
Systematic Approach to Find the Global Minimum of Relaxation Dispersion Data for Protein-Induced B–Z Transition of DNA

 , ,
, ,
Abstract
:1. Introduction
2. Results
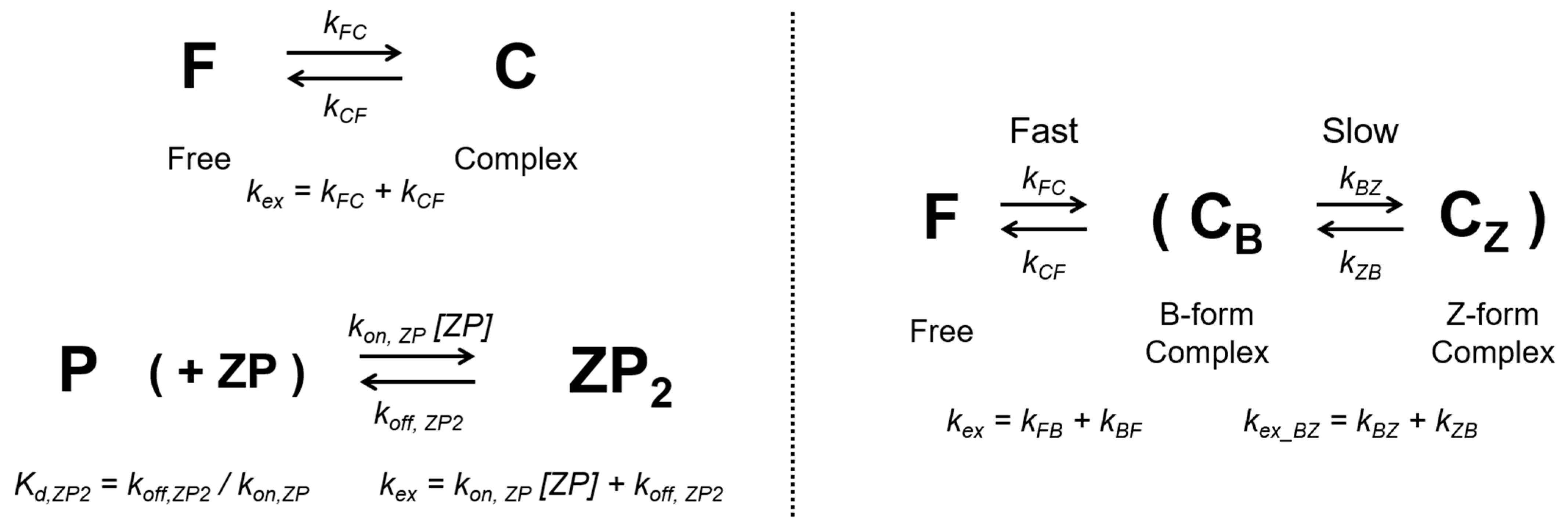
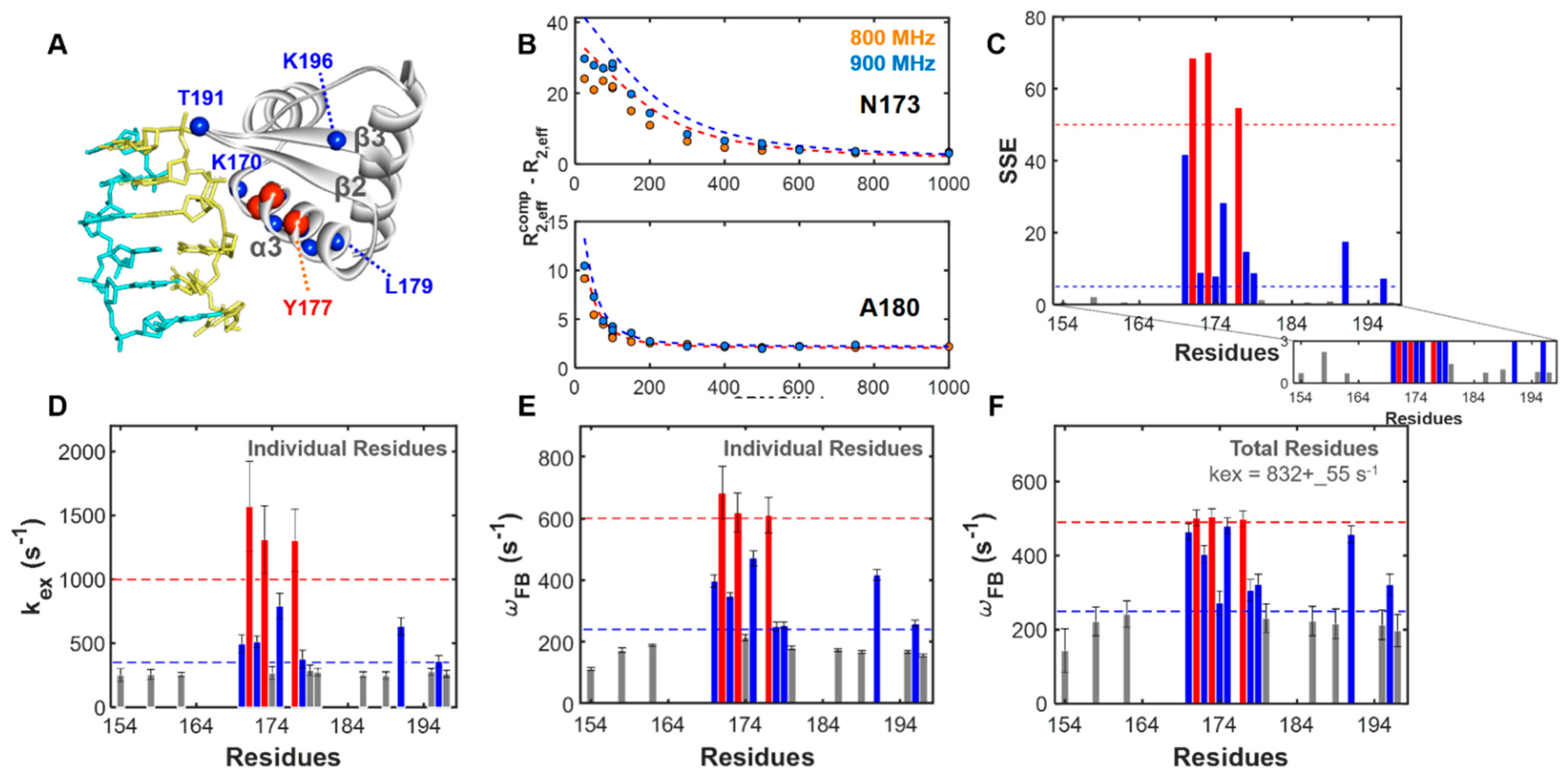
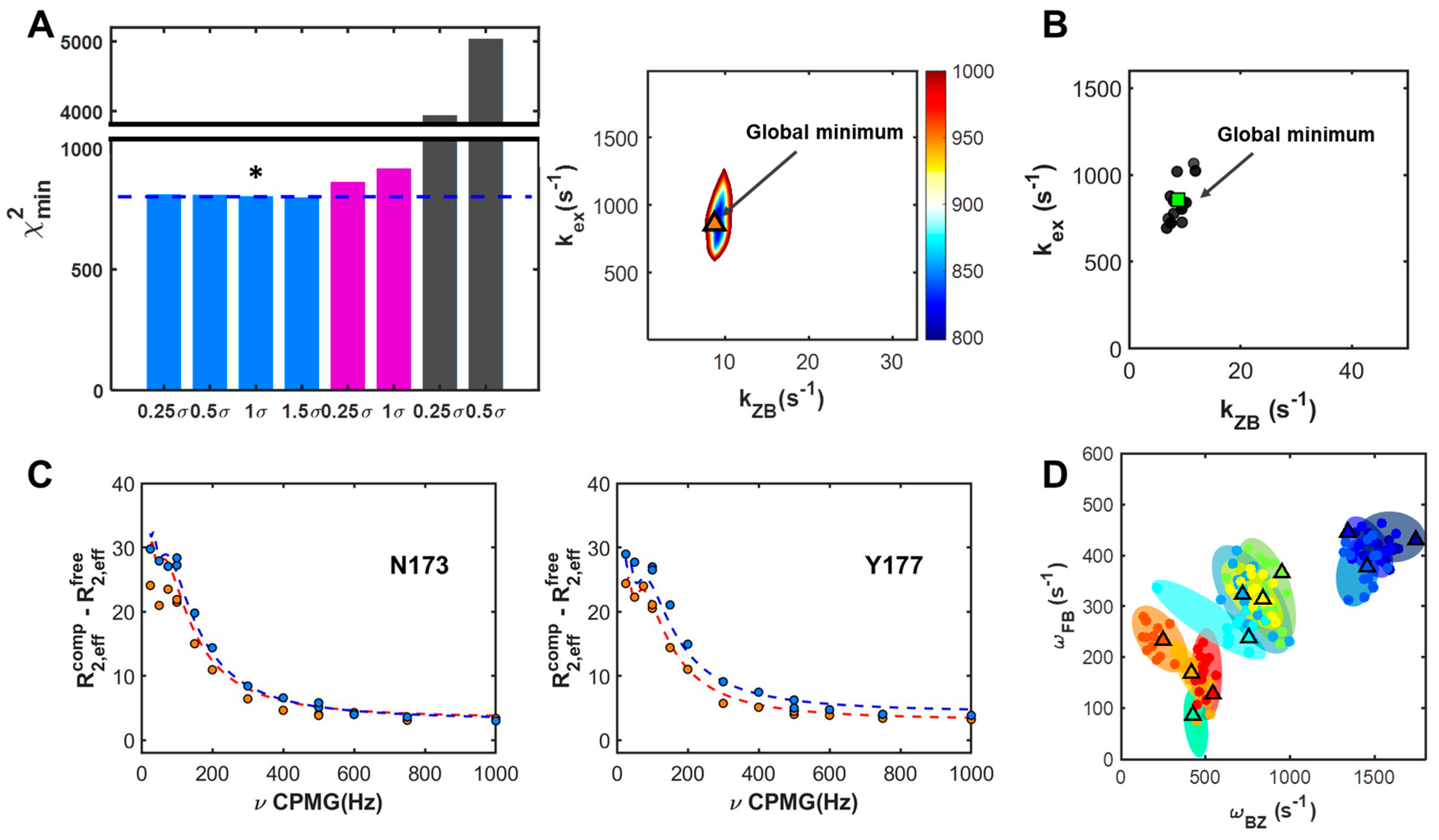
2.1. Classification of Three-State Conformational Transition of hZαADAR1
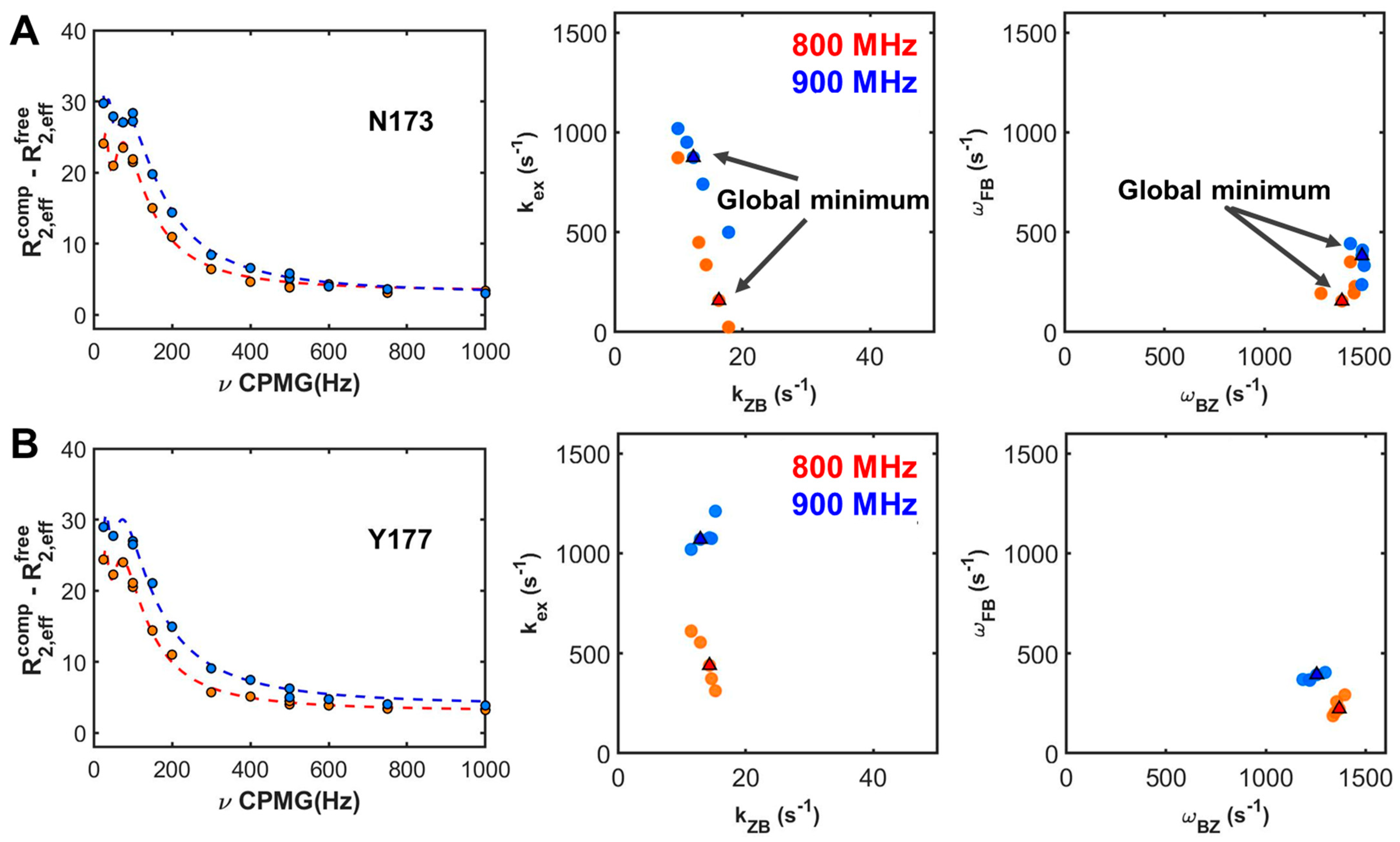
2.2. Site-Specific Analysis of Three-State Model Using Field-Dependent CPMG Profiles
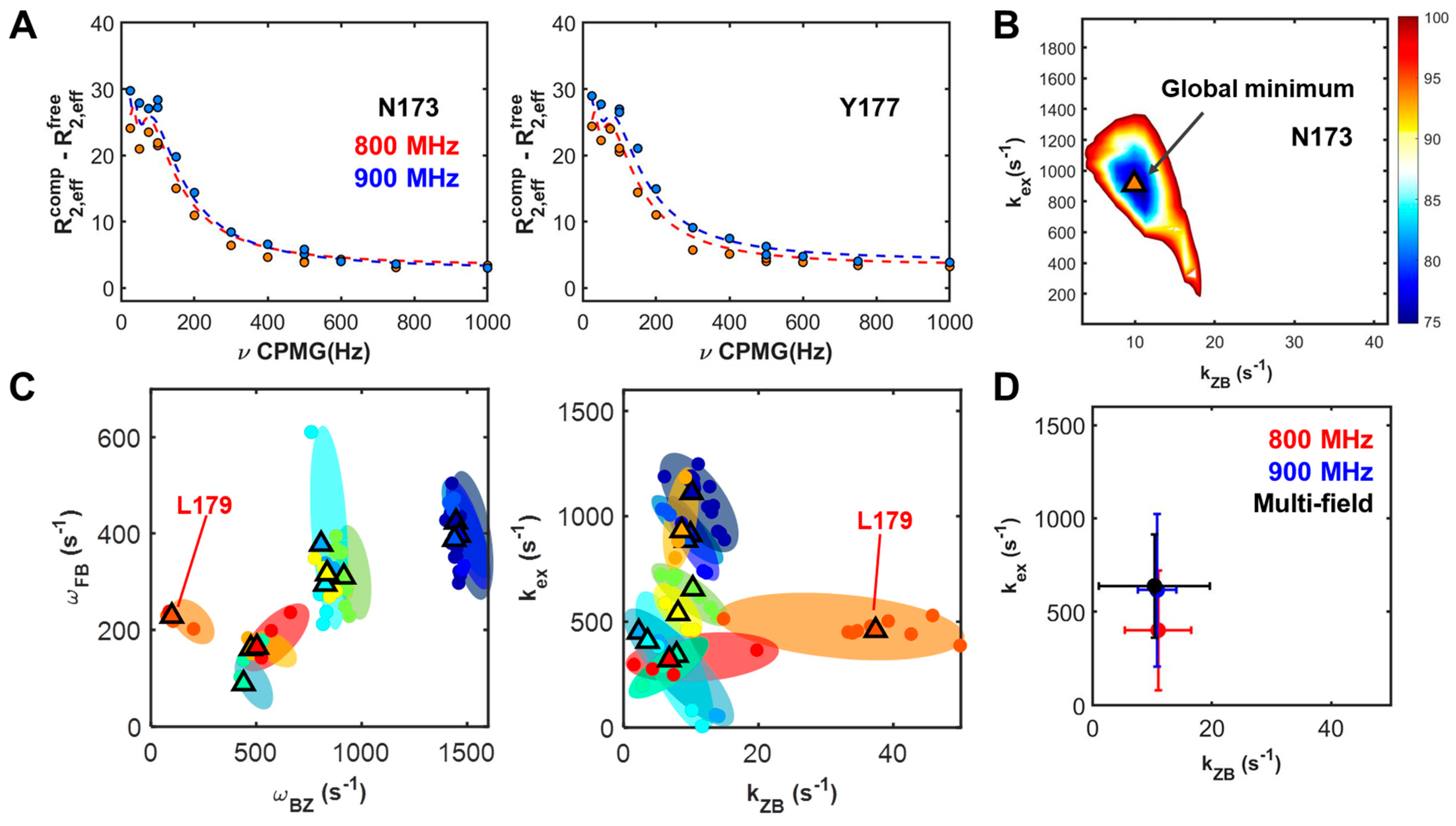
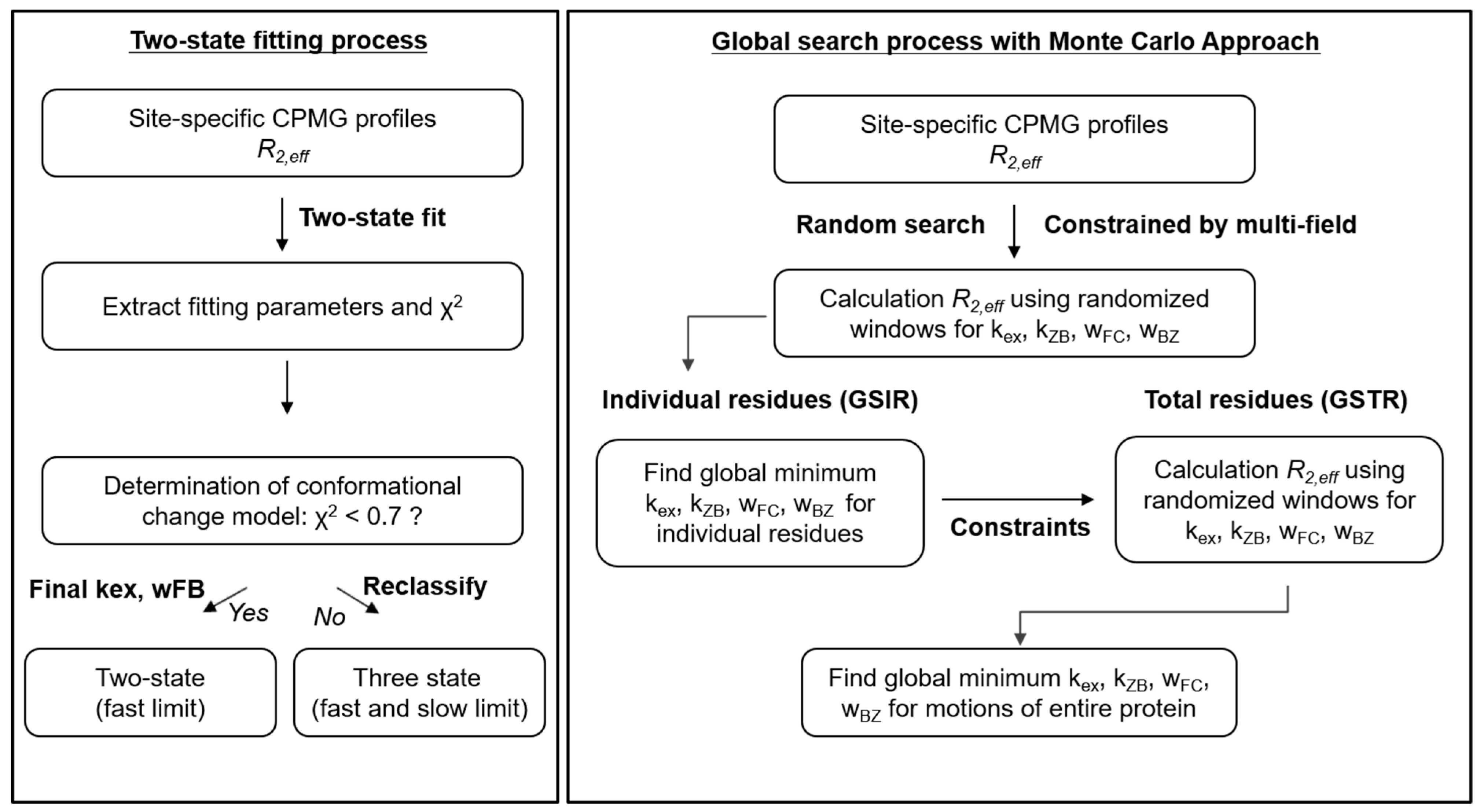
2.3. Dependencies of Parameters: Global Search for Total Residues (GSTR)
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. CPMG RD Results
4.3. Binding Models and Global Searches
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADAR1 | RNA-specific adenosine deaminase I |
| CPMG | Carr–Purcell–Meiboom–Gill |
| HSQC | Heteronuclear single quantum correlation |
| hZαADAR1 | The Zα domain of human ADAR1 |
| GSIR | Global search for individual residues |
| GSTR | Global search for total residues |
| RD | Relaxation dispersion |
| SSE | Sum of squared errors |
| ZBP | Z-DNA binding protein |
References
- Mizukoshi, Y.; Takeuchi, K.; Tokunaga, Y.; Matsuo, H.; Imai, M.; Fujisaki, M.; Kamoshida, H.; Takizawa, T.; Hanzawa, H.; Shimada, I. Targeting the cryptic sites: NMR-based strategy to improve protein druggability by controlling the conformational equilibrium. Sci. Adv. 2020, 6, eabd0480. [Google Scholar] [CrossRef]
