
Skeletal Aging and Osteoporosis: Mechanisms and Therapeutics

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Biology of Skeletal Aging
3. Senescence
3.1. Inducers of Cellular Senescence and Osteoporosis
3.1.1. DNA Damage and Genomic Instability
3.1.2. Telomere Dysfunction
3.1.3. Epigenetic Alterations
3.1.4. Loss of Proteostasis
3.1.5. Mitochondria and ROS
4. Cellular Senescence and Skeletal Aging
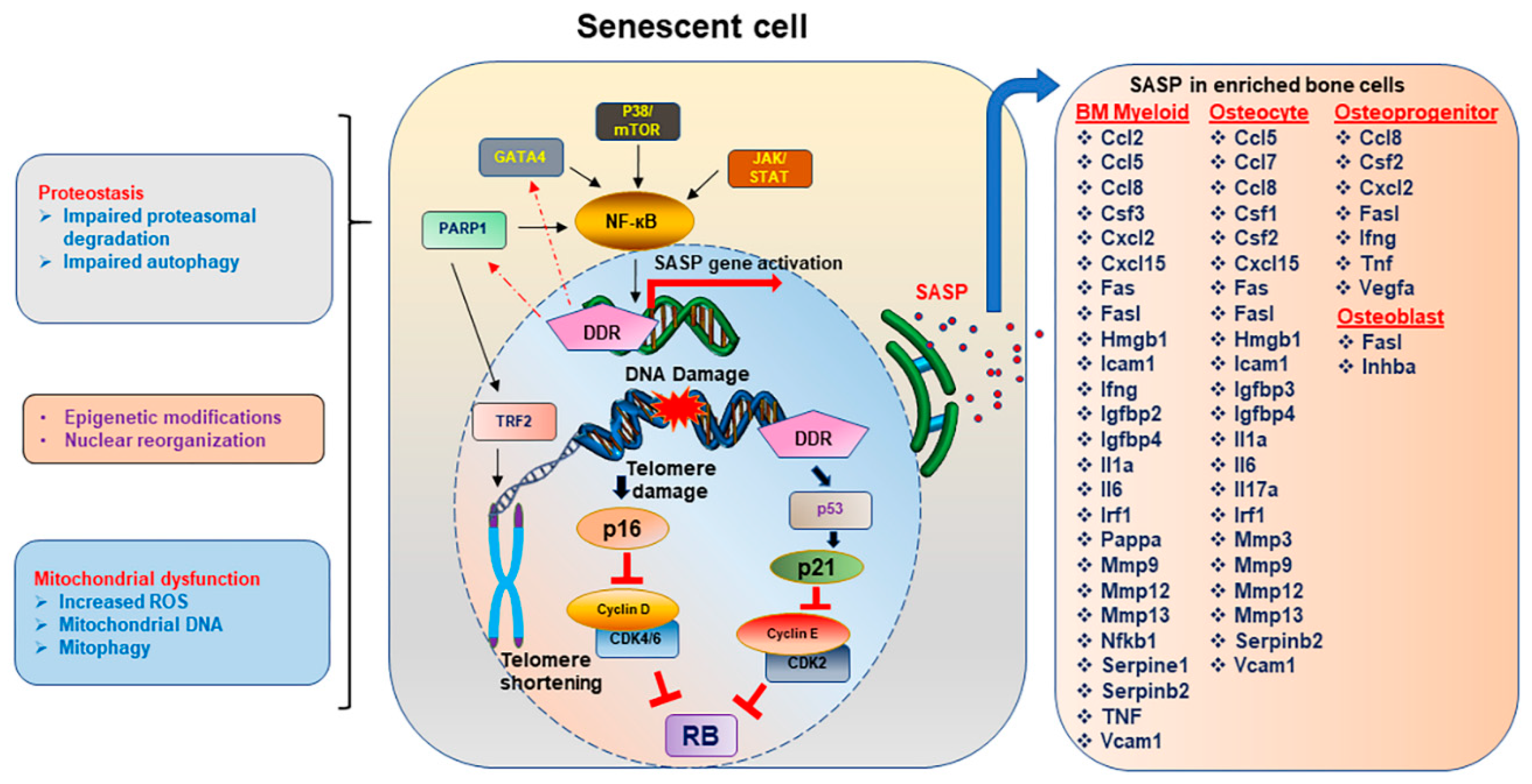
4.1. Senescence Associated Secretory Phenotype (SASP)
4.2. PARP1: Role in Senescence and Skeletal Aging
4.2.1. PARP1 in Senescence
4.2.2. PARP1 Role in Metabolism and Effects on Cellular Aging
4.2.3. PARP1 Role in Skeletal Aging
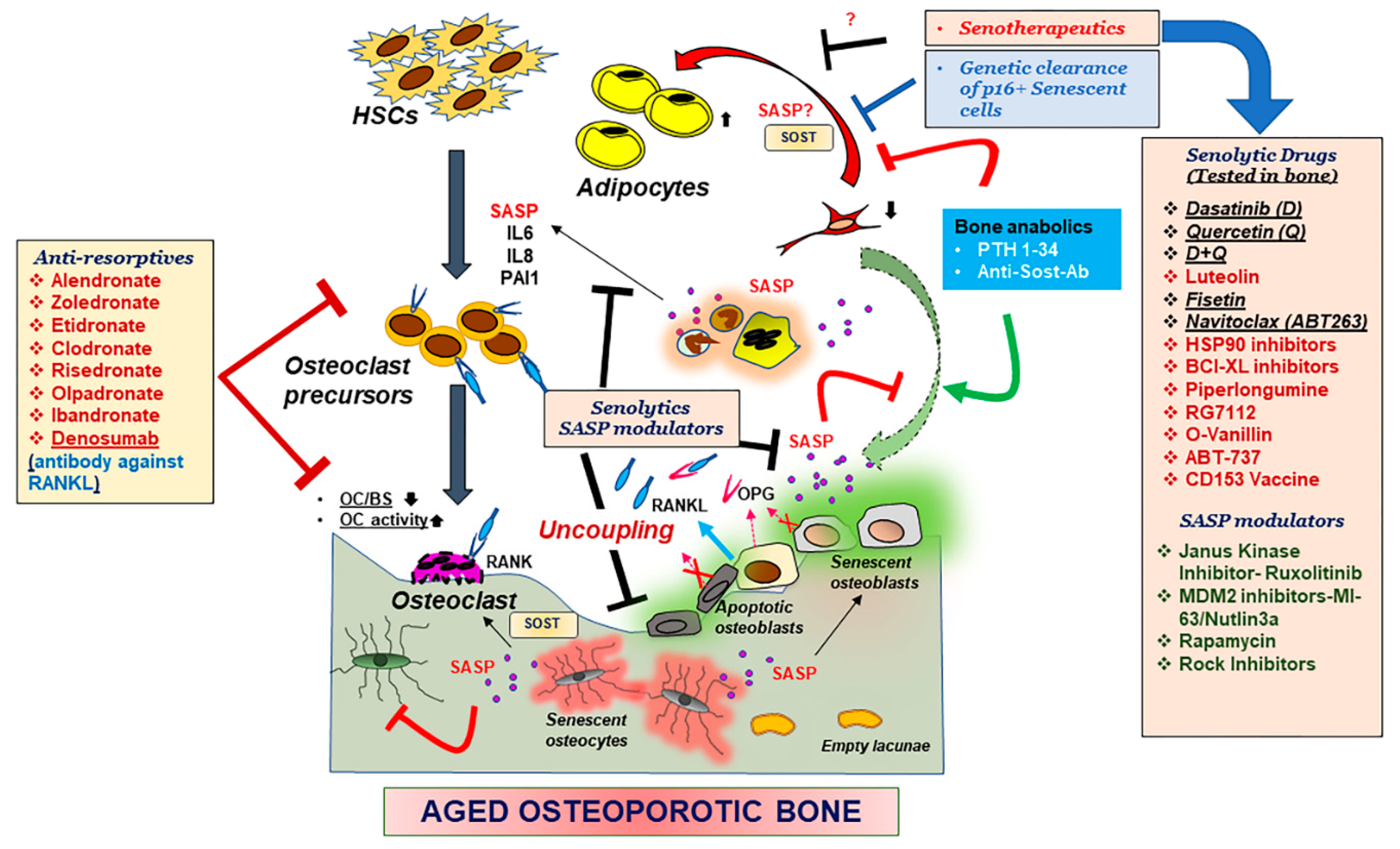
5. Therapeutics for Aging Bone
5.1. Parathyroid Hormone (PTH)
5.2. Anti-Sclerostin Antibody
5.3. Anti-Resorptives
5.4. Senolytics and SASP Modulators
6. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sozen, T.; Ozisik, L.; Basaran, N.C. An overview and management of osteoporosis. Eur. J. Rheumatol. 2017, 4, 46–56. [Google Scholar] [CrossRef]
- Manolagas, S.C. From estrogen-centric to aging and oxidative stress: A revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010, 31, 266–300. [Google Scholar] [CrossRef] [Green Version]
- Riggs, B.L.; Khosla, S.; Melton, L.J., 3rd. A unitary model for involutional osteoporosis: Estrogen deficiency causes both type I and type II osteoporosis in postmenopausal women and contributes to bone loss in aging men. J. Bone Miner. Res. 1998, 13, 763–773. [Google Scholar] [CrossRef]
- Farr, J.N.; Rowsey, J.L.; Eckhardt, B.A.; Thicke, B.S.; Fraser, D.G.; Tchkonia, T.; Kirkland, J.L.; Monroe, D.G.; Khosla, S. Independent Roles of Estrogen Deficiency and Cellular Senescence in the Pathogenesis of Osteoporosis: Evidence in Young Adult Mice and Older Humans. J. Bone Miner. Res. 2019, 34, 1407–1418. [Google Scholar] [CrossRef]
- Khosla, S.; Pacifici, R. Chapter 46–Estrogen Deficiency, Postmenopausal Osteoporosis, and Age-Related Bone Loss. In Osteoporosis, 4st ed.; Marcus, R., Feldman, D., Dempster, D.W., Luckey, M., Cauley, J.A., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 1113–1136. [Google Scholar]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Mochizuki, S.I.; Yano, K.; Fujise, N.; Sato, Y.; Goto, M.; Yamaguchi, K.; Kuriyama, M.; et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): A mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology 1998, 139, 1329–1337. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sarosi, I.; Yan, X.Q.; Morony, S.; Capparelli, C.; Tan, H.L.; McCabe, S.; Elliott, R.; Scully, S.; Van, G.; et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 1566–1571. [Google Scholar] [CrossRef] [Green Version]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Jilka, R.L. Biology of the basic multicellular unit and the pathophysiology of osteoporosis. Med. Pediatr. Oncol. 2003, 41, 182–185. [Google Scholar] [CrossRef]
- Mansour, A.; Mezour, M.A.; Badran, Z.; Tamimi, F. Extracellular Matrices for Bone Regeneration: A Literature Review. Tissue Eng. Part. A 2017, 23, 1436–1451. [Google Scholar] [CrossRef]
- Dallas, S.L.; Park-Snyder, S.; Miyazono, K.; Twardzik, D.; Mundy, G.R.; Bonewald, L.F. Characterization and autoregulation of latent transforming growth factor beta (TGF beta) complexes in osteoblast-like cell lines. Production of a latent complex lacking the latent TGF beta-binding protein. J. Biol. Chem. 1994, 269, 6815–6821. [Google Scholar] [CrossRef]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, G.A.; Bottemiller, B.L.; Turner, R.T.; Rader, J.I.; Baylink, D.J. Parathyroid hormone stimulates bone formation and resorption in organ culture: Evidence for a coupling mechanism. Proc. Natl. Acad. Sci. USA 1981, 78, 3204–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xian, L.; Wu, X.; Pang, L.; Lou, M.; Rosen, C.J.; Qiu, T.; Crane, J.; Frassica, F.; Zhang, L.; Rodriguez, J.P.; et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat. Med. 2012, 18, 1095–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000, 21, 115–137. [Google Scholar]
- Buenzli, P.R.; Sims, N.A. Quantifying the osteocyte network in the human skeleton. Bone 2015, 75, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Lin, C.; Stavre, Z.; Greenblatt, M.B.; Shim, J.H. Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells 2020, 9, 2073. [Google Scholar] [CrossRef]
- Vashishth, D.; Verborgt, O.; Divine, G.; Schaffler, M.B.; Fyhrie, D.P. Decline in osteocyte lacunar density in human cortical bone is associated with accumulation of microcracks with age. Bone 2000, 26, 375–380. [Google Scholar] [CrossRef]
- Jilka, R.L.; Almeida, M.; Ambrogini, E.; Han, L.; Roberson, P.K.; Weinstein, R.S.; Manolagas, S.C. Decreased oxidative stress and greater bone anabolism in the aged, when compared to the young, murine skeleton with parathyroid hormone administration. Aging Cell 2010, 9, 851–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdallah, B.M.; Haack-Sorensen, M.; Fink, T.; Kassem, M. Inhibition of osteoblast differentiation but not adipocyte differentiation of mesenchymal stem cells by sera obtained from aged females. Bone 2006, 39, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Fraser, D.G.; Wang, H.; Jaehn, K.; Ogrodnik, M.B.; Weivoda, M.M.; Drake, M.T.; Tchkonia, T.; LeBrasseur, N.K.; Kirkland, J.L.; et al. Identification of Senescent Cells in the Bone Microenvironment. J. Bone Miner. Res. 2016, 31, 1920–1929. [Google Scholar] [CrossRef] [PubMed]
- Kassem, M.; Marie, P.J. Senescence-associated intrinsic mechanisms of osteoblast dysfunctions. Aging Cell 2011, 10, 191–197. [Google Scholar] [CrossRef]
- Singh, L.; Brennan, T.A.; Russell, E.; Kim, J.H.; Chen, Q.; Brad Johnson, F.; Pignolo, R.J. Aging alters bone-fat reciprocity by shifting in vivo mesenchymal precursor cell fate towards an adipogenic lineage. Bone 2016, 85, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Duncan, E.L. Gene Testing in Everyday Clinical Use: Lessons from the Bone Clinic. J. Endocr. Soc. 2021, 5, bvaa200. [Google Scholar] [CrossRef]
- Patsch, J.M.; Kohler, T.; Berzlanovich, A.; Muschitz, C.; Bieglmayr, C.; Roschger, P.; Resch, H.; Pietschmann, P. Trabecular bone microstructure and local gene expression in iliac crest biopsies of men with idiopathic osteoporosis. J. Bone Miner. Res. 2011, 26, 1584–1592. [Google Scholar] [CrossRef]
- Farr, J.N.; Roforth, M.M.; Fujita, K.; Nicks, K.M.; Cunningham, J.M.; Atkinson, E.J.; Therneau, T.M.; McCready, L.K.; Peterson, J.M.; Drake, M.T.; et al. Effects of Age and Estrogen on Skeletal Gene Expression in Humans as Assessed by RNA Sequencing. PLoS ONE 2015, 10, e0138347. [Google Scholar] [CrossRef] [Green Version]
