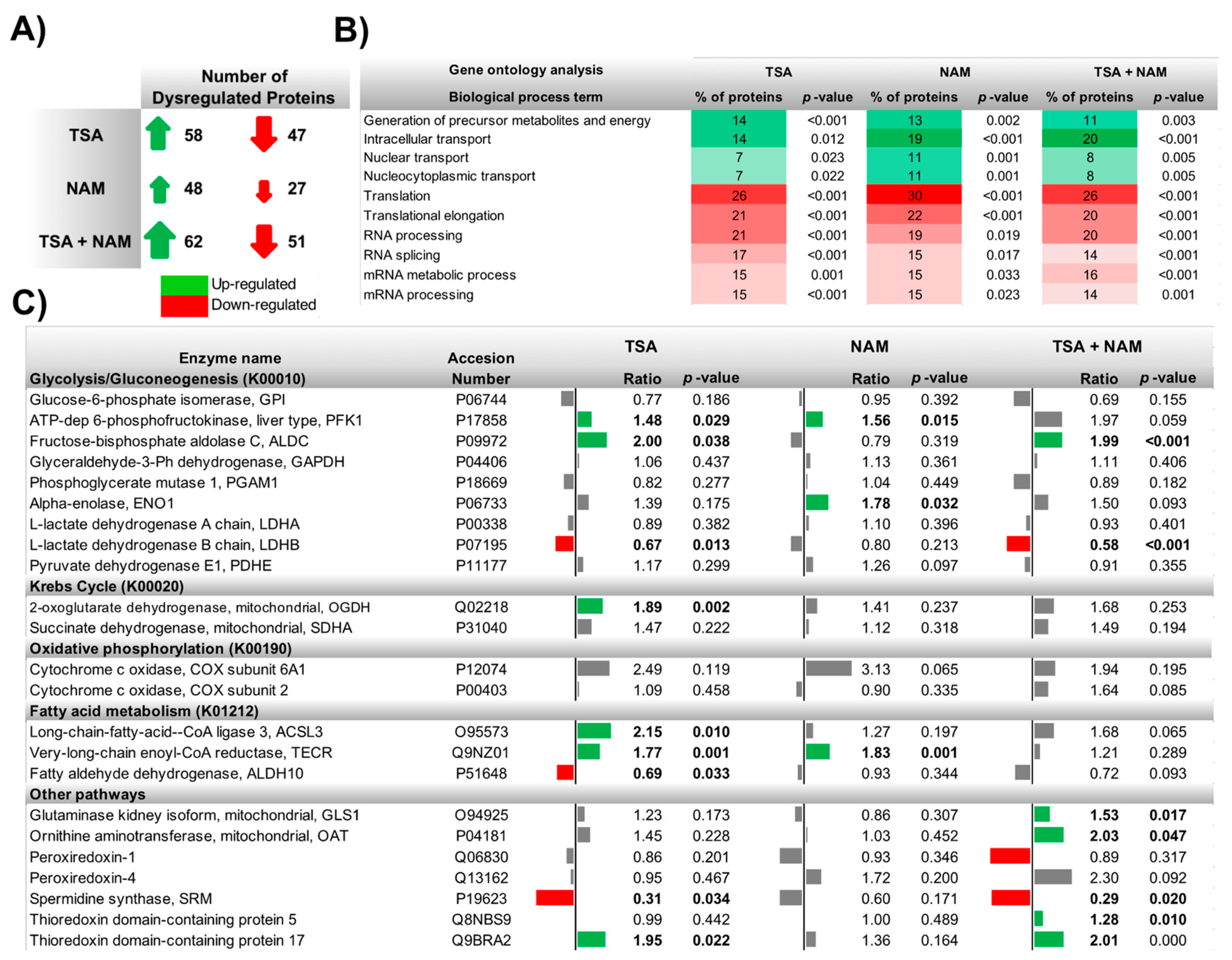
2.3. The Metabolic Changes Observed in KDAC Inhibition Are Enhanced under Hypoxia
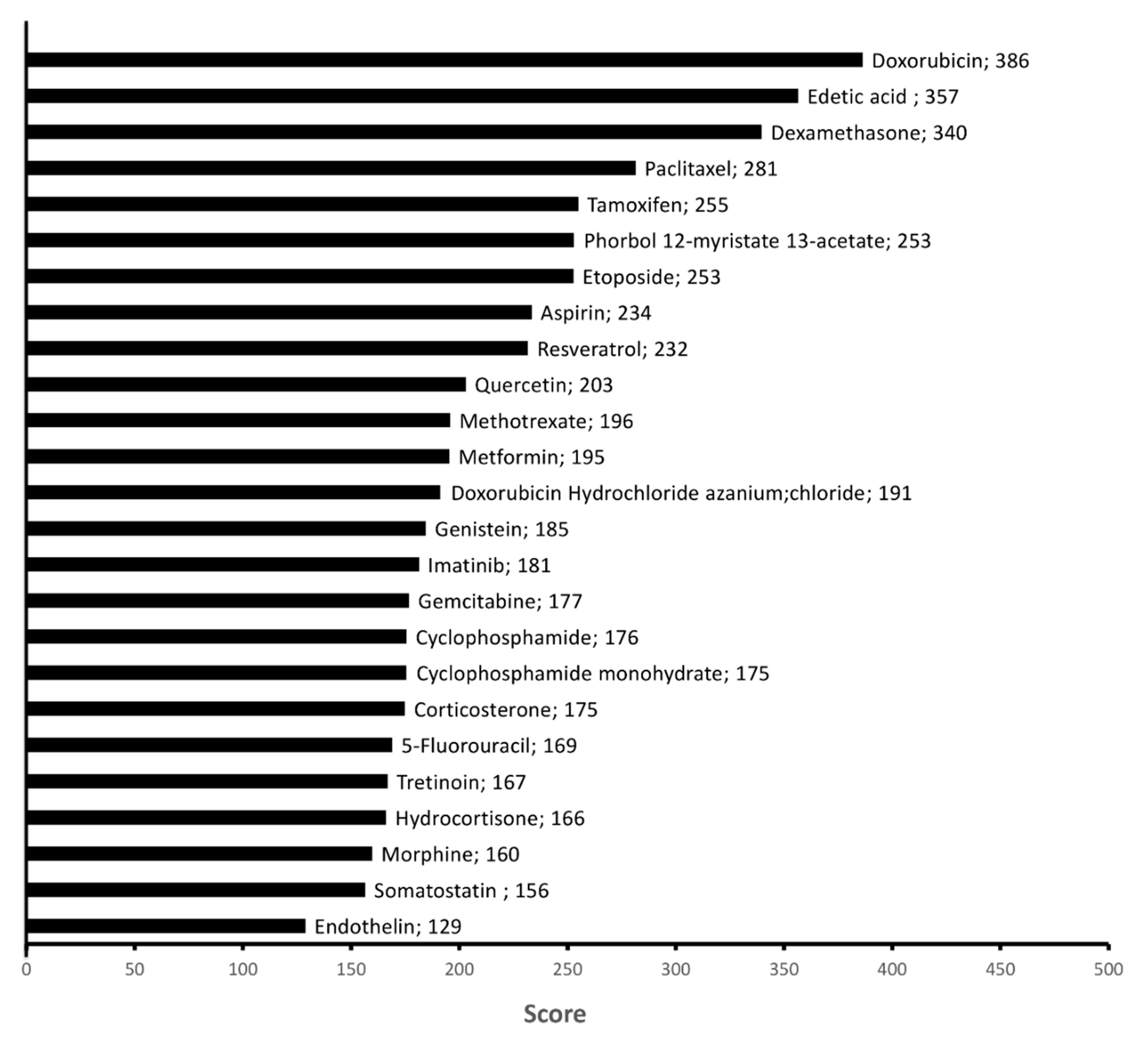
2.4. Chemicals Targeting Proteins Affected by KDAC Inhibition under Hypoxia
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability Assay
4.3. Cell Proliferation Assay
4.4. Apoptosis Assay
4.5. Cell Cycle Analysis
4.6. Measurement of Extracellular Metabolites
4.7. Determination of Intracellular ROS levels
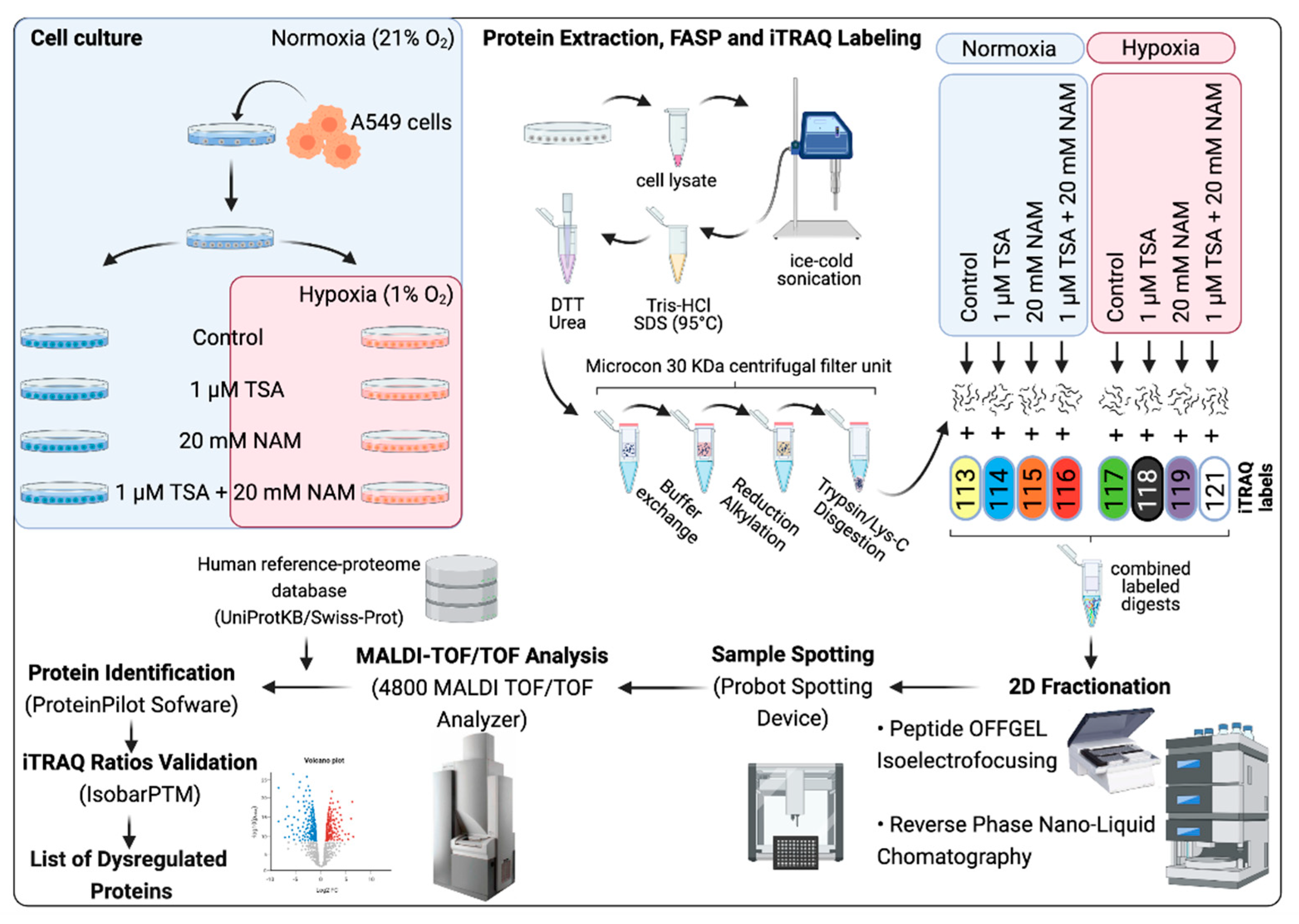
4.8. Proteomic Analysis
4.9. Filter-Aided Sample Preparation for iTRAQ Quantitation
4.9.1. iTRAQ Labeling
4.9.2. Two-dimensional (2D) fractionation: Peptide OFFGEL Isoelectrofocusing and Reversed Phase Nano-liquid Chromatography
4.9.3. MALDI-TOF/TOF Analysis
4.9.4. Database Search and Quantitative iTRAQ Analysis
4.10. Gene Ontology Enrichment Analysis
4.11. Western Blot Analysis
4.12. Enzyme Activities
4.13. Network Analysis by Node Embeddings
4.14. Experimental Design and Statistical Rationale
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AcCoA | Acetyl-CoA |
| ACSL3 | Long-chain-fatty-acid-CoA ligase 3 |
| ALDC | Fructose-bisphosphate aldolase C |
| ALDH10 | Fatty aldehyde dehydrogenase |
| ALK | Anaplastic lymphoma kinase |
| CID | Collision-induced dissociation |
| COX6A1 | Cytochrome c oxidase subunit 6A1 |
| COX2 | Cytochrome c oxidase subunit 2 |
| EGFR | Epidermal growth factor receptor |
| ENO1 | Alpha-enolase |
| ER | Estrogen receptor |
| FDR | False discovery rate |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| Glc | Glucose |
| Gln | Glutamine |
| GLS | Glutaminase |
| GLS1 | Glutaminase 1 |
| GO | Gene ontology |
| GPI | Glucose-6-phosphate isomerase |
| HIF | Hypoxia-inducible factor |
| HK | Hexokinase |
| IPG | Immobilized pH gradient |
| iTRAQ | Isobaric tags for relative and absolute quantitation |
| KDAC | Lysine deacetylase |
| KDAC6 | Lysine deacetylase 6 |
| KDACI | Lysine deacetylase inhibitor |
| Lac | Lactate |
| LC | Liquid chromatography |
| LDH | Lactate dehydrogenase |
| NAM | Nicotinamide |
| NSCLC | Non-small cell lung cancer |
| MMTS | Methylmethanethiosulfate |
| OAT | Ornithine aminotransferase |
| OGDH | 2-oxoglutarate dehydrogenase |
| OXPHOS | Oxidative phosphorylation |
| PFK1 | ATP-dependent 6-phosphofructokinase |
| PDHE | Pyruvate dehydrogenase subunit E |
| PGI | Glucose-6-phosphate isomerase |
| Pyr | Pyruvate |
| pI | Isoelectric point |
| PI | Propidium iodide |
| PIR | Protein information resource |
| PPP | Pentose phosphate pathway |
| PRDX1 | Peroxiredoxin-1 |
| PRDX4 | Peroxiredoxin-4 |
| PSME1 | Proteasome activator complex subunit 1 |
| PSME2 | Proteasome activator complex subunit 2 |
| ROS | Reactive oxygen species |
| SAHA | Suberoylanilide hydroxamic acid |
| SDHA | Succinate dehydrogenase complex subunit A |
| SIRT | Sirtuin |
| SIRT1 | Sirtuin 1 |
| SRM | Spermidine synthase |
| RP | Reversed phase |
| TCA | Tricarboxylic acid |
| TEAB | Triethylammonium bicarbonate buffer |
| TECR | Very-long-chain enoyl-CoA reductase |
| TKI | Tyrosine kinase inhibitor |
| TRX | Thioredoxin |
| TSA | Trichostatin A |
| TXNDC17 | Thioredoxin domain-containing protein 17 |
| TXNDC5 | Thioredoxin domain-containing protein 5 |
| UBA1 | Ubiquitin-like modifier-activating enzyme 1 |
| Wb | Western blot |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Lim, Z.-F.; Ma, P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019, 12, 1–18. [Google Scholar] [CrossRef]
- Jeannot, V.; Busser, B.; Brambilla, E.; Wislez, M.; Robin, B.; Cadranel, J.; Coll, J.-L.; Hurbin, A. The PI3K/AKT pathway promotes gefitinib resistance in mutant KRAS lung adenocarcinoma by a deacetylase-dependent mechanism. Int. J. Cancer 2013, 134, 2560–2571. [Google Scholar] [CrossRef]
- Saliani, M.; Jalal, R.; Ahmadian, M.R. From basic researches to new achievements in therapeutic strategies of KRAS-driven cancers. Cancer Biol. Med. 2019, 16, 435–461. [Google Scholar]