- Roos, M.; Ott, M.; Hofmann, M.; Link, S.; Rössler, E.; Balbach, J.; Krushelnitsky, A.G.; Saalwächter, K.; Roessler, E.A.; Saalwaechter, K. Coupling and Decoupling of Rotational and Translational Diffusion of Proteins under Crowding Conditions. J. Am. Chem. Soc. 2016, 138, 10365–10372. [Google Scholar] [CrossRef] [Green Version]
- Esmaili, N.; Leroy, J.L. i-motif solution structure and dynamics of the d(AACCCC) and d(CCCCAA) tetrahymena telomeric repeats. Nucleic Acids Res. 2005, 33, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otten, R.; Villali, J.; Kern, D.; Mulder, F.A.A. Probing Microsecond Time Scale Dynamics in Proteins by Methyl 1H Carr−Purcell−Meiboom−Gill Relaxation Dispersion NMR Measurements. Application to Activation of the Signaling Protein NtrC r. J. Am. Chem. Soc. 2010, 132, 17004–17014. [Google Scholar] [CrossRef] [PubMed]
- Korzhnev, D.M.; Bezsonova, I.; Lee, S.; Chalikian, T.V.; Kay, L.E. Alternate Binding Modes for a Ubiquitin–SH3 Domain Interaction Studied by NMR Spectroscopy. J. Mol. Biol. 2009, 386, 391–405. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Hansen, D.F.; Vallurupalli, P.; Kay, L.E. Measurement of Methyl Axis Orientations in Invisible, Excited States of Proteins by Relaxation Dispersion NMR Spectroscopy. J. Am. Chem. Soc. 2009, 131, 11939–11948. [Google Scholar] [CrossRef] [PubMed]