- Di Nisio, A.; Rocca, M.S.; Ghezzi, M.; Ponce, M.R.; Taglianetti, S.; Plebani, M.; Ferlin, A.; Foresta, C. Calcium-sensing receptor polymorphisms increase the risk of osteoporosis in ageing males. Endocrine 2018, 61, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Aitkulova, A.; Akilzhanova, A.; Abilova, Z.; Zhumatova, Z.; Akilzhanova, G.; Zholdybayeva, E. Collagen Type I alpha1 (COL1A1) Gene Polymorphism and Bone Mineral Density in Postmenopausal Kazakh Women. Cent. Asian J. Glob. Health 2014, 3, 144. [Google Scholar] [CrossRef]
- Bustamante, M.; Nogues, X.; Enjuanes, A.; Elosua, R.; Garcia-Giralt, N.; Perez-Edo, L.; Caceres, E.; Carreras, R.; Mellibovsky, L.; Balcells, S.; et al. COL1A1, ESR1, VDR and TGFB1 polymorphisms and haplotypes in relation to BMD in Spanish postmenopausal women. Osteoporos. Int. 2007, 18, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Rojano-Mejia, D.; Coral-Vazquez, R.M.; Espinosa, L.C.; Lopez-Medina, G.; Aguirre-Garcia, M.C.; Coronel, A.; Canto, P. JAG1 and COL1A1 polymorphisms and haplotypes in relation to bone mineral density variations in postmenopausal Mexican-Mestizo Women. Age 2013, 35, 471–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luther, J.; Yorgan, T.A.; Rolvien, T.; Ulsamer, L.; Koehne, T.; Liao, N.; Keller, D.; Vollersen, N.; Teufel, S.; Neven, M.; et al. Wnt1 is an Lrp5-independent bone-anabolic Wnt ligand. Sci. Transl. Med. 2018, 10, eaau7137. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Rajarajacholan, U.K.; Riabowol, K. Aging with ING: A comparative study of different forms of stress induced premature senescence. Oncotarget 2015, 6, 34118–34127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kural, K.C.; Tandon, N.; Skoblov, M.; Kel-Margoulis, O.V.; Baranova, A.V. Pathways of aging: Comparative analysis of gene signatures in replicative senescence and stress induced premature senescence. BMC Genom. 2016, 17 (Suppl. 14), 1030. [Google Scholar] [CrossRef] [Green Version]
- Cristofalo, V.J.; Pignolo, R.J. Replicative senescence of human fibroblast-like cells in culture. Physiol. Rev. 1993, 73, 617–638. [Google Scholar] [CrossRef]
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- el-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulic, V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- de Boer, J.; Andressoo, J.O.; de Wit, J.; Huijmans, J.; Beems, R.B.; van Steeg, H.; Weeda, G.; van der Horst, G.T.; van Leeuwen, W.; Themmen, A.P.; et al. Premature aging in mice deficient in DNA repair and transcription. Science 2002, 296, 1276–1279. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, N.; Wang, X.; Niu, Q.T.; Yeh, J.; Li, B. Atm-deficient mice: An osteoporosis model with defective osteoblast differentiation and increased osteoclastogenesis. Hum. Mol. Genet. 2006, 15, 1938–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruzankina, Y.; Pinzon-Guzman, C.; Asare, A.; Ong, T.; Pontano, L.; Cotsarelis, G.; Zediak, V.P.; Velez, M.; Bhandoola, A.; Brown, E.J. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 2007, 1, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Liu, K.; Robinson, A.R.; Clauson, C.L.; Blair, H.C.; Robbins, P.D.; Niedernhofer, L.J.; Ouyang, H. DNA damage drives accelerated bone aging via an NF-kappaB-dependent mechanism. J. Bone Miner. Res. 2013, 28, 1214–1228. [Google Scholar] [CrossRef] [PubMed]
- Misra, J.; Mohanty, S.T.; Madan, S.; Fernandes, J.A.; Hal Ebetino, F.; Russell, R.G.; Bellantuono, I. Zoledronate Attenuates Accumulation of DNA Damage in Mesenchymal Stem Cells and Protects Their Function. Stem Cells 2016, 34, 756–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, A.; Lin, T.; Zhu, J.; Tong, W.; Huo, Y.; Jia, H.; Zhang, Y.; Liu, X.S.; Cengel, K.; Xia, B.; et al. PTH1-34 blocks radiation-induced osteoblast apoptosis by enhancing DNA repair through canonical Wnt pathway. J. Biol. Chem. 2015, 290, 157–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, A.; Lin, T.; Young, T.; Tong, W.; Ma, X.; Tseng, W.J.; Kramer, I.; Kneissel, M.; Levine, M.A.; Zhang, Y.; et al. Suppression of Sclerostin Alleviates Radiation-Induced Bone Loss by Protecting Bone-Forming Cells and Their Progenitors Through Distinct Mechanisms. J. Bone Miner. Res. 2017, 32, 360–372. [Google Scholar] [CrossRef]
- Chandra, A.; Wang, L.; Young, T.; Zhong, L.; Tseng, W.J.; Levine, M.A.; Cengel, K.; Liu, X.S.; Zhang, Y.; Pignolo, R.J.; et al. Proteasome inhibitor bortezomib is a novel therapeutic agent for focal radiation-induced osteoporosis. FASEB J. 2018, 32, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Olovnikov, A.M. Principle of marginotomy in template synthesis of polynucleotides. Dokl. Akad. Nauk SSSR 1971, 201, 1496–1499. [Google Scholar]
- Watson, J.D. Origin of concatemeric T7 DNA. Nat. New Biol. 1972, 239, 197–201. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Chandra, A.; Lagnado, A.B.; Farr, J.N.; Monroe, D.G.; Park, S.; Hachfeld, C.; Tchkonia, T.; Kirkland, J.L.; Khosla, S.; Passos, J.F.; et al. Targeted Reduction of Senescent Cell Burden Alleviates Focal Radiotherapy-Related Bone Loss. J. Bone Miner. Res. 2020, 35, 1119–1131. [Google Scholar] [CrossRef]
- Baxter, M.A.; Wynn, R.F.; Jowitt, S.N.; Wraith, J.E.; Fairbairn, L.J.; Bellantuono, I. Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells 2004, 22, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Uhrhammer, N.A.; Lafarge, L.; Dos Santos, L.; Domaszewska, A.; Lange, M.; Yang, Y.; Aractingi, S.; Bessis, D.; Bignon, Y.J. Werner syndrome and mutations of the WRN and LMNA genes in France. Hum. Mutat. 2006, 27, 718–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignolo, R.J.; Suda, R.K.; McMillan, E.A.; Shen, J.; Lee, S.H.; Choi, Y.; Wright, A.C.; Johnson, F.B. Defects in telomere maintenance molecules impair osteoblast differentiation and promote osteoporosis. Aging Cell 2008, 7, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, T.A.; Egan, K.P.; Lindborg, C.M.; Chen, Q.; Sweetwyne, M.T.; Hankenson, K.D.; Xie, S.X.; Johnson, F.B.; Pignolo, R.J. Mouse models of telomere dysfunction phenocopy skeletal changes found in human age-related osteoporosis. Dis. Model. Mech. 2014, 7, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Vulliamy, T.; Beswick, R.; Kirwan, M.; Marrone, A.; Digweed, M.; Walne, A.; Dokal, I. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc. Natl. Acad. Sci. USA 2008, 105, 8073–8078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.A.; Tsai, P.C.; Joehanes, R.; Zheng, J.; Trajanoska, K.; Soerensen, M.; Forgetta, V.; Castillo-Fernandez, J.E.; Frost, M.; Spector, T.D.; et al. Epigenome-wide Association of DNA Methylation in Whole Blood With Bone Mineral Density. J. Bone Miner. Res. 2017, 32, 1644–1650. [Google Scholar] [CrossRef]