- Kerr, E.M.; Martins, C.P. Metabolic rewiring in mutant Kras lung cancer. FEBS J. 2018, 285, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-Driven Metabolic Rewiring Reveals Novel Actionable Targets in Cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Bober, P.; Alexovič, M.; Tomková, Z.; Kilík, R.; Sabo, J. RHOA and mDia1 promotes apoptosis of breast cancer cells via a high dose of doxorubicin treatment. Open Life Sci. 2019, 14, 619–627. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Benito, A.; Diaz-Moralli, S.; Coy, J.F.; Centelles, J.J.; Cascante, M. Role of the Pentose Phosphate Pathway in Tumour Metabolism. In Tumor Cell Metabolism: Pathways, Regulation and Biology; Mazurek, S., Shoshan, M., Eds.; Springer: Wien, Austria, 2015; pp. 143–163. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wu, M. Regulation of the pentose phosphate pathway in cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Martín-Bernabé, A.; Cortés, R.; Lehmann, S.G.; Seve, M.; Cascante, M.; Bourgoin-Voillard, S. Quantitative Proteomic Approach to Understand Metabolic Adaptation in Non-Small Cell Lung Cancer. J. Proteome Res. 2014, 13, 4695–4704. [Google Scholar] [CrossRef] [PubMed]
- Minakata, K.; Takahashi, F.; Nara, T.; Hashimoto, M.; Tajima, K.; Murakami, A.; Nurwidya, F.; Yae, S.; Koizumi, F.; Moriyama, H.; et al. Hypoxia induces gefitinib resistance in non-small-cell lung cancer with both mutant and wild-type epidermal growth factor receptors. Cancer Sci. 2012, 103, 1946–1954. [Google Scholar] [CrossRef]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Liu, X.; Chi, W.; Liu, Y.; Wei, L.; Wang, X.; Yu, J. Hypoxia-induced resistance to cisplatin and doxorubicin in non-small cell lung cancer is inhibited by silencing of HIF-1α gene. Cancer Chemother. Pharmacol. 2006, 58, 776–784. [Google Scholar] [CrossRef]
- Wu, H.-M.; Jiang, Z.-F.; Ding, P.-S.; Shao, L.-J.; Liu, R.-Y. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci. Rep. 2015, 5, 12291. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 Is Required for AMPK Activation and the Beneficial Effects of Resveratrol on Mitochondrial Function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Xu, W.-S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell. Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef]
- Verza, F.A.; Das, U.; Fachin, A.L.; Dimmock, J.R.; Marins, M. Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy. Cancers 2020, 12, 1664. [Google Scholar] [CrossRef] [PubMed]
- Martín-Bernabé, A.; Balcells, C.; Tarragó-Celada, J.; Foguet, C.; Bourgoin-Voillard, S.; Seve, M.; Cascante, M. The importance of post-translational modifications in systems biology approaches to identify therapeutic targets in cancer metabolism. Curr. Opin. Syst. Biol. 2017, 3, 161–169. [Google Scholar] [CrossRef]
- Huang, M.; Geng, M. Exploiting histone deacetylases for cancer therapy: from hematological malignancies to solid tumors. Sci. China Life Sci. 2016, 60, 94–97. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy with Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Tomos, I.; Garmpis, N.; Karakatsani, A.; Dimitroulis, D.; Garmpi, A.; Spartalis, E.; Kampolis, C.F.; Tsagkari, E.; A Loukeri, A.; et al. Histone Deacetylase Inhibitors as a Novel Targeted Therapy Against Non-small Cell Lung Cancer: Where Are We Now and What Should We Expect? Anticancer Res. 2018, 38, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.W.; Sequist, L.V. Complex Role of Histone Deacetylase Inhibitors in the Treatment of Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2012, 30, 2280–2282. [Google Scholar] [CrossRef]
- Wang, L.; Li, H.; Ren, Y.; Zou, S.; Fang, W.; Jiang, X.; Jia, L.; Li, M.; Liu, X.; Yuan, X.; et al. Targeting HDAC with a novel inhibitor effectively reverses paclitaxel resistance in non-small cell lung cancer via multiple mechanisms. Cell Death Dis. 2016, 7, e2063. [Google Scholar] [CrossRef] [PubMed]
- Witta, S. Histone Deacetylase Inhibitors in Non–Small-Cell Lung Cancer. J. Thorac. Oncol. 2012, 7, S404–S406. [Google Scholar] [CrossRef]
- Mukhopadhyay, N.K.; Weisberg, E.; Gilchrist, D.; Bueno, R.; Sugarbaker, D.J.; Jaklitsch, M.T. Effectiveness of Trichostatin A as a Potential Candidate for Anticancer Therapy in Non–Small-Cell Lung Cancer. Ann. Thorac. Surg. 2006, 81, 1034–1042. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Indispensable for p53 Activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Guan, K.-L.; Xiong, Y. Regulation of intermediary metabolism by protein acetylation. Trends Biochem. Sci. 2011, 36, 108–116. [Google Scholar] [CrossRef]