- Beach, H.; Cole, R.; Gill, M.L.; Loria, J.P. Conservation of mus-ms enzyme motions in the apo- and substrate-mimicked state. J. Am. Chem. Soc. 2005, 127, 9167–9176. [Google Scholar] [CrossRef]
- Grey, M.J.; Wang, C.; Palmer, A.G. Disulfide Bond Isomerization in Basic Pancreatic Trypsin Inhibitor: Multisite Chemical Exchange Quantified by CPMG Relaxation Dispersion and Chemical Shift Modeling. J. Am. Chem. Soc. 2003, 125, 14324–14335. [Google Scholar] [CrossRef]
- Farber, P.J.; Mittermaier, A. Relaxation dispersion NMR spectroscopy for the study of protein allostery. Biophys. Rev. 2015, 7, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Demers, J.-P.; Mittermaier, A. Binding Mechanism of an SH3 Domain Studied by NMR and ITC. J. Am. Chem. Soc. 2009, 131, 4355–4367. [Google Scholar] [CrossRef]
- Sugase, K.; Lansing, J.C.; Dyson, A.H.J.; Wright, P.E. Tailoring Relaxation Dispersion Experiments for Fast-Associating Protein Complexes. J. Am. Chem. Soc. 2007, 129, 13406–13407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jae, L.T.; Raaben, M.; Riemersma, M.; Van Beusekom, E.; Blomen, V.A.; Velds, A.; Kerkhoven, R.M.; Carette, J.E.; Topaloglu, H.; Meinecke, P.; et al. Deciphering the Glycosylome of Dystroglycanopathies Using Haploid Screens for Lassa Virus Entry. Science 2013, 340, 479–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.