- Fernandez-Rebollo, E.; Eipel, M.; Seefried, L.; Hoffmann, P.; Strathmann, K.; Jakob, F.; Wagner, W. Primary Osteoporosis Is Not Reflected by Disease-Specific DNA Methylation or Accelerated Epigenetic Age in Blood. J. Bone Miner. Res. 2018, 33, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Jintaridth, P.; Tungtrongchitr, R.; Preutthipan, S.; Mutirangura, A. Hypomethylation of Alu elements in post-menopausal women with osteoporosis. PLoS ONE 2013, 8, e70386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reppe, S.; Lien, T.G.; Hsu, Y.H.; Gautvik, V.T.; Olstad, O.K.; Yu, R.; Bakke, H.G.; Lyle, R.; Kringen, M.K.; Glad, I.K.; et al. Distinct DNA methylation profiles in bone and blood of osteoporotic and healthy postmenopausal women. Epigenetics 2017, 12, 674–687. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Wei, W.; Dechow, P.C.; Wan, Y. HDAC7 inhibits osteoclastogenesis by reversing RANKL-triggered beta-catenin switch. Mol. Endocrinol. 2013, 27, 325–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.; Han, L.; Bartell, S.M.; Kim, H.N.; Gubrij, I.; de Cabo, R.; O’Brien, C.A.; Manolagas, S.C.; Almeida, M. Sirtuin1 (Sirt1) promotes cortical bone formation by preventing beta-catenin sequestration by FoxO transcription factors in osteoblast progenitors. J. Biol. Chem. 2014, 289, 24069–24078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, T.M.; Westendorf, J.J. Histone deacetylase inhibitors promote osteoblast maturation. J. Bone Miner. Res. 2005, 20, 2254–2263. [Google Scholar] [CrossRef]
- Dudakovic, A.; Camilleri, E.T.; Paradise, C.R.; Samsonraj, R.M.; Gluscevic, M.; Paggi, C.A.; Begun, D.L.; Khani, F.; Pichurin, O.; Ahmed, F.S.; et al. Enhancer of zeste homolog 2 (Ezh2) controls bone formation and cell cycle progression during osteogenesis in mice. J. Biol. Chem. 2018, 293, 12894–12907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudakovic, A.; Camilleri, E.T.; Riester, S.M.; Paradise, C.R.; Gluscevic, M.; O’Toole, T.M.; Thaler, R.; Evans, J.M.; Yan, H.; Subramaniam, M.; et al. Enhancer of Zeste Homolog 2 Inhibition Stimulates Bone Formation and Mitigates Bone Loss Caused by Ovariectomy in Skeletally Mature Mice. J. Biol. Chem. 2016, 291, 24594–24606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudakovic, A.; Samsonraj, R.M.; Paradise, C.R.; Galeano-Garces, C.; Mol, M.O.; Galeano-Garces, D.; Zan, P.; Galvan, M.L.; Hevesi, M.; Pichurin, O.; et al. Inhibition of the epigenetic suppressor EZH2 primes osteogenic differentiation mediated by BMP2. J. Biol. Chem. 2020, 295, 7877–7893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemming, S.; Cakouros, D.; Vandyke, K.; Davis, M.J.; Zannettino, A.C.; Gronthos, S. Identification of Novel EZH2 Targets Regulating Osteogenic Differentiation in Mesenchymal Stem Cells. Stem Cells Dev. 2016, 25, 909–921. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Calle, J.; Fernandez, A.F.; Sainz, J.; Zarrabeitia, M.T.; Sanudo, C.; Garcia-Renedo, R.; Perez-Nunez, M.I.; Garcia-Ibarbia, C.; Fraga, M.F.; Riancho, J.A. Genome-wide profiling of bone reveals differentially methylated regions in osteoporosis and osteoarthritis. Arthritis Rheum. 2013, 65, 197–205. [Google Scholar] [CrossRef]
- Sun, J.; Feng, H.; Xing, W.; Han, Y.; Suo, J.; Yallowitz, A.R.; Qian, N.; Shi, Y.; Greenblatt, M.B.; Zou, W. Histone demethylase LSD1 is critical for endochondral ossification during bone fracture healing. Sci. Adv. 2020, 6, eaaz1410. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ermann, J.; Niu, N.; Yan, G.; Yang, Y.; Shi, Y.; Zou, W. Histone demethylase LSD1 regulates bone mass by controlling WNT7B and BMP2 signaling in osteoblasts. Bone Res. 2018, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Fan, Z.; Yu, B.; Chang, J.; Al Hezaimi, K.; Zhou, X.; Park, N.H.; Wang, C.Y. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell 2012, 11, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Vrtacnik, P.; Zupan, J.; Mlakar, V.; Kranjc, T.; Marc, J.; Kern, B.; Ostanek, B. Epigenetic enzymes influenced by oxidative stress and hypoxia mimetic in osteoblasts are differentially expressed in patients with osteoporosis and osteoarthritis. Sci. Rep. 2018, 8, 16215. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, Y.; Jin, C.; Zhang, M.; Lv, L.; Zhang, X.; Liu, H.; Zhou, Y. Histone H3K9 Acetyltransferase PCAF Is Essential for Osteogenic Differentiation Through Bone Morphogenetic Protein Signaling and May Be Involved in Osteoporosis. Stem Cells 2016, 34, 2332–2341. [Google Scholar] [CrossRef]
- Bedene, A.; Mencej Bedrac, S.; Jese, L.; Marc, J.; Vrtacnik, P.; Prezelj, J.; Kocjan, T.; Kranjc, T.; Ostanek, B. MiR-148a the epigenetic regulator of bone homeostasis is increased in plasma of osteoporotic postmenopausal women. Wien. Klin. Wochenschr. 2016, 128 (Suppl. 7), 519–526. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, C.; Balmayor, E.R.; van Griensven, M. miRNAs Related to Skeletal Diseases. Stem Cells Dev. 2016, 25, 1261–1281. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kim, H.W.; Matsu-ura, K.; Wang, Y.G.; Xu, M.; Ashraf, M. Abrogation of Age-Induced MicroRNA-195 Rejuvenates the Senescent Mesenchymal Stem Cells by Reactivating Telomerase. Stem Cells 2016, 34, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, M.I.; Silva, A.M.; Vasconcelos, D.M.; Almeida, C.R.; Caires, H.; Pinto, M.T.; Calin, G.A.; Santos, S.G.; Barbosa, M.A. miR-195 in human primary mesenchymal stromal/stem cells regulates proliferation, osteogenesis and paracrine effect on angiogenesis. Oncotarget 2016, 7, 7–22. [Google Scholar] [CrossRef] [Green Version]
- Aquino-Martinez, R.; Farr, J.N.; Weivoda, M.M.; Negley, B.A.; Onken, J.L.; Thicke, B.S.; Fulcer, M.M.; Fraser, D.G.; van Wijnen, A.J.; Khosla, S.; et al. miR-219a-5p Regulates Rorbeta During Osteoblast Differentiation and in Age-related Bone Loss. J. Bone Miner. Res. 2019, 34, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.M.; Park, S.J.; Jung, S.H.; Kim, E.J.; Jogeswar, G.; Ajita, J.; Rhee, Y.; Kim, C.H.; Lim, S.K. miR-182 is a negative regulator of osteoblast proliferation, differentiation, and skeletogenesis through targeting FoxO1. J. Bone Miner. Res. 2012, 27, 1669–1679. [Google Scholar] [CrossRef]
