
Management of Myeloma Bone Lesions




Abstract
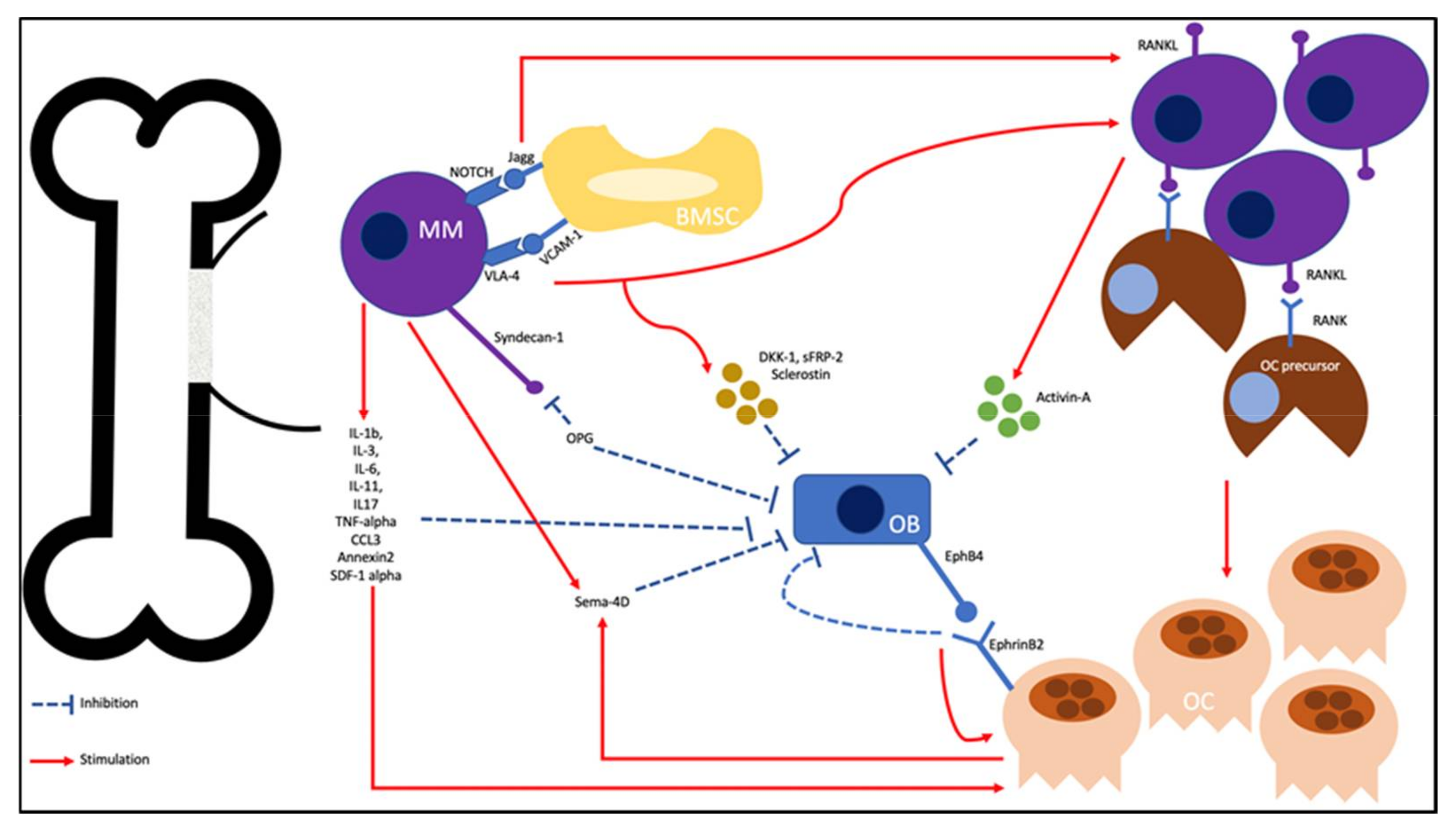
:1. Introduction
2. Pathophysiology
2.1. Increasing OC Activity
2.2. Enhancing OB Inhibition
2.3. Bidirectional Signaling in the Uncoupling of Osteoclastogenesis and Osteoblastogenesis
2.4. MM and the Bone Microenvironment
3. Predictors or Biomarkers
4. Treatment Overview for MBD
4.1. BPs
4.2. Denosumab
4.3. ASCT
4.4. Bortezomib-Based Regimens
5. Novel Agents
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASCT | autologous stem cell transplantation |
| BMA | bone-modifying agents |
| BMSC | bone marrow stromal cell |
| BP | bisphosphonate |
| CCL3 | chemokine C-C motif ligand 3 |
| CTX | C-telopeptide of type I collagen |
| DEXA | dual-energy x-ray absorptiometry |
| DKK-1 | dickkopf-1 |
| IL | interleukin |
| MBD | MM bone disease |
| Mibi | technetium-99m-sestamibi |
| MM | multiple myeloma |
| MRI | magnetic resonance imaging |
| OB | osteoblast |
| OC | osteoclast |
| OPG | osteoprotegerin |
| PC | prostate cancer |
| PET | positron emission tomography |
| RANKL | receptor activator of nuclear factor-kappa B ligand |
| Scl | sclerostin |
| sRANKL | soluble receptor activator of nuclear factor-kappa B ligand |
| SREs | skeletal-related events |
| TRACP-5b | tartrate-resistant acid phosphatase isoform-5b |
| Wnt | Wingless-type |
| ZA | zoledronic acid |
References
- Panaroni, C.; Yee, A.J.; Raje, N.S. Myeloma and Bone Disease. Curr. Osteoporos. Rep. 2017, 15, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callander, N.S.; Roodman, G.D. Myeloma bone disease. Semin. Hematol. 2001, 38, 276–285. [Google Scholar] [CrossRef]
- Raje, N.; Roodman, G.D. Advances in the Biology and Treatment of Bone Disease in Multiple Myeloma. Clin. Cancer Res. 2011, 17, 1278–1286. [Google Scholar] [CrossRef] [Green Version]
- Webb, S.L.; Edwards, C.M. Novel therapeutic targets in myeloma bone disease. Br. J. Pharmacol. 2014, 171, 3765–3776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corso, A.; Arcaini, L.; Mangiacavalli, S.; Astori, C.; Orlandi, E.; Lorenzi, A.; Passamonti, F.; Klersy, C.; Pascutto, C.; Canevari-Sciorati, A.; et al. Biochemical markers of bone disease in asymptomatic early stage multiple myeloma. A study on their role in identifying high risk patients. Haematology 2001, 86, 394–398. [Google Scholar]
- Terpos, E.; Politou, M.; Rahemtulla, A. New insights into the pathophysiology and management of bone disease in multiple myeloma. Br. J. Haematol. 2003, 123, 758–769. [Google Scholar] [CrossRef]
- Araki, K.; Sangai, T.; Miyamoto, S.; Maeda, H.; Zhang, S.-C.; Nakamura, M.; Ishii, G.; Hasebe, T.; Kusaka, H.; Akiyama, T.; et al. Inhibition of bone-derived insulin-like growth factors by a ligand-specific antibody suppresses the growth of human multiple myeloma in the human adult bone explanted in NOD/SCID mouse. Int. J. Cancer 2006, 118, 2602–2608. [Google Scholar] [CrossRef]
- Giuliani, N.; Morandi, F.; Tagliaferri, S.; Rizzoli, V. Targeting pathways mediating bone disease. Curr. Pharm. Biotechnol. 2006, 7, 423–429. [Google Scholar] [CrossRef]
- Heider, U.; Fleissner, C.; Zavrski, I.; Kaiser, M.; Hecht, M.; Jakob, C.; Sezer, O. Bone markers in multiple myeloma. Eur. J. Cancer 2006, 42, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E. Biochemical markers of bone metabolism in multiple myeloma. Cancer Treat. Rev. 2006, 32, 15–19. [Google Scholar] [CrossRef]