- Cheung, C.H.Y.; Juan, H.-F. Quantitative proteomics in lung cancer. J. Biomed. Sci. 2017, 24, 1–11. [Google Scholar] [CrossRef]
- Vinaiphat, A.; Low, J.K.; Yeoh, K.W.; Chng, W.J.; Sze, S.K. Application of Advanced Mass Spectrometry-Based Proteomics to Study Hypoxia Driven Cancer Progression. Front. Oncol. 2021, 11, 98. [Google Scholar] [CrossRef]
- Bush, J.T.; Chan, M.C.; Mohammed, S.; Schofield, C.J. Quantitative MS-Based Proteomics: Comparing the MCF-7 Cellular Response to Hypoxia and a 2-Oxoglutarate Analogue. ChemBioChem 2020, 21, 1647–1655. [Google Scholar] [CrossRef]
- Song, Z.; Pearce, M.C.; Jiang, Y.; Yang, L.; Goodall, C.; Miranda, C.L.; Milovancev, M.; Bracha, S.; Kolluri, S.K.; Maier, C.S. Delineation of hypoxia-induced proteome shifts in osteosarcoma cells with different metastatic propensities. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, P.; Sowers, J.L.; Machuca, D.F.; Mirfattah, B.; Herring, J.; Tang, H.; Chen, Y.; Tian, B.; Brasier, A.R.; et al. Proteome Analysis of Hypoxic Glioblastoma Cells Reveals Sequential Metabolic Adaptation of One-Carbon Metabolic Pathways. Mol. Cell. Proteom. 2017, 16, 1906–1921. [Google Scholar] [CrossRef] [PubMed]
- Djidja, M.-C.; Chang, J.; Hadjiprocopis, A.; Schmich, F.; Sinclair, J.; Mršnik, M.; Schoof, E.M.; Barker, H.E.; Linding, R.; Jorgensen, C.B.; et al. Identification of Hypoxia-Regulated Proteins Using MALDI-Mass Spectrometry Imaging Combined with Quantitative Proteomics. J. Proteome Res. 2014, 13, 2297–2313. [Google Scholar] [CrossRef]
- Bousquet, P.A.; Sandvik, J.A.; Arntzen, M.Ø.; Edin, N.F.J.; Christoffersen, S.; Krengel, U.; Pettersen, E.O.; Thiede, B. Hypoxia Strongly Affects Mitochondrial Ribosomal Proteins and Translocases, as Shown by Quantitative Proteomics of HeLa Cells. Int. J. Proteom. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, F.; Zhao, Y.; Moreira, V.M.; Tagliaferri, R.; Kere, J.; D’Amato, M.; Greco, D. Drug repositioning: a machine-learning approach through data integration. J. Cheminform. 2013, 5, 30. [Google Scholar] [CrossRef]
- Aliper, A.; Plis, S.; Artemov, A.; Ulloa, A.; Mamoshina, P.; Zhavoronkov, A. Deep Learning Applications for Predicting Pharmacological Properties of Drugs and Drug Repurposing Using Transcriptomic Data. Mol. Pharm. 2016, 13, 2524–2530. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Puhl, A.C.; Zorn, K.M.; Lane, T.R.; Russo, D.P.; Klein, J.J.; Hickey, A.J.; Clark, A.M. Exploiting machine learning for end-to-end drug discovery and development. Nat. Mater. 2019, 18, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Li, F.-L.; Cheng, Z.-L.; Lei, Q.-Y. Impact of acetylation on tumor metabolism. Mol. Cell. Oncol. 2014, 1, e963452. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.P.; Iliopoulos, D.; Rao, A.S.; Druck, T.; Semba, S.; Han, S.-Y.; McCorkell, K.A.; Lakshman, T.V.; Collins, J.E.; Wachsberger, P.; et al. Epigenetic modulation of endogenous tumor suppressor expression in lung cancer xenografts suppresses tumorigenicity. Int. J. Cancer 2006, 120, 24–31. [Google Scholar] [CrossRef]
- Chang, J.; Varghese, D.S.; Gillam, M.C.; Peyton, M.; Modi, B.; Schiltz, R.L.; Girard, L.; Martinez, E.D. Differential response of cancer cells to HDAC inhibitors trichostatin A and depsipeptide. Br. J. Cancer 2011, 106, 116–125. [Google Scholar] [CrossRef]
- Miyanaga, A.; Gemma, A.; Noro, R.; Kataoka, K.; Matsuda, K.; Nara, M.; Okano, T.; Seike, M.; Yoshimura, A.; Kawakami, A.; et al. Antitumor activity of histone deacetylase inhibitors in non-small cell lung cancer cells: development of a molecular predictive model. Mol. Cancer Ther. 2008, 7, 1923–1930. [Google Scholar] [CrossRef]
- Rosato, R.R.; Almenara, J.A.; Dai, Y.; Grant, S. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol. Cancer Ther. 2003, 2, 1273–1284. [Google Scholar]