- Chen, R.; Qiu, H.; Tong, Y.; Liao, F.; Hu, X.; Qiu, Y.; Liao, Y. MiRNA-19a-3p alleviates the progression of osteoporosis by targeting HDAC4 to promote the osteogenic differentiation of hMSCs. BioChem. Biophys Res. Commun. 2019, 516, 666–672. [Google Scholar] [CrossRef]
- Gao, G.C.; Yang, D.W.; Liu, W. LncRNA TERC alleviates the progression of osteoporosis by absorbing miRNA-217 to upregulate RUNX2. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 526–534. [Google Scholar]
- Li, K.; Chen, S.; Cai, P.; Chen, K.; Li, L.; Yang, X.; Yi, J.; Luo, X.; Du, Y.; Zheng, H. MiRNA-483-5p is involved in the pathogenesis of osteoporosis by promoting osteoclast differentiation. Mol. Cell Probes 2020, 49, 101479. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, W.; Huang, Y. MiRNA-133a is involved in the regulation of postmenopausal osteoporosis through promoting osteoclast differentiation. Acta Biochim. Biophys. Sin. 2018, 50, 273–280. [Google Scholar] [CrossRef] [Green Version]
- Gong, N.; Zhu, W.; Xu, R.; Teng, Z.; Deng, C.; Zhou, H.; Xia, M.; Zhao, M. Keratinocytes-derived exosomal miRNA regulates osteoclast differentiation in middle ear cholesteatoma. BioChem. Biophys. Res. Commun. 2020, 525, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Zhang, D.; Wu, H.; Zhu, Q.; Yang, C.; Zhu, J. MiRNA-199a-5p positively regulated RANKL-induced osteoclast differentiation by target Mafb protein. J. Cell BioChem. 2018. [Google Scholar] [CrossRef]
- Kocijan, R.; Muschitz, C.; Geiger, E.; Skalicky, S.; Baierl, A.; Dormann, R.; Plachel, F.; Feichtinger, X.; Heimel, P.; Fahrleitner-Pammer, A.; et al. Circulating microRNA Signatures in Patients With Idiopathic and Postmenopausal Osteoporosis and Fragility Fractures. J. Clin. Endocrinol. Metab. 2016, 101, 4125–4134. [Google Scholar] [CrossRef] [PubMed]
- Feichtinger, X.; Muschitz, C.; Heimel, P.; Baierl, A.; Fahrleitner-Pammer, A.; Redl, H.; Resch, H.; Geiger, E.; Skalicky, S.; Dormann, R.; et al. Bone-related Circulating MicroRNAs miR-29b-3p, miR-550a-3p, and miR-324-3p and their Association to Bone Microstructure and Histomorphometry. Sci. Rep. 2018, 8, 4867. [Google Scholar] [CrossRef] [PubMed]
- Hrdlicka, H.C.; Pereira, R.C.; Shin, B.; Yee, S.P.; Deymier, A.C.; Lee, S.K.; Delany, A.M. Inhibition of miR-29-3p isoforms via tough decoy suppresses osteoblast function in homeostasis but promotes intermittent parathyroid hormone-induced bone anabolism. Bone 2021, 143, 115779. [Google Scholar] [CrossRef]
- Laxman, N.; Rubin, C.J.; Mallmin, H.; Nilsson, O.; Pastinen, T.; Grundberg, E.; Kindmark, A. Global miRNA expression and correlation with mRNA levels in primary human bone cells. RNA 2015, 21, 1433–1443. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.Y.; Li, N.; Lin, S.; Wang, B.; Lan, H.Y.; Li, G. miRNA-29b improves bone healing in mouse fracture model. Mol. Cell Endocrinol. 2016, 430, 97–107. [Google Scholar] [CrossRef]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Li, H.; Li, D.; Ma, Z.; Qian, Z.; Kang, X.; Jin, X.; Li, F.; Wang, X.; Chen, Q.; Sun, H.; et al. Defective autophagy in osteoblasts induces endoplasmic reticulum stress and causes remarkable bone loss. Autophagy 2018, 14, 1726–1741. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Qi, M.; An, Y.; Zhang, L.; Yang, R.; Doro, D.H.; Liu, W.; Jin, Y. Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging Cell 2018, 17, e12709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chondrogianni, N.; Trougakos, I.P.; Kletsas, D.; Chen, Q.M.; Gonos, E.S. Partial proteasome inhibition in human fibroblasts triggers accelerated M1 senescence or M2 crisis depending on p53 and Rb status. Aging Cell 2008, 7, 717–732. [Google Scholar] [CrossRef] [PubMed]
- Kretowski, R.; Borzym-Kluczyk, M.; Cechowska-Pasko, M. Hypoxia enhances the senescence effect of bortezomib--the proteasome inhibitor--on human skin fibroblasts. Biomed. Res. Int. 2014, 2014, 196249. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Di Filippo, C.; Laieta, M.T.; Vestini, R.; Barbieri, M.; Sangiulo, P.; Crescenzi, B.; Ferraraccio, F.; Rossi, F.; D’Amico, M.; et al. Effects of ubiquitin-proteasome system deregulation on the vascular senescence and atherosclerosis process in elderly patients. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 200–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett, I.R.; Chen, D.; Gutierrez, G.; Zhao, M.; Escobedo, A.; Rossini, G.; Harris, S.E.; Gallwitz, W.; Kim, K.B.; Hu, S.; et al. Selective inhibitors of the osteoblast proteasome stimulate bone formation in vivo and in vitro. J. Clin. Investig. 2003, 111, 1771–1782. [Google Scholar] [CrossRef] [Green Version]
- Mundy, G.; Gutierrez, G.; Garrett, R.; Gallwitz, W.; Rossini, G.; Christiansen, C.; Langenberg, A. Proteasome inhibitors stimulate both bone formation and hair growth by similar mechanisms. Ann. N. Y Acad. Sci. 2007, 1117, 298–301. [Google Scholar] [CrossRef]
- Legesse-Miller, A.; Raitman, I.; Haley, E.M.; Liao, A.; Sun, L.L.; Wang, D.J.; Krishnan, N.; Lemons, J.M.; Suh, E.J.; Johnson, E.L.; et al. Quiescent fibroblasts are protected from proteasome inhibition-mediated toxicity. Mol. Biol. Cell 2012, 23, 3566–3581. [Google Scholar] [CrossRef]
- Von Zglinicki, T.; Saretzki, G.; Docke, W.; Lotze, C. Mild hyperoxia shortens telomeres and inhibits proliferation of fibroblasts: A model for senescence? Exp. Cell Res. 1995, 220, 186–193. [Google Scholar] [CrossRef]
- Vizioli, M.G.; Liu, T.; Miller, K.N.; Robertson, N.A.; Gilroy, K.; Lagnado, A.B.; Perez-Garcia, A.; Kiourtis, C.; Dasgupta, N.; Lei, X.; et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020, 34, 428–445. [Google Scholar] [CrossRef]
- Domazetovic, V.; Marcucci, G.; Iantomasi, T.; Brandi, M.L.; Vincenzini, M.T. Oxidative stress in bone remodeling: Role of antioxidants. Clin. Cases Miner. Bone Metab. 2017, 14, 209–216. [Google Scholar] [CrossRef]
- Agidigbi, T.S.; Kim, C. Reactive Oxygen Species in Osteoclast Differentiation and Possible Pharmaceutical Targets of ROS-Mediated Osteoclast Diseases. Int. J. Mol. Sci. 2019, 20, 3576. [Google Scholar] [CrossRef] [Green Version]
- Altindag, O.; Erel, O.; Soran, N.; Celik, H.; Selek, S. Total oxidative/anti-oxidative status and relation to bone mineral density in osteoporosis. Rheumatol. Int. 2008, 28, 317–321. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhu, L.; Zhang, D.; Li, N.; Li, Q.; Dai, P.; Mao, Y.; Li, X.; Ma, J.; Huang, S. Oxidative Stress-Related Biomarkers in Postmenopausal Osteoporosis: A Systematic Review and Meta-Analyses. Dis. Markers 2016, 2016, 7067984. [Google Scholar] [CrossRef] [PubMed]