-R.; Hwang, J.; Hur, J.H.; Ryu, K.-S.; Kim, K.K.; Choi, B.-S.; Kim, N.-K.; Lee, J.-H. NMR Dynamics Study Reveals the Zα Domain of Human ADAR1 Associates with and Dissociates from Z-RNA More Slowly than Z-DNA. ACS Chem. Biol. 2018, 14, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Rennella, E.; Sekhar, A.; Kay, L.E. Self-Assembly of Human Profilin-1 Detected by Carr–Purcell–Meiboom–Gill Nuclear Magnetic Resonance (CPMG NMR) Spectroscopy. Biochemistry 2017, 56, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Farber, P.J.; Slager, J.; Mittermaier, A.K. Local Folding and Misfolding in the PBX Homeodomain from a Three-State Analysis of CPMG Relaxation Dispersion NMR Data. J. Phys. Chem. B 2012, 116, 10317–10329. [Google Scholar] [CrossRef] [PubMed]
- Neudecker, P.; Korzhnev, D.M.; Kay, L.E. Assessment of the Effects of Increased Relaxation Dispersion Data on the Extraction of 3-site Exchange Parameters Characterizing the Unfolding of an SH3 Domain. J. Biomol. NMR 2006, 34, 129–135. [Google Scholar] [CrossRef]
- Korzhnev, D.M.; Neudecker, P.; Mittermaier, A.; Orekhov, V.Y.; Kay, L.E. Multiple-Site Exchange in Proteins Studied with a Suite of Six NMR Relaxation Dispersion Experiments: An Application to the Folding of a Fyn SH3 Domain Mutant. J. Am. Chem. Soc. 2005, 127, 15602–15611. [Google Scholar] [CrossRef]