- Amoedo, N.D.; Rodrigues, M.F.; Pezzuto, P.; Galina, A.; Da Costa, R.M.; De Almeida, F.C.L.; El-Bacha, T.; Rumjanek, F.D. Energy Metabolism in H460 Lung Cancer Cells: Effects of Histone Deacetylase Inhibitors. PLoS ONE 2011, 6, e22264. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.H. Induction of apoptosis by trichostatin A, a histone deacetylase inhibitor, is associated with inhibition of cyclooxygenase-2 activity in human non-small cell lung cancer cells. Int. J. Oncol. 2005, 27, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shao, K.; Sun, T. SIRT1 Regulates the Human Alveolar Epithelial A549 Cell Apoptosis Induced byPseudomonas AeruginosaLipopolysaccharide. Cell. Physiol. Biochem. 2013, 31, 92–101. [Google Scholar] [CrossRef]
- Platta, C.S.; Greenblatt, D.Y.; Kunnimalaiyaan, M.; Chen, H. The HDAC Inhibitor Trichostatin A Inhibits Growth of Small Cell Lung Cancer Cells. J. Surg. Res. 2007, 142, 219–226. [Google Scholar] [CrossRef]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of Cellular Metabolism by Protein Lysine Acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Cheng, Z.; Zhu, J.; Xu, W.; Peng, X.; Chen, C.; Li, W.; Wang, F.; Cao, L.; Yi, X.; et al. Suberoylanilide Hydroxamic Acid Treatment Reveals Crosstalks among Proteome, Ubiquitylome and Acetylome in Non-Small Cell Lung Cancer A549 Cell Line. Sci. Rep. 2015, 5, 9520. [Google Scholar] [CrossRef]
- Narayan, S.; Bader, G.D.; Reimand, J. Frequent mutations in acetylation and ubiquitination sites suggest novel driver mechanisms of cancer. Genome Med. 2016, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2012, 280, 775–793. [Google Scholar] [CrossRef] [PubMed]
- Kulka, L.A.M.; Fangmann, P.-V.; Panfilova, D.; Olzscha, H. Impact of HDAC Inhibitors on Protein Quality Control Systems: Consequences for Precision Medicine in Malignant Disease. Front. Cell Dev. Biol. 2020, 8, 425. [Google Scholar] [CrossRef] [PubMed]
- Heider, U.; Rademacher, J.; Lamottke, B.; Mieth, M.; Moebs, M.; Von Metzler, I.; Assaf, C.; Sezer, O. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in cutaneous T cell lymphoma. Eur. J. Haematol. 2009, 82, 440–449. [Google Scholar] [CrossRef]
- Laporte, A.N.; Poulin, N.M.; Barrott, J.J.; Wang, X.Q.; Lorzadeh, A.; Werff, R.V.; Jones, K.B.; Underhill, T.M.; Nielsen, T.O. Death by HDAC Inhibition in Synovial Sarcoma Cells. Mol. Cancer Ther. 2017, 16, 2656–2667. [Google Scholar] [CrossRef]
- Park, M.-T.; Kim, M.-J.; Suh, Y.; Kim, R.-K.; Kim, H.; Lim, E.-J.; Yoo, K.-C.; Lee, G.-H.; Kim, Y.-H.; Hwang, S.-G.; et al. Novel signaling axis for ROS generation during K-Ras-induced cellular transformation. Cell Death Differ. 2014, 21, 1185–1197. [Google Scholar] [CrossRef]
- Ralph, S.J.; Rodríguez-Enríquez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sánchez, R. The causes of cancer revisited: “Mitochondrial malignancy” and ROS-induced oncogenic transformation – Why mitochondria are targets for cancer therapy. Mol. Asp. Med. 2010, 31, 145–170. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Gong, K.; Xie, J.; Yi, H.; Li, W. CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS-dependent apoptosis and differentiation in human leukaemia cells. Biochem. J. 2012, 443, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.R.; Almenara, J.A.; Maggio, S.C.; Coe, S.; Atadja, P.; Dent, P.; Grant, S. Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol. Cancer Ther. 2008, 7, 3285–3297. [Google Scholar] [CrossRef] [PubMed]
- You, B.R.; Park, W.H. Trichostatin A induces apoptotic cell death of HeLa cells in a Bcl-2 and oxidative stress-dependent manner. Int. J. Oncol. 2012, 42, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Kinnula, V.L.; Pääkkö, P.; Soini, Y. Antioxidant enzymes and redox regulating thiol proteins in malignancies of human lung. FEBS Lett. 2004, 569, 1–6. [Google Scholar] [CrossRef]
- Zhang, S.-F.; Wang, X.-Y.; Fu, Z.-Q.; Peng, Q.-H.; Zhang, J.-Y.; Ye, F.; Fu, Y.-F.; Zhou, C.-Y.; Lu, W.-G.; Cheng, X.-D.; et al. TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy 2015, 11, 225–238. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, A.; Li, H.; Zhi, H.; Lu, F. RETRACTED: STAT3-dependent TXNDC17 expression mediates Taxol resistance through inducing autophagy in human colorectal cancer cells. Gene 2016, 584, 75–82. [Google Scholar] [CrossRef]