- Dobson, P.F.; Dennis, E.P.; Hipps, D.; Reeve, A.; Laude, A.; Bradshaw, C.; Stamp, C.; Smith, A.; Deehan, D.J.; Turnbull, D.M.; et al. Mitochondrial dysfunction impairs osteogenesis, increases osteoclast activity, and accelerates age related bone loss. Sci. Rep. 2020, 10, 11643. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Tsuboyama, T.; Kasai, R.; Okumura, H.; Yamamuro, T.; Higuchi, K.; Higuchi, K.; Kohno, A.; Yonezu, T.; Utani, A.; et al. Age-related changes in bone mass in the senescence-accelerated mouse (SAM). SAM-R/3 and SAM-P/6 as new murine models for senile osteoporosis. Am. J. Pathol. 1986, 125, 276–283. [Google Scholar] [PubMed]
- Takeda, T. Senescence-accelerated mouse (SAM): A biogerontological resource in aging research. Neuro. Biol. Aging 1999, 20, 105–110. [Google Scholar] [CrossRef]
- Battmann, A.; Schulz, A.; Stahl, U. Cellular senescence: A mechanism of the development of osteoporosis? Orthopade 2001, 30, 405–411. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef]
- Kim, H.N.; Chang, J.; Iyer, S.; Han, L.; Campisi, J.; Manolagas, S.C.; Zhou, D.; Almeida, M. Elimination of senescent osteoclast progenitors has no effect on the age-associated loss of bone mass in mice. Aging Cell 2019, 18, e12923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Kim, H.N.; Chang, J.; Shao, L.; Han, L.; Iyer, S.; Manolagas, S.C.; O’Brien, C.A.; Jilka, R.L.; Zhou, D.; Almeida, M. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell 2017, 16, 693–703. [Google Scholar] [CrossRef]
- Coppe, J.P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.Y.; Campisi, J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.C.; Sangesland, M.; Kaeberlein, M.; Rabinovitch, P.S. Modulating mTOR in aging and health. Interdiscip Top. Gerontol. 2015, 40, 107–127. [Google Scholar] [PubMed]
- Zhang, B.; Fu, D.; Xu, Q.; Cong, X.; Wu, C.; Zhong, X.; Ma, Y.; Lv, Z.; Chen, F.; Han, L.; et al. The senescence-associated secretory phenotype is potentiated by feedforward regulatory mechanisms involving Zscan4 and TAK1. Nat. Commun. 2018, 9, 1723. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Zhou, L.; Sarantos, M.R.; Rodier, F.; Freund, A.; de Keizer, P.L.; Liu, S.; Demaria, M.; Cong, Y.S.; Kapahi, P.; et al. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell 2012, 11, 569–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Wang, Y.; Wang, J.; Zhai, J.; He, F.; Zhu, G. Irradiation-induced senescence of bone marrow mesenchymal stem cells aggravates osteogenic differentiation dysfunction via paracrine signaling. Am. J. Physiol. Cell Physiol. 2020, 318, C1005–C1017. [Google Scholar] [CrossRef]
- Aquino-Martinez, R.; Rowsey, J.L.; Fraser, D.G.; Eckhardt, B.A.; Khosla, S.; Farr, J.N.; Monroe, D.G. LPS-induced premature osteocyte senescence: Implications in inflammatory alveolar bone loss and periodontal disease pathogenesis. Bone 2020, 132, 115220. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, L.; Lu, A.; Han, Y.; Colangelo, D.; Bukata, C.; Scibetta, A.; Yousefzadeh, M.J.; Li, X.; Gurkar, A.U.; et al. ATM is a key driver of NF-kappaB-dependent DNA-damage-induced senescence, stem cell dysfunction and aging. Aging 2020, 12, 4688–4710. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Liu, Z.; Peng, L.; Tang, X.; Meng, F.; Ao, Y.; Zhou, M.; Wang, M.; Cao, X.; Qin, B.; et al. Boosting ATM activity alleviates aging and extends lifespan in a mouse model of progeria. Elife 2018, 7, e34836. [Google Scholar] [CrossRef]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghesan, M.; Fusilli, C.; Rappa, F.; Panebianco, C.; Rizzo, G.; Oben, J.A.; Mazzoccoli, G.; Faulkes, C.; Pata, I.; Agodi, A.; et al. DNA Hypomethylation and Histone Variant macroH2A1 Synergistically Attenuate Chemotherapy-Induced Senescence to Promote Hepatocellular Carcinoma Progression. Cancer Res. 2016, 76, 594–606. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Shin, Y.; Lee, S.; Kim, M.; Punj, V.; Lu, J.F.; Shin, H.; Kim, K.; Ulmer, T.S.; Koh, J.; et al. Regulation of Breast Cancer-Induced Osteoclastogenesis by MacroH2A1.2 Involving EZH2-Mediated H3K27me3. Cell Rep. 2018, 24, 224–237. [Google Scholar] [CrossRef]
- Burkle, A. DNA repair and PARP in aging. Free Radic. Res. 2006, 40, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Gomez, M.; Wu, J.; Schreiber, V.; Dunlap, J.; Dantzer, F.; Wang, Y.; Liu, Y. PARP1 Is a TRF2-associated poly(ADP-ribose)polymerase and protects eroded telomeres. Mol. Biol. Cell 2006, 17, 1686–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beneke, S.; Cohausz, O.; Malanga, M.; Boukamp, P.; Althaus, F.; Burkle, A. Rapid regulation of telomere length is mediated by poly(ADP-ribose) polymerase-1. Nucleic Acids Res. 2008, 36, 6309–6317. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F.; Hande, M.P.; Tong, W.M.; Lansdorp, P.M.; Wang, Z.Q.; Jackson, S.P. Functions of poly(ADP-ribose) polymerase in controlling telomere length and chromosomal stability. Nat. Genet. 1999, 23, 76–80. [Google Scholar] [CrossRef]
- O’Connor, M.S.; Safari, A.; Liu, D.; Qin, J.; Songyang, Z. The human Rap1 protein complex and modulation of telomere length. J. Biol. Chem. 2004, 279, 28585–28591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grube, K.; Burkle, A. Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with species-specific life span. Proc. Natl. Acad. Sci. USA 1992, 89, 11759–11763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunzmann, A.; Dedoussis, G.; Jajte, J.; Malavolta, M.; Mocchegiani, E.; Burkle, A. Effect of zinc on cellular poly(ADP-ribosyl)ation capacity. Exp. Gerontol. 2008, 43, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muiras, M.L.; Muller, M.; Schachter, F.; Burkle, A. Increased poly(ADP-ribose) polymerase activity in lymphoblastoid cell lines from centenarians. J. Mol. Med. 1998, 76, 346–354. [Google Scholar] [CrossRef]
- Chevanne, M.; Calia, C.; Zampieri, M.; Cecchinelli, B.; Caldini, R.; Monti, D.; Bucci, L.; Franceschi, C.; Caiafa, P. Oxidative DNA damage repair and parp 1 and parp 2 expression in Epstein-Barr virus-immortalized B lymphocyte cells from young subjects, old subjects, and centenarians. Rejuvenation Res. 2007, 10, 191–204. [Google Scholar] [CrossRef]
- Spina Purrello, V.; Cormaci, G.; Denaro, L.; Reale, S.; Costa, A.; Lalicata, C.; Sabbatini, M.; Marchetti, B.; Avola, R. Effect of growth factors on nuclear and mitochondrial ADP-ribosylation processes during astroglial cell development and aging in culture. Mech. Ageing Dev. 2002, 123, 511–520. [Google Scholar] [CrossRef]