- Miyahara, T.; Nakatsuji, H.; Sugiyama, H. Similarities and Differences between RNA and DNA Double-Helical Structures in Circular Dichroism Spectroscopy: A SAC-CI Study. J. Phys. Chem. A 2016, 120, 9008–9018. [Google Scholar] [CrossRef]
- Pan, F.; Roland, C.; Sagui, C. Ion distributions around left- and right-handed DNA and RNA duplexes: A comparative study. Nucleic Acids Res. 2014, 42, 13981–13996. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.; Kim, D.; Kim, K.K.; Kim, Y.-G.; Hohng, S. Intrinsic Z-DNA Is Stabilized by the Conformational Selection Mechanism of Z-DNA-Binding Proteins. J. Am. Chem. Soc. 2011, 133, 668–671. [Google Scholar] [CrossRef]
- Rich, A.; Zhang, S. Z-DNA: The long road to biological function. Textb. Met. Struct. Chem. 2018, 4, 261–268. [Google Scholar] [CrossRef]
- Lee, A.-R.; Kim, N.-H.; Seo, Y.-J.; Choi, S.-R.; Lee, J.-H. Thermodynamic Model for B-Z Transition of DNA Induced by Z-DNA Binding Proteins. Molecules 2018, 23, 2748. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, T.; Rould, M.A.; Lowenhaupt, K.; Herbert, A.; Rich, A. Crystal Structure of the Zα Domain of the Human Editing Enzyme ADAR1 Bound to Left-Handed Z-DNA. Textb. Met. Struct. Chem. 2018, 284, 234–238. [Google Scholar] [CrossRef]
- Ha, S.C.; Lowenhaupt, K.; Rich, A.; Kim, Y.-G.; Kim, K.K. Crystal structure of a junction between B-DNA and Z-DNA reveals two extruded bases. Textb. Met. Struct. Chem. 2018, 437, 207–210. [Google Scholar] [CrossRef]