- Roodman, G.D. Targeting the bone microenvironment in multiple myeloma. J. Bone Miner. Metab. 2010, 28, 244–250. [Google Scholar] [CrossRef]
- Roodman, G. Erratum to? Pathogenesis of myeloma bone disease? Blood Cells Mol. Dis. 2004, 109, 283–291. [Google Scholar] [CrossRef]
- Labropoulou, V.; Theocharis, A.; Symeonidis, A.; Skandalis, S.; Karamanos, N.; Kalofonos, H. Pathophysiology and Pharmacological Targeting of Tumor-Induced Bone Disease: Current Status and Emerging Therapeutic Interventions. Curr. Med. Chem. 2011, 18, 1584–1598. [Google Scholar] [CrossRef]
- Gavriatopoulou, M.; Dimopoulos, M.A.; Kastritis, E.; Terpos, E. Emerging treatment approaches for myeloma-related bone disease. Expert Rev. Hematol. 2017, 10, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Calimeri, T.; Battista, E.; Conforti, F.; Neri, P.; di Martino, M.T.; Rossi, M.; Foresta, U.; Piro, E.; Ferrara, F.; Amorosi, A.; et al. A unique three-dimensional SCID-polymeric scaffold (SCID-synth-hu) model for in vivo expansion of human primary multiple myeloma cells. Leukemia 2011, 25, 707–711. [Google Scholar] [CrossRef]
- Gooding, S.; Edwards, C.M. New approaches to targeting the bone marrow microenvironment in multiple myeloma. Curr. Opin. Pharmacol. 2016, 28, 43–49. [Google Scholar] [CrossRef]
- Xi, H.; An, R.; Li, L.; Wang, G.; Tao, Y.; Gao, L. Myeloma bone disease: Progress in pathogenesis. Prog. Biophys. Mol. Biol. 2016, 122, 149–155. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, E.K.; Raje, N.S. Myeloma bone disease: Pathogenesis and treatment. Clin. Adv. Hematol. Oncol. H&O 2017, 15, 285–295. [Google Scholar]
- Yee, A.J.; Raje, N.S. Denosumab for the treatment of bone disease in solid tumors and multiple myeloma. Future Oncol. 2018, 14, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Dimopoulos, M.A. Myeloma bone disease: From biology findings to treatment ap-proaches. Blood 2019, 133, 1534–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berenson, J.R. Myeloma bone disease. Best Pract. Res. Clin. Haematol. 2005, 18, 653–672. [Google Scholar] [CrossRef]
- Huston, A.; Roodman, G.D. Role of the microenvironment in multiple myeloma bone disease. Future Oncol. 2006, 2, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.M.; Edwards, J.R.; Lwin, S.T.; Esparza, J.; Oyajobi, B.O.; McCluskey, B.; Munoz, S.; Grubbs, B.; Mundy, G.R. Increasing Wnt signaling in the bone marrow microenvironment inhibits the development of myeloma bone disease and reduces tumor burden in bone in vivo. Blood 2008, 111, 2833–2842. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, M.; Mieth, M.; Liebisch, P.; Oberländer, R.; Rademacher, J.; Jakob, C.; Kleeberg, L.; Fleissner, C.; Braendle, E.; Peters, M.; et al. Serum concentrations of DKK-1 correlate with the extent of bone disease in patients with multiple myeloma. Eur. J. Haematol. 2008, 80, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Efstathiou, E.; Christoulas, D.; Roussou, M.; Katodritou, E.; Dimopoulos, M.-A. RANKL inhibition: Clinical implications for the management of patients with multiple myeloma and solid tumors with bone metastases. Expert Opin. Biol. Ther. 2009, 9, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.K.; Stewart, J.P.; Bam, R.; Qu, P.; Barlogie, B.; van Rhee, F.; Shaughnessy, J.D.; Epstein, J.; Yaccoby, S. CYR61/CCN1 overexpression in the myeloma microenvironment is associated with superior survival and reduced bone disease. Blood 2014, 124, 2051–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Deng, S.; An, G.; Qin, X.; Sui, W.; Zou, D.; Qiu, L.; Xu, Y. Detection of serum DKK1 in multiple myeloma and myeloma bone disease. Zhonghua Xue Ye Xue Za Zhi 2015, 36, 682–685. [Google Scholar] [PubMed]