- Shall, S.; de Murcia, G. Poly(ADP-ribose) polymerase-1: What have we learned from the deficient mouse model? Mutat. Res. 2000, 460, 1–15. [Google Scholar] [CrossRef]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Cohausz, O.; Blenn, C.; Malanga, M.; Althaus, F.R. The roles of poly(ADP-ribose)-metabolizing enzymes in alkylation-induced cell death. Cell Mol. Life Sci. 2008, 65, 644–655. [Google Scholar] [CrossRef] [Green Version]
- Hassa, P.O.; Hottiger, M.O. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-kappaB in inflammatory disorders. Cell Mol. Life Sci. 2002, 59, 1534–1553. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [Green Version]
- Adler, A.S.; Kawahara, T.L.; Segal, E.; Chang, H.Y. Reversal of aging by NFkappaB blockade. Cell Cycle 2008, 7, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Berger, N.A.; Sims, J.L.; Catino, D.M.; Berger, S.J. Poly(ADP-ribose) polymerase mediates the suicide response to massive DNA damage: Studies in normal and DNA-repair defective cells. Princess Takamatsu Symp. 1983, 13, 219–226. [Google Scholar] [PubMed]
- Canto, C.; Auwerx, J. Calorie restriction: Is AMPK a key sensor and effector? Physiology 2011, 26, 214–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zha, S.; Li, Z.; Cao, Q.; Wang, F.; Liu, F. PARP1 inhibitor (PJ34) improves the function of aging-induced endothelial progenitor cells by preserving intracellular NAD(+) levels and increasing SIRT1 activity. Stem Cell Res. Ther. 2018, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, G.M.; Wise-Draper, T.M.; Morreale, R.J.; Morrison, M.A.; Gole, B.; Schwemberger, S.; Tichy, E.D.; Lu, L.; Babcock, G.F.; Wells, J.M.; et al. The human DEK oncogene regulates DNA damage response signaling and repair. Nucleic Acids Res. 2011, 39, 7465–7476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrer, J.; Popp, O.; Malanga, M.; Beneke, S.; Markovitz, D.M.; Ferrando-May, E.; Burkle, A.; Kappes, F. High-affinity interaction of poly(ADP-ribose) and the human DEK oncoprotein depends upon chain length. Biochemistry 2010, 49, 7119–7130. [Google Scholar] [CrossRef] [Green Version]
- Yao, H.; Sundar, I.K.; Gorbunova, V.; Rahman, I. P21-PARP-1 pathway is involved in cigarette smoke-induced lung DNA damage and cellular senescence. PLoS ONE 2013, 8, e80007. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gao, J.; Zhou, J.; Liu, H.; Xu, C. Olaparib induced senescence under P16 or P53 dependent manner in ovarian cancer. J. Gynecol. Oncol. 2019, 30, e26. [Google Scholar] [CrossRef]
- Martinez-Zamudio, R.I.; Ha, H.C. PARP1 enhances inflammatory cytokine expression by alteration of promoter chromatin structure in microglia. Brain. Behav. 2014, 4, 552–565. [Google Scholar] [CrossRef]
- Hurtado-Bages, S.; Guberovic, I.; Buschbeck, M. The MacroH2A1.1—PARP1 Axis at the Intersection between Stress Response and Metabolism. Front. Genet. 2018, 9, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, O.; Drews, L.F.; Nguyen, A.; Tatsuta, T.; Gkioni, L.; Hendrich, O.; Zhang, Q.; Langer, T.; Pletcher, S.; Wakelam, M.J.O.; et al. A nutritional memory effect counteracts benefits of dietary restriction in old mice. Nat. Metab. 2019, 1, 1059–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangerich, A.; Herbach, N.; Hanf, B.; Fischbach, A.; Popp, O.; Moreno-Villanueva, M.; Bruns, O.T.; Burkle, A. Inflammatory and age-related pathologies in mice with ectopic expression of human PARP-1. Mech. Ageing Dev. 2010, 131, 389–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS ONE 2012, 7, e42357. [Google Scholar] [CrossRef]
- Kam, T.I.; Mao, X.; Park, H.; Chou, S.C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [Green Version]
- Feng, F.Y.; de Bono, J.S.; Rubin, M.A.; Knudsen, K.E. Chromatin to Clinic: The Molecular Rationale for PARP1 Inhibitor Function. Mol. Cell 2015, 58, 925–934. [Google Scholar] [CrossRef] [Green Version]
- Fouquerel, E.; Goellner, E.M.; Yu, Z.; Gagne, J.P.; Barbi de Moura, M.; Feinstein, T.; Wheeler, D.; Redpath, P.; Li, J.; Romero, G.; et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 2014, 8, 1819–1831. [Google Scholar] [CrossRef] [Green Version]
- Andrabi, S.A.; Umanah, G.K.; Chang, C.; Stevens, D.A.; Karuppagounder, S.S.; Gagne, J.P.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 10209–10214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, M.M.; Kong, X.; Moncada, E.; Chen, Y.; Imamura, H.; Wang, P.; Berns, M.W.; Yokomori, K.; Digman, M.A. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell 2019, 30, 2584–2597. [Google Scholar] [CrossRef] [PubMed]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [Green Version]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef] [Green Version]
- Robaszkiewicz, A.; Qu, C.; Wisnik, E.; Ploszaj, T.; Mirsaidi, A.; Kunze, F.A.; Richards, P.J.; Cinelli, P.; Mbalaviele, G.; Hottiger, M.O. ARTD1 regulates osteoclastogenesis and bone homeostasis by dampening NF-kappaB-dependent transcription of IL-1beta. Sci. Rep. 2016, 6, 21131. [Google Scholar] [CrossRef] [Green Version]
- Robaszkiewicz, A.; Valko, Z.; Kovacs, K.; Hegedus, C.; Bakondi, E.; Bai, P.; Virag, L. The role of p38 signaling and poly(ADP-ribosyl)ation-induced metabolic collapse in the osteogenic differentiation-coupled cell death pathway. Free Radic. Biol. Med. 2014, 76, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, K.H.; Hayward, R.; Rajan, R.; Whitehead, M.; Cobb, A.M.; Ahmad, S.; Sun, M.; Goldberga, I.; Li, R.; Bashtanova, U.; et al. Poly(ADP-Ribose) Links the DNA Damage Response and Biomineralization. Cell Rep. 2019, 27, 3124–3138. [Google Scholar] [CrossRef] [Green Version]
- Duer, M.; Cobb, A.M.; Shanahan, C.M. DNA Damage Response: A Molecular Lynchpin in the Pathobiology of Arteriosclerotic Calcification. Arterioscler. Thromb. Vasc. Biol. 2020, 40, e193–e202. [Google Scholar] [CrossRef]
- Zengin, A.; Jarjou, L.M.; Janha, R.E.; Prentice, A.; Cooper, C.; Ebeling, P.R.; Ward, K.A. Sex-Specific Associations Between Cardiac Workload, Peripheral Vascular Calcification, and Bone Mineral Density: The Gambian Bone and Muscle Aging Study. J. Bone Miner. Res. 2020. [Google Scholar] [CrossRef]
- Nagy, E.; Caidahl, K.; Franco-Cereceda, A.; Back, M. Increased transcript level of poly(ADP-ribose) polymerase (PARP-1) in human tricuspid compared with bicuspid aortic valves correlates with the stenosis severity. BioChem. Biophys Res. Commun. 2012, 420, 671–675. [Google Scholar] [CrossRef]
- Wang, C.; Xu, W.; An, J.; Liang, M.; Li, Y.; Zhang, F.; Tong, Q.; Huang, K. Poly(ADP-ribose) polymerase 1 accelerates vascular calcification by upregulating Runx2. Nat. Commun. 2019, 10, 1203. [Google Scholar] [CrossRef] [Green Version]
- Forteo approved for osteoporosis treatment. FDA Consum. 2003, 37, 4.