- Wang, Z.; Choi, M.K.; Ban, T.; Yanai, H.; Negishi, H.; Lu, Y.; Tamura, T.; Takaoka, A.; Nishikura, K.; Taniguchi, T. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc. Natl. Acad. Sci. USA 2008, 105, 5477–5482. [Google Scholar] [CrossRef] [Green Version]
- Yanai, H.; Savitsky, D.; Tamura, T.; Taniguchi, T. Regulation of the cytosolic DNA-sensing system in innate immunity: A current view. Curr. Opin. Immunol. 2009, 21, 17–22. [Google Scholar] [CrossRef]
- Kim, Y.-G.; Lowenhaupt, K.; Oh, D.-B.; Kim, K.K.; Rich, A. Evidence that vaccinia virulence factor E3L binds to Z-DNA in vivo: Implications for development of a therapy for poxvirus infection. Textb. Met. Struct. Chem. 2018, 101, 244–248. [Google Scholar] [CrossRef]
- Kim, Y.-G.; Muralinath, M.; Brandt, T.; Pearcy, M.; Hauns, K.; Lowenhaupt, K.; Jacobs, B.L.; Rich, A. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 6974–6979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothenburg, S.; Deigendesch, N.; Dittmar, K.; Koch-Nolte, F.; Haag, F.; Lowenhaupt, K.; Rich, A. A PKR-like eukaryotic initiation factor 2 kinase from zebrafish contains Z-DNA binding domains instead of dsRNA binding domains. Proc. Natl. Acad. Sci. USA 2005, 102, 1602–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.-S.; Kim, N.-H.; Choi, S.-R.; Oh, K.-I.; Lee, J.-H. Protein-induced B-Z transition of DNA duplex containing a 2′-OMe guanosine. Biochem. Biophys. Res. Commun. 