- Xiong, Y.; Guan, K.-L. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J. Cell Biol. 2012, 198, 155–164. [Google Scholar] [CrossRef]
- Guarente, L. The Logic Linking Protein Acetylation and Metabolism. Cell Metab. 2011, 14, 151–153. [Google Scholar] [CrossRef]
- Smith, B.; Schafer, X.L.; Ambeskovic, A.; Spencer, C.M.; Land, H.; Munger, J. Addiction to Coupling of the Warburg Effect with Glutamine Catabolism in Cancer Cells. Cell Rep. 2016, 17, 821–836. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Zha, X.; Wang, F.; Wang, Y.; He, S.; Jing, Y.; Wu, X.; Zhang, H. Lactate Dehydrogenase B Is Critical for Hyperactive mTOR-Mediated Tumorigenesis. Cancer Res. 2011, 71, 13. [Google Scholar] [CrossRef] [PubMed]
- Brisson, L.; Bański, P.; Sboarina, M.; Dethier, C.; Danhier, P.; Fontenille, M.-J.; Van Hée, V.F.; Vazeille, T.; Tardy, M.; Falces, J.; et al. Lactate Dehydrogenase B Controls Lysosome Activity and Autophagy in Cancer. Cancer Cell 2016, 30, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty Acid Oxidation Mediated by Acyl-CoA Synthetase Long Chain 3 Is Required for Mutant KRAS Lung Tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar] [CrossRef] [PubMed]
- Saliakoura, M.; Reynoso-Moreno, I.; Pozzato, C.; Sebastiano, M.R.; Galié, M.; Gertsch, J.; Konstantinidou, G. The ACSL3-LPIAT1 signaling drives prostaglandin synthesis in non-small cell lung cancer. Oncogene 2020, 39, 2948–2960. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nat. Cell Biol. 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Hardie, D.G. Energy sensing by the AMP-activated protein kinase and its effects on muscle metabolism. Proc. Nutr. Soc. 2010, 70, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sánchez, R.; Rodriguez-Enriquez, S.; Saavedra, E.; Marin-Hernandez, A.; Gallardo-Perez, J.C. The bioenergetics of cancer: is glycolysis the main ATP supplier in all tumor cells? BioFactors 2009, 35, 209–225. [Google Scholar] [CrossRef]
- Li, P.; Zhang, D.; Shen, L.; Dong, K.; Wu, M.; Ou, Z.; Shi, D. Redox homeostasis protects mitochondria through accelerating ROS conversion to enhance hypoxia resistance in cancer cells. Sci. Rep. 2016, 6, 22831. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Kong, X.; Lin, Z.; Liang, D.; Fath, D.; Sang, N.; Caro, J. Histone Deacetylase Inhibitors Induce VHL and Ubiquitin-Independent Proteasomal Degradation of Hypoxia-Inducible Factor 1α. Mol. Cell. Biol. 2006, 26, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Emerling, B.M.; Ricoult, S.J.H.; Guarente, L.P. SirT3 suppresses hypoxia inducible factor 1α and tumor growth by inhibiting mitochondrial ROS production. Oncogene 2011, 30, 2986–2996. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-H.; Lee, Y.-M.; Chun, Y.-S.; Chen, J.; Kim, J.-E.; Park, J.-W. Sirtuin 1 Modulates Cellular Responses to Hypoxia by Deacetylating Hypoxia-Inducible Factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Faraji, A.; Manshadi, H.R.D.; Mobaraki, M.; Zare, M.; Houshmand, M. Association of ABCB1 and SLC22A16 Gene Polymorphisms with Incidence of Doxorubicin-Induced Febrile Neutropenia: A Survey of Iranian Breast Cancer Patients. PLoS ONE 2016, 11, e0168519. [Google Scholar] [CrossRef]
- Aniogo, E.C.; George, B.P.A.; Abrahamse, H. Phthalocyanine induced phototherapy coupled with Doxorubicin; a promising novel treatment for breast cancer. Expert Rev. Anticancer. Ther. 2017, 17, 693–702. [Google Scholar] [CrossRef]
- Nitiss, K.C.; Nitiss, J.L. Twisting and Ironing: Doxorubicin Cardiotoxicity by Mitochondrial DNA Damage. Clin. Cancer Res. 2014, 20, 4737–4739. [Google Scholar] [CrossRef]
- Sermeus, A.; Genin, M.; Maincent, A.; Fransolet, M.; Notte, A.; LeClere, L.; Riquier, H.; Arnould, T.; Michiels, C. Hypoxia-Induced Modulation of Apoptosis and BCL-2 Family Proteins in Different Cancer Cell Types. PLoS ONE 2012, 7, e47519. [Google Scholar] [CrossRef]
- Daurio, N.A.; Tuttle, S.W.; Worth, A.J.; Song, E.Y.; Davis, J.M.; Snyder, N.W.; Blair, I.A.; Koumenis, C. AMPK Activation and Metabolic Reprogramming by Tamoxifen through Estrogen Receptor–Independent Mechanisms Suggests New Uses for This Therapeutic Modality in Cancer Treatment. Cancer Res. 2016, 76, 3295–3306. [Google Scholar] [CrossRef]