- Gan, Z.Y.; Fitter, S.; van Dyke, K.; To, L.B.; Zannettino, A.C.; Martin, S.K. The effect of the dual PI3K and mTOR inhibitor BEZ235 on tumour growth and osteolytic bone disease in multiple myeloma. Eur. J. Haematol. 2014, 94, 343–354. [Google Scholar] [CrossRef]
- Abe, M. Mechanisms of myeloma-induced bone disease. Clin. Calcium 2016, 26, 699–706. [Google Scholar] [PubMed]
- Loredana, S.; Santo, L.; Wein, M.N.; Hu, D.Z.; Cirstea, D.D.; Nemani, N.; Tai, Y.-T.; Raines, S.E.; Kuhstoss, S.A.; Munshi, N.C.; et al. Regulation of Sclerostin Expression in Multiple Myeloma by Dkk-1: A Potential Therapeutic Strategy for Myeloma Bone Disease. J. Bone Miner. Res. 2016, 31, 1225–1234. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Peng, F.; Liu, Z.; Jiang, F.; Li, L.; Gao, S.; Wang, G.; Song, J.; Ruan, E.; Shao, Z.; et al. CYR61/CCN1 stimulates proliferation and differentiation of osteoblasts in vitro and contributes to bone remodelling in vivo in myeloma bone disease. Int. J. Oncol. 2016, 50, 631–639. [Google Scholar] [CrossRef] [Green Version]
- McDonald, M.M.; Delgado-Calle, J. Sclerostin: An Emerging Target for the Treatment of Cancer-Induced Bone Disease. Curr. Osteoporos. Rep. 2017, 15, 532–541. [Google Scholar] [CrossRef]
- Heusschen, R.; Muller, J.; Duray, E.; Withofs, N.; Bolomsky, A.; Baron, F.; Beguin, Y.; Menu, E.; Ludwig, H.; Caers, J. Molecular mechanisms, current management and next generation therapy in myeloma bone disease. Leuk. Lymphoma 2018, 59, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Christoulas, D.; Gavriatopoulou, M. Biology and treatment of myeloma related bone disease. Metabolism 2018, 80, 80–90. [Google Scholar] [CrossRef]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Dimopoulos, M.A. Pathogenesis of bone disease in multiple myeloma: From bench to bedside. Blood Cancer J. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiang, Y.-W.; Shaughnessy, J.D.; Yaccoby, S. Wnt3a signaling within bone inhibits multiple myeloma bone disease and tumor growth. Blood 2008, 112, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Berenson, J.; Raje, N.; Roodman, G.D. Management of bone disease in multiple myeloma. Expert Rev. Hematol. 2014, 7, 113–125. [Google Scholar] [CrossRef]
- Gavriatopoulou, M.; Dimopoulos, M.-A.; Christoulas, D.; Migkou, M.; Iakovaki, M.; Gkotzamanidou, M.; Terpos, E. Dickkopf-1: A suitable target for the management of myeloma bone disease. Expert Opin. Ther. Targets 2009, 13, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Condon, K.W.; Kuhstoss, S.A.; Plotkin, L.I.; Bellido, T.; Roodman, G.D. Genetic deletion of Sost or pharmacological inhibition of sclerostin prevent multiple myeloma-induced bone disease without affecting tumor growth. Leukemia 2017, 31, 2686–2694. [Google Scholar] [CrossRef] [PubMed]