2020, 533, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Placido, D.; Brown, B.A.; Lowenhaupt, K.; Rich, A.; Athanasiadis, A. A Left-Handed RNA Double Helix Bound by the Zα Domain of the RNA-Editing Enzyme ADAR1. Structure 2007, 15, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.-M.; Bang, J.; Lee, E.-H.; Ahn, H.-C.; Seo, Y.-J.; Kim, K.K.; Kim, Y.-G.; Choi, B.-S.; Lee, J.-H. NMR Spectroscopic Elucidation of the B−Z Transition of a DNA Double Helix Induced by the Zα Domain of Human ADAR1. J. Am. Chem. Soc. 2009, 131, 11485–11491. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| kex (s−1) | kZB (s−1) | |
|---|---|---|
| Two-state | 832 ± 55 | - |
| GSIR | 638 ± 277 | 10 ± 9 |
| GSIR (800 MHz) | 401 ± 318 | 11 ± 5.5 |
| GSIR (900 MHz) | 615 ± 408 | 11 ± 3.2 |
| GSTR 1 | 844 ± 41 (858) | 9.8 ± 0.8 (8.7) |
| GSIR | GSTR | |||||||
|---|---|---|---|---|---|---|---|---|
| Residue | kex (s−1) | ΔωFB (Hz) | kZB (s−1) | ΔωBZ (Hz) | kex (s−1) | ΔωFB (Hz) | kZB (s−1) | ΔωBZ (Hz) |
| K170 | 450 | 378 | 2.25 | 807 | 845 ± 41.3 (858) | 342 ± 14.0 (320) | 9.81 ± 0.75 (8.74) | 633 ± 62.1 (718) |
| E171 | 1110 | 424 | 10.1 | 1450 | 385 ± 36.3 (430) | 1590 ± 145 (1740) | ||
| I172 | 407 | 295 | 3.56 | 827 | 226 ± 22.9 (238) | 758 ± 83.9 (755) | ||
| N173 | 912 | 396 | 9.93 | 1460 | 407 ± 31.6 (446) | 1473 ± 128 (1340) | ||
| R174 | 343 | 89.3 | 7.86 | 440 | 85.8 ± 16.0 (85.9) | 419 ± 45.4 (424) | ||