- Deal, C.; Gideon, J. Recombinant human PTH 1-34 (Forteo): An anabolic drug for osteoporosis. Cleve Clin. J. Med. 2003, 70, 585–586, 589–590, 592–594 passim. [Google Scholar] [CrossRef]
- Hutton, S.F. Forteo (teriparatide): First approved medication to rebuild bone. S. D. J. Med. 2003, 56, 423–424. [Google Scholar] [PubMed]
- Vahle, J.L.; Long, G.G.; Sandusky, G.; Westmore, M.; Ma, Y.L.; Sato, M. Bone neoplasms in F344 rats given teriparatide [rhPTH(1-34)] are dependent on duration of treatment and dose. Toxicol. Pathol. 2004, 32, 426–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilsenan, A.; Midkiff, K.; Harris, D.; McQuay, L.; Hunter, S.; Kellier-Steele, N.; Andrews, E. Assessing the incidence of osteosarcoma among teriparatide users based on Medicare Part D and US State Cancer Registry Data. Pharmacoepidemiol. Drug Saf. 2020, 29, 1616–1626. [Google Scholar] [CrossRef]
- Schnoke, M.; Midura, S.B.; Midura, R.J. Parathyroid hormone suppresses osteoblast apoptosis by augmenting DNA repair. Bone 2009, 45, 590–602. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Zheng, L.; Fan, Y.; Zhang, J.; Xu, R.; Xie, J.; Zhou, X. Parathyroid hormone ameliorates temporomandibular joint osteoarthritic-like changes related to age. Cell Prolif. 2020, 53, e12755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardawi, M.S.; Rouzi, A.A.; Al-Sibiani, S.A.; Al-Senani, N.S.; Qari, M.H.; Mousa, S.A. High serum sclerostin predicts the occurrence of osteoporotic fractures in postmenopausal women: The Center of Excellence for Osteoporosis Research Study. J. Bone Miner. Res. 2012, 27, 2592–2602. [Google Scholar] [CrossRef] [PubMed]
- Ardawi, M.S.; Al-Kadi, H.A.; Rouzi, A.A.; Qari, M.H. Determinants of serum sclerostin in healthy pre- and postmenopausal women. J. Bone Miner. Res. 2011, 26, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Piemonte, S.; Romagnoli, E.; Bratengeier, C.; Woloszczuk, W.; Tancredi, A.; Pepe, J.; Cipriani, C.; Minisola, S. Serum sclerostin levels decline in post-menopausal women with osteoporosis following treatment with intermittent parathyroid hormone. J. Endocrinol. Investig. 2012, 35, 866–868. [Google Scholar]
- Modder, U.I.; Hoey, K.A.; Amin, S.; McCready, L.K.; Achenbach, S.J.; Riggs, B.L.; Melton, L.J., 3rd; Khosla, S. Relation of age, gender, and bone mass to circulating sclerostin levels in women and men. J. Bone Miner. Res. 2011, 26, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Bhattoa, H.P.; Wamwaki, J.; Kalina, E.; Foldesi, R.; Balogh, A.; Antal-Szalmas, P. Serum sclerostin levels in healthy men over 50 years of age. J. Bone Miner. Metab. 2013, 31, 579–584. [Google Scholar] [CrossRef]
- Roforth, M.M.; Fujita, K.; McGregor, U.I.; Kirmani, S.; McCready, L.K.; Peterson, J.M.; Drake, M.T.; Monroe, D.G.; Khosla, S. Effects of age on bone mRNA levels of sclerostin and other genes relevant to bone metabolism in humans. Bone 2014, 59, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Dovjak, P.; Dorfer, S.; Foger-Samwald, U.; Kudlacek, S.; Marculescu, R.; Pietschmann, P. Serum levels of sclerostin and dickkopf-1: Effects of age, gender and fracture status. Gerontology 2014, 60, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Reppe, S.; Noer, A.; Grimholt, R.M.; Halldorsson, B.V.; Medina-Gomez, C.; Gautvik, V.T.; Olstad, O.K.; Berg, J.P.; Datta, H.; Estrada, K.; et al. Methylation of bone SOST, its mRNA, and serum sclerostin levels correlate strongly with fracture risk in postmenopausal women. J. Bone Miner. Res. 2015, 30, 249–256. [Google Scholar] [CrossRef]
- Gombos, G.C.; Bajsz, V.; Pek, E.; Schmidt, B.; Sio, E.; Molics, B.; Betlehem, J. Direct effects of physical training on markers of bone metabolism and serum sclerostin concentrations in older adults with low bone mass. BMC Musculoskelet Disord. 2016, 17, 254. [Google Scholar] [CrossRef] [Green Version]
- Janik, M.; Stuss, M.; Michalska-Kasiczak, M.; Jegier, A.; Sewerynek, E. Effects of physical activity on sclerostin concentrations. Endokrynol. Pol. 2018, 69, 142–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ota, K.; Quint, P.; Ruan, M.; Pederson, L.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Sclerostin is expressed in osteoclasts from aged mice and reduces osteoclast-mediated stimulation of mineralization. J. Cell BioChem. 2013, 114, 1901–1907. [Google Scholar] [CrossRef] [Green Version]
- Balemans, W.; Ebeling, M.; Patel, N.; Van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; Van Den Ende, J.; Willems, P.; et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balemans, W.; Van Den Ende, J.; Freire Paes-Alves, A.; Dikkers, F.G.; Willems, P.J.; Vanhoenacker, F.; de Almeida-Melo, N.; Alves, C.F.; Stratakis, C.A.; Hill, S.C.; et al. Localization of the gene for sclerosteosis to the van Buchem disease-gene region on chromosome 17q12-q21. Am. J. Hum. Genet. 1999, 64, 1661–1669. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ominsky, M.S.; Niu, Q.T.; Sun, N.; Daugherty, B.; D’Agostin, D.; Kurahara, C.; Gao, Y.; Cao, J.; Gong, J.; et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 2008, 23, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ominsky, M.S.; Warmington, K.S.; Morony, S.; Gong, J.; Cao, J.; Gao, Y.; Shalhoub, V.; Tipton, B.; Haldankar, R.; et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J. Bone Miner. Res. 2009, 24, 578–588. [Google Scholar] [CrossRef] [PubMed]
- McClung, M.R.; Grauer, A.; Boonen, S.; Bolognese, M.A.; Brown, J.P.; Diez-Perez, A.; Langdahl, B.L.; Reginster, J.Y.; Zanchetta, J.R.; Wasserman, S.M.; et al. Romosozumab in postmenopausal women with low bone mineral density. N. Engl. J. Med. 2014, 370, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langdahl, B.L.; Libanati, C.; Crittenden, D.B.; Bolognese, M.A.; Brown, J.P.; Daizadeh, N.S.; Dokoupilova, E.; Engelke, K.; Finkelstein, J.S.; Genant, H.K.; et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: A randomised, open-label, phase 3 trial. Lancet 2017, 390, 1585–1594. [Google Scholar] [CrossRef]
- Chouinard, L.; Felx, M.; Mellal, N.; Varela, A.; Mann, P.; Jolette, J.; Samadfam, R.; Smith, S.Y.; Locher, K.; Buntich, S.; et al. Carcinogenicity risk assessment of romosozumab: A review of scientific weight-of-evidence and findings in a rat lifetime pharmacology study. Regul. Toxicol. Pharmacol. 2016, 81, 212–222. [Google Scholar] [CrossRef] [Green Version]
- McClung, M.R.; Brown, J.P.; Diez-Perez, A.; Resch, H.; Caminis, J.; Meisner, P.; Bolognese, M.A.; Goemaere, S.; Bone, H.G.; Zanchetta, J.R.; et al. Effects of 24 Months of Treatment With Romosozumab Followed by 12 Months of Denosumab or Placebo in Postmenopausal Women With Low Bone Mineral Density: A Randomized, Double-Blind, Phase 2, Parallel Group Study. J. Bone Miner. Res. 2018, 33, 1397–1406. [Google Scholar] [CrossRef]