- Niikawa, H.; Suzuki, T.; Miki, Y.; Suzuki, S.; Nagasaki, S.; Akahira, J.; Honma, S.; Evans, D.B.; Hayashi, S.-I.; Kondo, T.; et al. Intratumoral Estrogens and Estrogen Receptors in Human Non–Small Cell Lung Carcinoma. Clin. Cancer Res. 2008, 14, 4417–4426. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.-C.; Chiu, H.-C.; Syu, J.-J.; Jian, Y.-J.; Chen, C.-Y.; Jian, Y.-T.; Huang, Y.-J.; Wo, T.-Y.; Lin, Y.-W. Tamoxifen enhances erlotinib-induced cytotoxicity through down-regulating AKT-mediated thymidine phosphorylase expression in human non-small-cell lung cancer cells. Biochem. Pharmacol. 2014, 88, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Yuan, Y.; Sun, J.; Gao, W.; Shu, Y.-Q. Combined tamoxifen and gefitinib in non-small cell lung cancer shows antiproliferative effects. Biomed. Pharmacother. 2010, 64, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Kizaka-Kondoh, S.; Itasaka, S.; Xie, X.; Inoue, M.; Tanimoto, K.; Shibuya, K.; Hiraoka, M. Hypoxia inducible factor-1 influences sensitivity to paclitaxel of human lung cancer cell lines under normoxic conditions. Cancer Sci. 2007, 98, 1394–1401. [Google Scholar] [CrossRef]
- Cosse, J.-P.; Sermeus, A.; Vannuvel, K.; Ninane, N.; Raes, M.; Michiels, C. Differential effects of hypoxia on etoposide-induced apoptosis according to the cancer cell lines. Mol. Cancer 2007, 6, 61. [Google Scholar] [CrossRef]
- Ahmed, Z.S.O.; Dou, Q.P. Updated Review and Perspective on 20S Proteasome Inhibitors in the Treatment of Lung Cancer. Curr. Cancer Drug Targets 2020, 20, 392–409. [Google Scholar] [CrossRef]
- García-Fernández, C.; Fornaguera, C.; Borrós, S. Nanomedicine in Non-Small Cell Lung Cancer: From Conventional Treatments to Immunotherapy. Cancers 2020, 12, 1609. [Google Scholar] [CrossRef]
- Duffaud, F.; Le Cesne, A. Imatinib in the treatment of solid tumours. Target. Oncol. 2009, 4, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, K.; Nakagawa, K.; Hamada, J.-I.; Hida, Y.; Tada, M.; Kondo, S.; Moriuchi, T. Imatinib mesylate inhibits the proliferation-stimulating effect of human lung cancer-associated stromal fibroblasts on lung cancer cells. Int. J. Oncol. 2010, 37, 869–877. [Google Scholar] [CrossRef]
- Aono, Y.; Nishioka, Y.; Inayama, M.; Ugai, M.; Kishi, J.; Uehara, H.; Izumi, K.; Sone, S. Imatinib as a Novel Antifibrotic Agent in Bleomycin-induced Pulmonary Fibrosis in Mice. Am. J. Respir. Crit. Care Med. 2005, 171, 1279–1285. [Google Scholar] [CrossRef]
- Abdollahi, A.; Li, M.; Ping, G.; Plathow, C.; Domhan, S.; Kiessling, F.; Lee, L.B.; McMahon, G.; Gröne, H.-J.; Lipson, K.E.; et al. Inhibition of platelet-derived growth factor signaling attenuates pulmonary fibrosis. J. Exp. Med. 2005, 201, 925–935. [Google Scholar] [CrossRef]
- Vitiello, G.A.; Medina, B.D.; Zeng, S.; Bowler, T.G.; Zhang, J.Q.; Loo, J.K.; Param, N.J.; Liu, M.; Moral, A.J.; Zhao, J.N.; et al. Mitochondrial Inhibition Augments the Efficacy of Imatinib by Resetting the Metabolic Phenotype of Gastrointestinal Stromal Tumor. Clin. Cancer Res. 2017, 24, 972–984. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Calderon, F.; Gregory, M.A.; Pham-Danis, C.; DeRyckere, D.; Stevens, B.M.; Zaberezhnyy, V.; Hill, A.A.; Gemta, L.; Kumar, A.; Kumar, V.; et al. Tyrosine Kinase Inhibition in Leukemia Induces an Altered Metabolic State Sensitive to Mitochondrial Perturbations. Clin. Cancer Res. 2015, 21, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; A Mancuso, A.; Bui, T.V.; Tong, X.; Gruber, J.J.; Swider, C.R.; Sanchez, P.V.; Lum, J.J.; Sayed, N.; Melo, J.V.; et al. Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1α-induced metabolic reprograming. Oncogene 2010, 29, 2962–2972. [Google Scholar] [CrossRef] [PubMed]
- Kluza, J.; Jendoubi, M.; Ballot, C.; Dammak, A.; Jonneaux, A.; Idziorek, T.; Joha, S.; Dauphin, V.; Malet-Martino, M.; Balayssac, S.; et al. Exploiting Mitochondrial Dysfunction for Effective Elimination of Imatinib-Resistant Leukemic Cells. PLoS ONE 2011, 6, e21924. [Google Scholar] [CrossRef]