| V175 | 656 | 310 | 10.3 | 915 | 345 ± 14.1 (366) | 900 ± 79.7 (951) | ||
| Y177 | 885 | 388 | 9.29 | 1440 | 367 ± 45.8 (378) | 1404 ± 165 (1450) | ||
| S178 | 932 | 161 | 8.62 | 474 | 143 ± 19.0 (169) | 456 ± 54.5 (416) | ||
| L179 | 458 | 229 | 37.4 | 99.5 | 264 ± 23.3 (233) | 176 ± 53.6 (248) | ||
| T191 | 538 | 316 | 8.08 | 836 | 334 ± 27.7 (314) | 764 ± 90.1 (838) | ||
| K196 | 321 | 164 | 6.79 | 503 | 144 ± 19.5 (128) | 483 ± 89.6 (542) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, K.-I.; Lee, A.-R.; Choi, S.-R.; Go, Y.; Ryu, K.-S.; Kim, E.-H.; Lee, J.-H. Systematic Approach to Find the Global Minimum of Relaxation Dispersion Data for Protein-Induced B–Z Transition of DNA. Int. J. Mol. Sci. 2021, 22, 3517. https://doi.org/10.3390/ijms22073517
Oh K-I, Lee A-R, Choi S-R, Go Y, Ryu K-S, Kim E-H, Lee J-H. Systematic Approach to Find the Global Minimum of Relaxation Dispersion Data for Protein-Induced B–Z Transition of DNA. International Journal of Molecular Sciences. 2021; 22(7):3517. https://doi.org/10.3390/ijms22073517
Chicago/Turabian StyleOh, Kwang-Im, Ae-Ree Lee, Seo-Ree Choi, Youyeon Go, Kyoung-Seok Ryu, Eun-Hee Kim, and Joon-Hwa Lee. 2021. "Systematic Approach to Find the Global Minimum of Relaxation Dispersion Data for Protein-Induced B–Z Transition of DNA" International Journal of Molecular Sciences 22, no. 7: 3517. https://doi.org/10.3390/ijms22073517