- McClung, M.R.; Bolognese, M.A.; Brown, J.P.; Reginster, J.Y.; Langdahl, B.L.; Maddox, J.; Shi, Y.; Rojeski, M.; Meisner, P.D.; Grauer, A. A single dose of zoledronate preserves bone mineral density for up to 2 years after a second course of romosozumab. Osteoporos. Int. 2020, 31, 2231–2241. [Google Scholar] [CrossRef]
- Van Beek, E.; Lowik, C.; van der Pluijm, G.; Papapoulos, S. The role of geranylgeranylation in bone resorption and its suppression by bisphosphonates in fetal bone explants in vitro: A clue to the mechanism of action of nitrogen-containing bisphosphonates. J. Bone Miner. Res. 1999, 14, 722–729. [Google Scholar] [CrossRef]
- Kimmel, D.B. Mechanism of action, pharmacokinetic and pharmacodynamic profile, and clinical applications of nitrogen-containing bisphosphonates. J. Dent. Res. 2007, 86, 1022–1033. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Sun, K.; Sun, Y.; Li, H.; Han, D.; Bai, Y.; Zhao, R.; Guo, Z. Anti-Ageing Effect of Physalis alkekengi Ethyl Acetate Layer on a d-galactose-Induced Mouse Model through the Reduction of Cellular Senescence and Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Chang, J.; Liu, X.; Zhang, X.; Zhang, S.; Zhang, X.; Zhou, D.; Zheng, G. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging 2016, 8, 2915–2926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherif, H.; Bisson, D.G.; Mannarino, M.; Rabau, O.; Ouellet, J.A.; Haglund, L. Senotherapeutic drugs for human intervertebral disc degeneration and low back pain. Elife 2020, 9, e54693. [Google Scholar] [CrossRef]
- Ritschka, B.; Knauer-Meyer, T.; Goncalves, D.S.; Mas, A.; Plassat, J.L.; Durik, M.; Jacobs, H.; Pedone, E.; Di Vicino, U.; Cosma, M.P.; et al. The senotherapeutic drug ABT-737 disrupts aberrant p21 expression to restore liver regeneration in adult mice. Genes Dev. 2020, 34, 489–494. [Google Scholar] [CrossRef]
- Yoshida, S.; Nakagami, H.; Hayashi, H.; Ikeda, Y.; Sun, J.; Tenma, A.; Tomioka, H.; Kawano, T.; Shimamura, M.; Morishita, R.; et al. The CD153 vaccine is a senotherapeutic option for preventing the accumulation of senescent T cells in mice. Nat. Commun. 2020, 11, 2482. [Google Scholar] [CrossRef]
- Bode-Boger, S.M.; Martens-Lobenhoffer, J.; Tager, M.; Schroder, H.; Scalera, F. Aspirin reduces endothelial cell senescence. BioChem. Biophys. Res. Commun. 2005, 334, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Kim, J.; Field, K.; Reid, C.; Chatzistamou, I.; Shim, M. Aspirin ameliorates the long-term adverse effects of doxorubicin through suppression of cellular senescence. FASEB BioAdv. 2019, 1, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Jurk, D.; Khosla, S.; Kirkland, J.L.; LeBrasseur, N.K.; Miller, J.D.; Passos, J.F.; Pignolo, R.J.; Tchkonia, T.; Niedernhofer, L.J. Senolytic Drugs: Reducing Senescent Cell Viability to Extend Health Span. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 779–803. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Schaum, N.; Alimirah, F.; Lopez-Dominguez, J.A.; Orjalo, A.V.; Scott, G.; Desprez, P.Y.; Benz, C.; Davalos, A.R.; Campisi, J. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci. Rep. 2018, 8, 2410. [Google Scholar] [CrossRef]
- Houssaini, A.; Breau, M.; Kebe, K.; Abid, S.; Marcos, E.; Lipskaia, L.; Rideau, D.; Parpaleix, A.; Huang, J.; Amsellem, V.; et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight 2018, 3, e93203. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef]
- Lai, P.; Song, Q.; Yang, C.; Li, Z.; Liu, S.; Liu, B.; Li, M.; Deng, H.; Cai, D.; Jin, D.; et al. Loss of Rictor with aging in osteoblasts promotes age-related bone loss. Cell Death Dis. 2016, 7, e2408. [Google Scholar] [CrossRef]
- Ho, L.; Wang, L.; Roth, T.M.; Pan, Y.; Verdin, E.M.; Hsiao, E.C.; Nissenson, R.A. Sirtuin-3 Promotes Adipogenesis, Osteoclastogenesis, and Bone Loss in Aging Male Mice. Endocrinology 2017, 158, 2741–2753. [Google Scholar] [CrossRef]
- An, J.Y.; Quarles, E.K.; Mekvanich, S.; Kang, A.; Liu, A.; Santos, D.; Miller, R.A.; Rabinovitch, P.S.; Cox, T.C.; Kaeberlein, M. Rapamycin treatment attenuates age-associated periodontitis in mice. Geroscience 2017, 39, 457–463. [Google Scholar] [CrossRef] [Green Version]
- Mao, K.; Chen, J.; Yu, H.; Li, H.; Ren, Y.; Wu, X.; Wen, Y.; Zou, F.; Li, W. Poly (ADP-ribose) polymerase 1 inhibition prevents neurodegeneration and promotes alpha-synuclein degradation via transcription factor EB-dependent autophagy in mutant alpha-synucleinA53T model of Parkinson’s disease. Aging Cell 2020, 19, e13163. [Google Scholar] [CrossRef]
- Mohamed, J.S.; Wilson, J.C.; Myers, M.J.; Sisson, K.J.; Alway, S.E. Dysregulation of SIRT-1 in aging mice increases skeletal muscle fatigue by a PARP-1-dependent mechanism. Aging 2014, 6, 820–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.K.; Roberts, R.L.; Benson, R.D., Jr.; Pierce, J.L.; Yu, K.; Hamrick, M.W.; McGee-Lawrence, M.E. The Senolytic Drug Navitoclax (ABT-263) Causes Trabecular Bone Loss and Impaired Osteoprogenitor Function in Aged Mice. Front. Cell Dev. Biol. 2020, 8, 354. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.W.; Son, Y.J.; Yun, J.M.; Kim, S.H. Fisetin Inhibits Osteoclast Differentiation via Downregulation of p38 and c-Fos-NFATc1 Signaling Pathways. Evid. Based Complement. Alternat. Med. 2012, 2012, 810563. [Google Scholar] [CrossRef] [PubMed]
- Leotoing, L.; Wauquier, F.; Guicheux, J.; Miot-Noirault, E.; Wittrant, Y.; Coxam, V. The polyphenol fisetin protects bone by repressing NF-kappaB and MKP-1-dependent signaling pathways in osteoclasts. PLoS ONE 2013, 8, e68388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chandra, A.; Rajawat, J. Skeletal Aging and Osteoporosis: Mechanisms and Therapeutics. Int. J. Mol. Sci. 2021, 22, 3553. https://doi.org/10.3390/ijms22073553
Chandra A, Rajawat J. Skeletal Aging and Osteoporosis: Mechanisms and Therapeutics. International Journal of Molecular Sciences. 2021; 22(7):3553. https://doi.org/10.3390/ijms22073553
Chicago/Turabian StyleChandra, Abhishek, and Jyotika Rajawat. 2021. "Skeletal Aging and Osteoporosis: Mechanisms and Therapeutics" International Journal of Molecular Sciences 22, no. 7: 3553. https://doi.org/10.3390/ijms22073553
APA StyleChandra, A., & Rajawat, J. (2021). Skeletal Aging and Osteoporosis: Mechanisms and Therapeutics. International Journal of Molecular Sciences, 22(7), 3553. https://doi.org/10.3390/ijms22073553