- Levy, A.; Doyen, J. Metformin for non-small cell lung cancer patients: Opportunities and pitfalls. Crit. Rev. Oncol. 2018, 125, 41–47. [Google Scholar] [CrossRef]
- Wink, K.C.J.; Belderbos, J.S.A.; Dieleman, E.M.T.; Rossi, M.; Rasch, C.R.N.; Damhuis, R.A.M.; Houben, R.M.A.; Troost, E.G.C. Improved progression free survival for patients with diabetes and locally advanced non-small cell lung cancer (NSCLC) using metformin during concurrent chemoradiotherapy. Radiother. Oncol. 2016, 118, 453–459. [Google Scholar] [CrossRef]
- Li, L.; Han, R.; Xiao, H.; Lin, C.; Wang, Y.; Liu, H.; Li, K.; Chen, H.; Sun, F.; Yang, Z.; et al. Metformin Sensitizes EGFR-TKI–Resistant Human Lung Cancer Cells In Vitro and In Vivo through Inhibition of IL-6 Signaling and EMT Reversal. Clin. Cancer Res. 2014, 20, 2714. [Google Scholar] [CrossRef]
- Troncone, M.; Cargnelli, S.M.; Villani, L.A.; Isfahanian, N.; Broadfield, L.A.; Zychla, L.; Wright, J.; Pond, G.; Steinberg, G.R.; Tsakiridis, T. Targeting metabolism and AMP-activated kinase with metformin to sensitize non-small cell lung cancer (NSCLC) to cytotoxic therapy: translational biology and rationale for current clinical trials. Oncotarget 2017, 8, 57733–57754. [Google Scholar] [CrossRef] [PubMed]
- Storozhuk, Y.; Hopmans, S.N.; Sanli, T.; Barron, C.C.; Tsiani, E.; Cutz, J.-C.; Pond, G.R.; Wright, J.L.; Singh, G.; Tsakiridis, T. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br. J. Cancer 2013, 108, 2021–2032. [Google Scholar] [CrossRef]
- Lee, B.B.; Kim, Y.; Kim, D.; Cho, E.Y.; Han, J.; Kim, H.K.; Shim, Y.M.; Kim, D.-H. Metformin and tenovin-6 synergistically induces apoptosis through LKB1-independent SIRT1 down-regulation in non-small cell lung cancer cells. J. Cell. Mol. Med. 2019, 23, 2872–2889. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Q.-C.; Jiang, S.-J. Effect of trichostatin A and paclitaxel on the proliferation and apoptosis of lung adenocarcinoma cells. Chin. Med. J. 2013, 126, 129–134. [Google Scholar]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; De Álava, E.; Hajji, N.; García-Domínguez, D.J. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front. Genet. 2020, 11, 1113. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutellingsperger, C. A novel assay for apoptosis Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef]
- Gutmann I, L-Lactate. Determination with lactate dehydrogenase and NAD. In Methods of Enzymatic Analysis, 2nd ed.; Bergmeyer, H., Ed.; Academic Press: Cambridge, MA, USA, 1963; pp. 1464–1468. [Google Scholar]
- Kunst, A.; Draeger, B.; Ziegenhorn, J. UV methods with hexokinase and glucose-6-phosphate dehydrogenase. In Methods in Enzymatic Analysis; H.U. Bergmeyer: Basel, Switzerland, 1984; pp. 162–172. [Google Scholar]
- Tarrado-Castellarnau, M.; de Atauri, P.; Tarrago-Celada, J.; Perarnau, J.; Yuneva, M.; Thomson, T.M.; Cascante, M. De novo MYC addiction as an adaptive response of cancer cells to CDK4/6 inhibition. Mol. Syst. Biol. 2017, 13. [Google Scholar] [CrossRef]
- Lehmann, S.G.; Seve, M.; VanWonterghem, L.; Michelland, S.; Cunin, V.; Coll, J.-L.; Hurbin, A.; Bourgoin-Voillard, S. A large scale proteome analysis of the gefitinib primary resistance overcome by KDAC inhibition in KRAS mutated adenocarcinoma cells overexpressing amphiregulin. J. Proteom. 2019, 195, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Askri, D.; Cunin, V.; Béal, D.; Berthier, S.; Chovelon, B.; Arnaud, J.; Rachidi, W.; Sakly, M.; Amara, S.; Sève, M.; et al. Investigating the toxic effects induced by iron oxide nanoparticles on neuroblastoma cell line: an integrative study combining cytotoxic, genotoxic and proteomic tools. Nanotoxicology 2019, 13, 1021–1040. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Campone, M.; Valo, I.; Jézéquel, P.; Moreau, M.; Boissard, A.; Campion, L.; Loussouarn, D.; Verriele, V.; Coqueret, O.; Guette, C. Prediction of Recurrence and Survival for Triple-Negative Breast Cancer (TNBC) by a Protein Signature in Tissue Samples. Mol. Cell. Proteom. 2015, 14, 2936–2946. [Google Scholar] [CrossRef]