- Pitari, M.R.; Rossi, M.; Amodio, N.; Botta, C.; Morelli, E.; Federico, C.; Gullà, A.; Caracciolo, D.; di Martino, M.T.; Arbitrio, M.; et al. Inhibition of miR-21 restores RANKL/OPG ratio in multiple myeloma-derived bone marrow stromal cells and impairs the resorbing activity of mature osteoclasts. Oncotarget 2015, 6, 27343–27358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, C.-H.; Hsiao, H.-H. NRF2 Is One of the Players Involved in Bone Marrow Mediated Drug Resistance in Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 3503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, C.-H.; Hsu, C.-M.; Hsiao, S.Y.; Hsiao, H.-H. Pathogenic Mechanisms of Myeloma Bone Disease and Possible Roles for NRF2. Int. J. Mol. Sci. 2020, 21, 6723. [Google Scholar] [CrossRef]
- Pennisi, A.; Ling, W.; Li, X.; Khan, S.; Shaughnessy, J.D., Jr.; Barlogie, B.; Yaccoby, S. The ephrinB2/EphB4 axis is dysreg-ulated in osteoprogenitors from myeloma patients and its activation affects myeloma bone disease and tumor growth. Blood 2009, 114, 1803–1812. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Abe, M.; Hiasa, M.; Oda, A.; Amou, H.; Kido, S.; Harada, T.; Tanaka, O.; Miki, H.; Nakamura, S.; et al. Tgf-Beta inhibition restores terminal osteoblast differentiation to suppress myeloma growth. PLoS ONE 2010, 5, e9870. [Google Scholar] [CrossRef]
- Negishi-Koga, T.; Shinohara, M.; Komatsu, N.; Bito, H.; Kodama, T.; Friedel, R.H.; Takayanagi, H. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat. Med. 2011, 17, 1473–1480. [Google Scholar] [CrossRef]
- Garcia-Gomez, A.; Quwaider, D.; Canavese, M.; Ocio, E.M.; Tian, Z.; Blanco, J.F.; Berger, A.J.; Ortiz-De-Solorzano, C.; Hernández-Iglesias, T.; Martens, A.C.; et al. Preclinical Activity of the Oral Proteasome Inhibitor MLN9708 in Myeloma Bone Disease. Clin. Cancer Res. 2014, 20, 1542–1554. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, R.; Nickel, N.; Godau, J.; Willie, B.M.; Duda, G.N.; Cirovic, B.; Leutz, A.; A Manz, R.; Bogen, B.; Dorken, B.; et al. Notch pathway inhibition controls myeloma bone disease in the murine MOPC315.BM model. Blood Cancer J. 2014, 4, e217. [Google Scholar] [CrossRef]
- Hideshima, T.; Chauhan, D.; Schlossman, R.; Richardson, P.; Anderson, K.C. The role of tumor necrosis factor α in the pathophysiology of human multiple myeloma: Therapeutic applications. Oncogene 2001, 20, 4519–4527. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Pivonka, P.; Buenzli, P.R.; Smith, D.W.; Dunstan, C.R. Computational modeling of interactions between mul-tiple myeloma and the bone microenvironment. PLoS ONE 2011, 6, e27494. [Google Scholar]
- Weng, W.-W.; Dong, M.-J.; Zhang, J.; Yang, J.; Xu, Q.; Zhu, Y.-J.; Liu, N.-H. A systematic review of MRI, scintigraphy, FDG-PET and PET/CT for diagnosis of multiple myeloma related bone disease-which is best? Asian Pac. J. Cancer Prev. 2014, 15, 9879–9884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, W.; Da, Y.; Lin, Q.; Li, H.; Gao, X.; Zhou, D.; Zhuang, J. The value of chest computerized tomography in evaluation of bone disease and clinical prognosis of multiple myeloma. Zhonghua Nei ke za Zhi 2015, 54, 711–715. [Google Scholar]
