
Chronically Elevated Exogenous Glucose Elicits Antipodal Effects on the Proteome Signature of Differentiating Human iPSC-Derived Pancreatic Progenitors

 ,
,  , ,
, ,  , ,
, , 
Abstract
:1. Introduction
2. Results
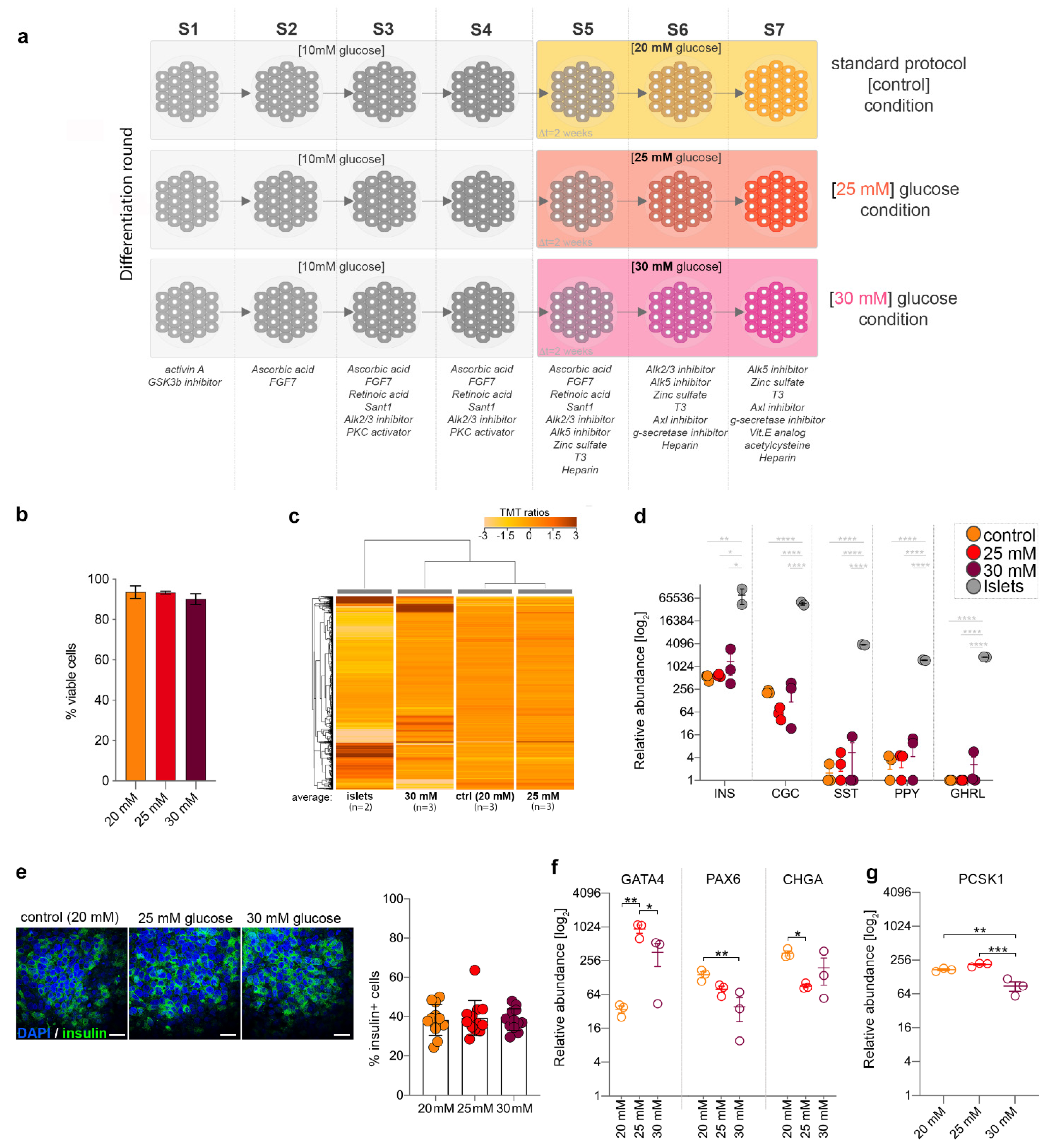