- Unwin, R.D.; Griffiths, J.R.; Whetton, A.D. Simultaneous analysis of relative protein expression levels across multiple samples using iTRAQ isobaric tags with 2D nano LC–MS/MS. Nat. Protoc. 2010, 5, 1574–1582. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Müller, A.; Dayon, L.; Köcher, T.; Hainard, A.; Pichler, P.; Schmidt-Erfurth, U.; Superti-Furga, G.; Sanchez, J.-C.; Mechtler, K.; et al. General Statistical Modeling of Data from Protein Relative Expression Isobaric Tags. J. Proteome Res. 2011, 10, 2758–2766. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Leskovec, J. node2vec: Scalable Feature Learning for Networks. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Association for Computing Machinery, San Francisco, CA, USA, 13–17 August 2016; pp. 855–864. [Google Scholar]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Batchelor, C.; Dumontier, M.; Hastings, J.; Willighagen, E.; Bolton, E. PubChemRDF: towards the semantic annotation of PubChem compound and substance databases. J. Cheminform. 2015, 7, 34. [Google Scholar] [CrossRef]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. Comparative Toxicogenomics Database (CTD): update 2021. Nucleic Acids Res. 2021, 49, D1138–D1143. [Google Scholar] [CrossRef] [PubMed]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug–Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef]
- Licata, L.; Surdo, P.L.; Iannuccelli, M.; Palma, A.; Micarelli, E.; Perfetto, L.; Peluso, D.; Calderone, A.; Castagnoli, L.; Cesareni, G. SIGNOR 2.0, the SIGnaling Network Open Resource 2.0: 2019 update. Nucleic Acids Res. 2019, 48, D504–D510. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar]
- Oughtred, R.; Stark, C.; Breitkreutz, B.-J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef] [PubMed]
- Meldal, B.H.M.; Bye-A-Jee, H.; Gajdoš, L.; Hammerová, Z.; Horáčková, A.; Melicher, F.; Perfetto, L.; Pokorný, D.; Lopez, M.R.; Türková, A.; et al. Complex Portal 2018: extended content and enhanced visualization tools for macromolecular complexes. Nucleic Acids Res. 2019, 47, D550–D558. [Google Scholar] [CrossRef]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; Del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef]
- Calderone, A.; Castagnoli, L.; Cesareni, G. mentha: a resource for browsing integrated protein-interaction networks. Nat. Methods 2013, 10, 690–691. [Google Scholar] [CrossRef]
- Calderone, A.; Iannuccelli, M.; Peluso, D.; Licata, L. Using the MINT Database to Search Protein Interactions. Curr. Protoc. Bioinform. 2020, 69, e93. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Bernabé, A.; Tarragó-Celada, J.; Cunin, V.; Michelland, S.; Cortés, R.; Poignant, J.; Boyault, C.; Rachidi, W.; Bourgoin-Voillard, S.; Cascante, M.; et al. Quantitative Proteomic Approach Reveals Altered Metabolic Pathways in Response to the Inhibition of Lysine Deacetylases in A549 Cells under Normoxia and Hypoxia. Int. J. Mol. Sci. 2021, 22, 3378. https://doi.org/10.3390/ijms22073378
Martín-Bernabé A, Tarragó-Celada J, Cunin V, Michelland S, Cortés R, Poignant J, Boyault C, Rachidi W, Bourgoin-Voillard S, Cascante M, et al. Quantitative Proteomic Approach Reveals Altered Metabolic Pathways in Response to the Inhibition of Lysine Deacetylases in A549 Cells under Normoxia and Hypoxia. International Journal of Molecular Sciences. 2021; 22(7):3378. https://doi.org/10.3390/ijms22073378
Chicago/Turabian StyleMartín-Bernabé, Alfonso, Josep Tarragó-Celada, Valérie Cunin, Sylvie Michelland, Roldán Cortés, Johann Poignant, Cyril Boyault, Walid Rachidi, Sandrine Bourgoin-Voillard, Marta Cascante, and et al. 2021. "Quantitative Proteomic Approach Reveals Altered Metabolic Pathways in Response to the Inhibition of Lysine Deacetylases in A549 Cells under Normoxia and Hypoxia" International Journal of Molecular Sciences 22, no. 7: 3378. https://doi.org/10.3390/ijms22073378
APA StyleMartín-Bernabé, A., Tarragó-Celada, J., Cunin, V., Michelland, S., Cortés, R., Poignant, J., Boyault, C., Rachidi, W., Bourgoin-Voillard, S., Cascante, M., & Seve, M. (2021). Quantitative Proteomic Approach Reveals Altered Metabolic Pathways in Response to the Inhibition of Lysine Deacetylases in A549 Cells under Normoxia and Hypoxia. International Journal of Molecular Sciences, 22(7), 3378. https://doi.org/10.3390/ijms22073378