- Wang, Y.; Yee, A.J.; Sirard, C.; Landau, S.; Raje, N.; Mahmood, U. Sodium fluoride PET imaging as a quantitative phar-macodynamic biomarker for bone homeostasis during anti-DKK1 therapy for multiple myeloma. Blood Cancer J. 2017, 7, e615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Walker, B.A.; Brewer, D.; Gregory, W.M.; Ashcroft, J.; Ross, F.M.; Jackson, G.H.; Child, A.J.; Davies, F.E.; Morgan, G.J. A Gene Expression–Based Predictor for Myeloma Patients at High Risk of Developing Bone Disease on Bisphosphonate Treatment. Clin. Cancer Res. 2011, 17, 6347–6355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berenson, J.R.; Lichtenstein, A.; Porter, L.; Dimopoulos, M.A.; Bordoni, R.; George, S.; Lipton, A.; Keller, A.; Ballester, O.; Kovacs, M.J.; et al. Efficacy of Pamidronate in Reducing Skeletal Events in Patients with Advanced Multiple Myeloma. N. Engl. J. Med. 1996, 334, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.S.; Gordon, D.; Kaminski, M.; Howell, A.; Belch, A.; Mackey, J.; Apffelstaedt, J.; Hussein, M.; E Coleman, R.; Reitsma, D.J.; et al. Zoledronic acid versus pamidronate in the treatment of skeletal metastases in patients with breast cancer or osteolytic lesions of multiple myeloma: A phase III, double-blind, comparative trial. Cancer J. 2001, 7, 377–387. [Google Scholar]
- Gimsing, P.; Carlson, K.; Turesson, I.; Fayers, P.; Waage, A.; Vangsted, A.; Mylin, A.; Gluud, C.; Juliusson, G.; Gregersen, H.; et al. Effect of pamidronate 30 mg versus 90 mg on physical function in patients with newly diagnosed multiple myeloma (Nordic Myeloma Study Group): A double-blind, randomised controlled trial. Lancet Oncol. 2010, 11, 973–982. [Google Scholar] [CrossRef] [Green Version]
- Morgan, G.J.; E Davies, F.; Gregory, W.M.; Cocks, K.; E Bell, S.; Szubert, A.J.; Navarro-Coy, N.; Drayson, M.T.; Owen, R.G.; Feyler, S.; et al. First-line treatment with zoledronic acid as compared with clodronic acid in multiple myeloma (MRC Myeloma IX): A randomised controlled trial. Lancet 2010, 376, 1989–1999. [Google Scholar] [CrossRef] [Green Version]
- Himelstein, A.L.; Foster, J.C.; Khatcheressian, J.L.; Roberts, J.D.; Seisler, D.K.; Novotny, P.J.; Qin, R.; Go, R.S.; Grubbs, S.S.; O’Connor, T.; et al. Effect of Longer-Interval vs Standard Dosing of Zoledronic Acid on Skeletal Events in Patients with Bone Metastases: A Randomized Clinical Trial. Jama 2017, 317, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Terpos, E.; Willenbacher, W.; Shimizu, K.; García-Sanz, R.; Durie, B.; Legieć, W.; Krejčí, M.; Laribi, K.; Zhu, L.; et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: An international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol. 2018, 19, 370–381. [Google Scholar] [CrossRef]
- Locke, F.L.; Morgan, G.J. What is the evidence for the use of bisphosphonate therapy in newly diagnosed multiple myeloma patients lacking bone disease? Hematology 2012, 2012, 350–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terpos, E. Bisphosphonate anticancer activity in multiple myeloma. Anti-Cancer Agents Med. Chem. 2012, 12, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Søe, K.; Plesner, T.; Jakobsen, E.H.; Hansen, C.T.; Jørgensen, H.B.; Delaisse, J.-M. Is retention of zoledronic acid onto bone different in multiple myeloma and breast cancer patients with bone metastasis? J. Bone Miner. Res. 2013, 28, 1738–1750. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T. Management of bone disease in multiple myeloma. Gan kagaku ryoho. Cancer Chemother. 2012, 39, 1187–1190. [Google Scholar]
- Jeon, H.-L.; Oh, I.-S.; Baek, Y.-H.; Yang, H.; Park, J.; Hong, S.; Shin, J.-Y. Zoledronic acid and skeletal-related events in patients with bone metastatic cancer or multiple myeloma. J. Bone Miner. Metab. 2020, 38, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.A. Denosumab for bone lesions in multiple myeloma—What is its value? Haematology 2018, 103, 753–754. [Google Scholar] [CrossRef] [Green Version]
- Alegre, A.; Gironella, M.; Bailén, A.; Giraldo, P. Zoledronic acid in the management of bone disease as a consequence of multiple myeloma: A review. Eur. J. Haematol. 2014, 92, 181–188. [Google Scholar] [CrossRef]
- Clark, R.E.; Fraser, W.D. Bone Turnover Following Autologous Transplantation in Multiple Myeloma. Leuk. Lymphoma 2002, 43, 511–516. [Google Scholar] [CrossRef]
- Terpos, E.; Politou, M.; Szydlo, R.; Nadal, E.; Avery, S.; Olavarria, E.; Kanfer, E.; Goldman, J.M.; Apperley, J.F.; Rahemtulla, A. Autologous stem cell transplantation normalizes abnormal bone remodeling and sRANKL/osteoprotegerin ratio in pa-tients with multiple myeloma. Leukemia 2004, 18, 1420–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, P.; Ren, L.; Gratton, K.; Stebner, E.; Johnson, J.; Klimowicz, A.; Duggan, P.; Tassone, P.; Mansoor, A.; Stewart, U.A.; et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011, 118, 6368–6379. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Heath, D.J.; Rahemtulla, A.; Zervas, K.; Chantry, A.; Anagnostopoulos, A.; Pouli, A.; Katodritou, E.; Verrou, E.; Vervessou, E.C.; et al. Bortezomib reduces serum dickkopf-1 and receptor activator of nuclear factor-kappaB ligand con-centrations and normalises indices of bone remodelling in patients with relapsed multiple myeloma. Br. J. Haematol. 2006, 135, 688–692. [Google Scholar] [CrossRef]
- Hamasaki, M.; Hideshima, T.; Tassone, P.; Neri, P.; Ishitsuka, K.; Yasui, H.; Shiraishi, N.; Raje, N.; Kumar, S.; Picker, D.H.; et al. Azaspirane (N-N-diethyl-8,8-dipropyl-2-azaspiro [4.5] decane-2-propanamine) inhibits human multiple myeloma cell growth in the bone marrow milieu in vitro and in vivo. Blood 2005, 105, 4470–4476. [Google Scholar] [CrossRef] [Green Version]
- Yasui, H.; Hideshima, T.; Hamasaki, M.; Roccaro, A.M.; Shiraishi, N.; Kumar, S.; Tassone, P.; Ishitsuka, K.; Raje, N.; Tai, Y.-T.; et al. SDX-101, the R-enantiomer of etodolac, induces cytotoxicity, overcomes drug resistance, and enhances the activity of dexamethasone in multiple myeloma. Blood 2005, 106, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Burger, R.; le Gouill, S.; Tai, Y.-T.; Shringarpure, R.; Tassone, P.; Neri, P.; Podar, K.; Catley, L.; Hideshima, T.; Chauhan, D.; et al. Janus kinase inhibitor INCB20 has antiproliferative and apoptotic effects on human myeloma cells in vitro and in vivo. Mol. Cancer Ther. 2009, 8, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Holien, T.; Sundan, A. Oncogene addiction to c-MYC in myeloma cells. Oncotarget 2012, 3, 739–740. [Google Scholar] [CrossRef] [Green Version]
- Maes, K.; de Smedt, E.; Lemaire, M.; de Raeve, H.; Menu, E.; van Valckenborgh, E.; McClue, S.; Vanderkerken, K.; de Bruyne, E. The role of DNA damage and repair in decitabine-mediated apoptosis in multiple myeloma. Oncotarget 2014, 5, 3115–3129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roodman, G.D. Mechanisms of Bone Metastasis. N. Engl. J. Med. 2004, 350, 1655–1664. [Google Scholar] [CrossRef]
- Deleu, S.; Lemaire, M.; Arts, J.; Menu, E.; van Valckenborgh, E.; Broek, I.V.; de Raeve, H.; Coulton, L.; van Camp, B.; Croucher, P.; et al. Bortezomib Alone or in Combination with the Histone Deacetylase Inhibitor JNJ-26481585: Effect on Myeloma Bone Disease in the 5T2MM Murine Model of Myeloma. Cancer Res. 2009, 69, 5307–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallet, S.; Raje, N.; Ishitsuka, K.; Hideshima, T.; Podar, K.; Chhetri, S.; Pozzi, S.; Breitkreutz, I.; Kiziltepe, T.; Yasui, H.; et al. MLN3897, a novel CCR1 inhibitor, impairs osteoclastogenesis and inhibits the interaction of multiple myeloma cells and osteoclasts. Blood 2007, 110, 3744–3752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, R.; Liu, H.; Zhao, S.; Wang, Y.; Li, L.; Gao, S.; Ruan, E.; Wang, G.; Wang, H.; Song, J.; et al. Osteoblast inhibition by chemokine cytokine ligand3 in myeloma-induced bone disease. Cancer Cell Int. 2014, 14, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scullen, T.; Santo, L.; Vallet, S.; Fulciniti, M.; Eda, H.; Cirstea, D.; Patel, K.; Nemani, N.; Yee, A.; Mahindra, A.; et al. Le-nalidomide in combination with an activin A-neutralizing antibody: Preclinical rationale for a novel anti-myeloma strategy. Leukemia 2013, 27, 1715–1721. [Google Scholar] [CrossRef] [PubMed]
- Abdulkadyrov, K.M.; Salogub, G.N.; Khuazheva, N.K.; Sherman, M.L.; Laadem, A.; Barger, R.; Knight, R.; Srinivasan, S.; Terpos, E. Sotatercept in patients with osteolytic lesions of multiple myeloma. Br. J. Haematol. 2014, 165, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Fulciniti, M.; Hideshima, T.; Vermot-Desroches, C.; Pozzi, S.; Nanjappa, P.; Shen, Z.; Patel, N.; Smith, E.S.; Wang, W.; Prabhala, R.; et al. A High-Affinity Fully Human Anti-IL-6 mAb, 1339, for the Treatment of Multiple Myeloma. Clin. Cancer Res. 2009, 15, 7144–7152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noonan, K.; Marchionni, L.; Anderson, J.; Pardoll, D.; Roodman, G.D.; Borrello, I. A novel role of IL-17-producing lym-phocytes in mediating lytic bone disease in multiple myeloma. Blood 2010, 116, 3554–3563. [Google Scholar] [CrossRef] [Green Version]
- Prabhala, R.H.; Fulciniti, M.; Pelluru, D.; Rashid, N.U.; Nigroiu, A.; Nanjappa, P.; Pai, C.; Lee, S.; Prabhala, N.S.; Bandi, R.L.; et al. Targeting IL-17A in multiple myeloma: A potential novel therapeutic approach in myeloma. Leukemia 2016, 30, 379–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobune, M.; Chiba, H.; Kato, J.; Kato, K.; Nakamura, K.; Kawano, Y.; Takada, K.; Takimoto, R.; Takayama, T.; Hamada, H.; et al. Wnt3/RhoA/ROCK signaling pathway is involved in adhesion-mediated drug resistance of multiple myeloma in an autocrine mechanism. Mol. Cancer Ther. 2007, 6, 1774–1784. [Google Scholar] [CrossRef] [Green Version]
- Kleber, M.; Ntanasis-Stathopoulos, I.; Dimopoulos, M.A.; Terpos, E. Monoclonal antibodies against RANKL and sclerostin for myeloma-related bone disease: Can they change the standard of care? Expert Rev. Hematol. 2019, 12, 651–663. [Google Scholar] [CrossRef]
- Kaveh, S.; Hosseinifard, H.; Ghadimi, N.; Vojdanian, M.; Aryankhesal, A. Efficacy and safety of Romosozumab in treat-ment for low bone mineral density: A systematic review and meta-analysis. Clin. Rheumatol. 2020, 39, 3261–3276. [Google Scholar] [CrossRef] [PubMed]
- Tian, E.; Zhan, F.; Walker, R.; Rasmussen, E.; Ma, Y.; Barlogie, B.; Shaughnessy, J.D., Jr. The Role of the Wnt-Signaling Antagonist DKK1 in the Development of Osteolytic Lesions in Multiple Myeloma. N. Engl. J. Med. 2003, 349, 2483–2494. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.; Filzmoser, J.-M.; Pecherstorfer, M.; Podar, K. Myeloma Bone Disease: Update on Pathogenesis and Novel Treatment Strategies. Pharmaceutics 2018, 10, 202. [Google Scholar] [CrossRef] [Green Version]
- Fowler, J.A.; Edwards, C.M.; Croucher, P.I. Tumor–host cell interactions in the bone disease of myeloma. Bone 2011, 48, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Study | Study Design | Patient Papulation | Treatment Drug | Treatment Schedule | Patients, n | Median Time to First SRE, months | SREs Incidence, % | ONJ Incidence, % | Renal Toxicity, % |
|---|---|---|---|---|---|---|---|---|---|
| Berenson et al. [55] | Randomization | Stage III myeloma | Pamidronate vs. placebo | 90 mg pamidronate 4 h IV infusion every 4 weeks for 9 cycles | 196 vs. 181 | Significantly less in placebo group (p = 0.01) | 24 vs. 41 (p < 0.01) | NR | NR |
| Rosen et al. [56] | Phase III, double-blind, comparative trial | Durie-Salmon Stage III myeloma | ZA vs. pamidronate | 4 or 8 mg ZA IV or 15 min or 90 mg pamidronate IV 2 h every 3–4 w for 12 months | 129 vs. 65 | 12.5 vs. 9.4 | NR | NR | NR |
| Gimsing et al. [57] | Double-blind, randomized, phase 3 trial | MM patients starting antimyeloma treatment | Pamidronate | 30 vs. 90 mg of pamidronate | 252 vs. 252 | 10.2 vs. 9.2 (p = 0.63) | 33.7 vs. 35.2 | 0.8 vs. 3.2 | NR |
| Morgan et al. [58] | Computer-generated randomization | Newly diagnosed MM | ZA vs. clodronate | 4 mg of ZA IV every 3–4 weeks or 1600 mg of clodronic acid orally daily | 981 vs. 979 | NR | 27 vs. 35 (p = 0.0004) | 4 vs. 1 | Similar for the two treatment groups (p = 0.55) |
| Himelstein et al. [59] | Randomized, open-label | MM with at least one site of bone involvement | ZA | ZA every 12 vs. every 4 weeks | 139 vs. 139 | NR | 55 vs. 60 | NR | NR |
| Raje et al. [60] | Double-blind, double-dummy, randomized, controlled, phase 3 | MM with at least one lytic bone lesion | Denosumab vs. ZA | 120 mg of denosumab SC plus placebo IV or ZA 4 mg IV plus placebo SC every 4 weeks | 859 vs. 859 | 22.8 vs. 24 (p = 0.01) | 43.8 vs. 44.6 | 4.1 vs. 2.8 | 10 vs. 17.1 |
| Molecular Target | Mechanism | Use in MM/Therapeutic Implication |
|---|---|---|
| Increased OC Activity | ||
| Inhibition of miR-21 [41] |
|
|
| CCL-3 (MIP-1α) [7,79,80] |
|
|
| Activin A [81,82] |
|
|
| IL-6 [83] |
|
|
| IL-17 [84,85] |
|
|
| Suppressed OB Activity | ||
| Wnt pathway [37,86] |
|
|
| Scl [15,33,87,88] |
|
|
| DKK1 [31,89,90] |
|
|
| EphrinB2/EphB4 signaling pathway [44] |
|
|
| Adiponectin [91] |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, J.-S.; Yen, C.-H.; Hsu, C.-M.; Hsiao, H.-H. Management of Myeloma Bone Lesions. Int. J. Mol. Sci. 2021, 22, 3389. https://doi.org/10.3390/ijms22073389
Du J-S, Yen C-H, Hsu C-M, Hsiao H-H. Management of Myeloma Bone Lesions. International Journal of Molecular Sciences. 2021; 22(7):3389. https://doi.org/10.3390/ijms22073389
Chicago/Turabian StyleDu, Jeng-Shiun, Chia-Hung Yen, Chin-Mu Hsu, and Hui-Hua Hsiao. 2021. "Management of Myeloma Bone Lesions" International Journal of Molecular Sciences 22, no. 7: 3389. https://doi.org/10.3390/ijms22073389
APA StyleDu, J.-S., Yen, C.-H., Hsu, C.-M., & Hsiao, H.-H. (2021). Management of Myeloma Bone Lesions. International Journal of Molecular Sciences, 22(7), 3389. https://doi.org/10.3390/ijms22073389