2.1. High Glucose Concentrations Dysregulate Key Factors with Role in Pancreatic Islet Cells Development
2.2. Mildly Increased Glucose Levels Impact the Growth and Developmental Profile of the In Vitro Differentiating Cells
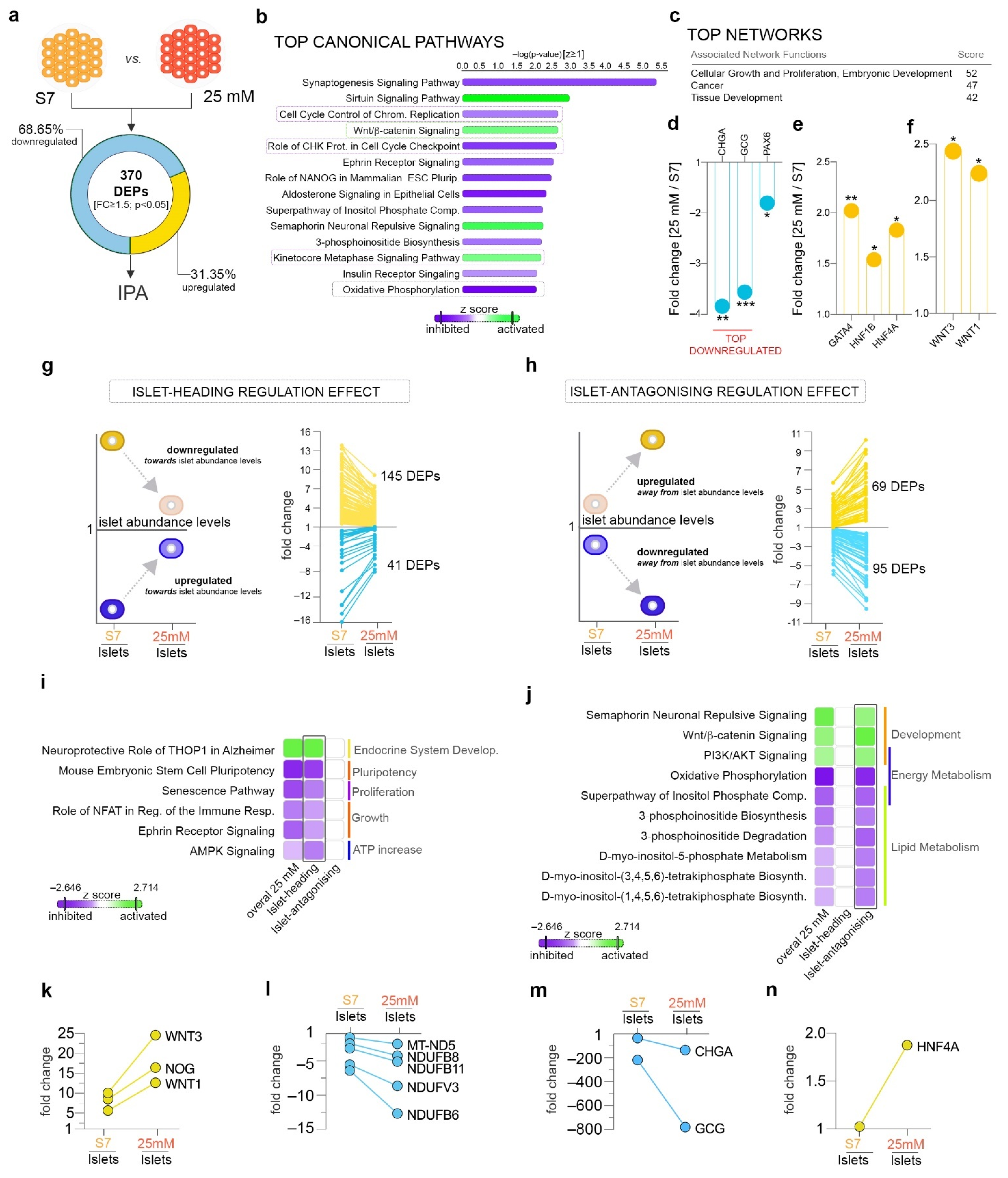
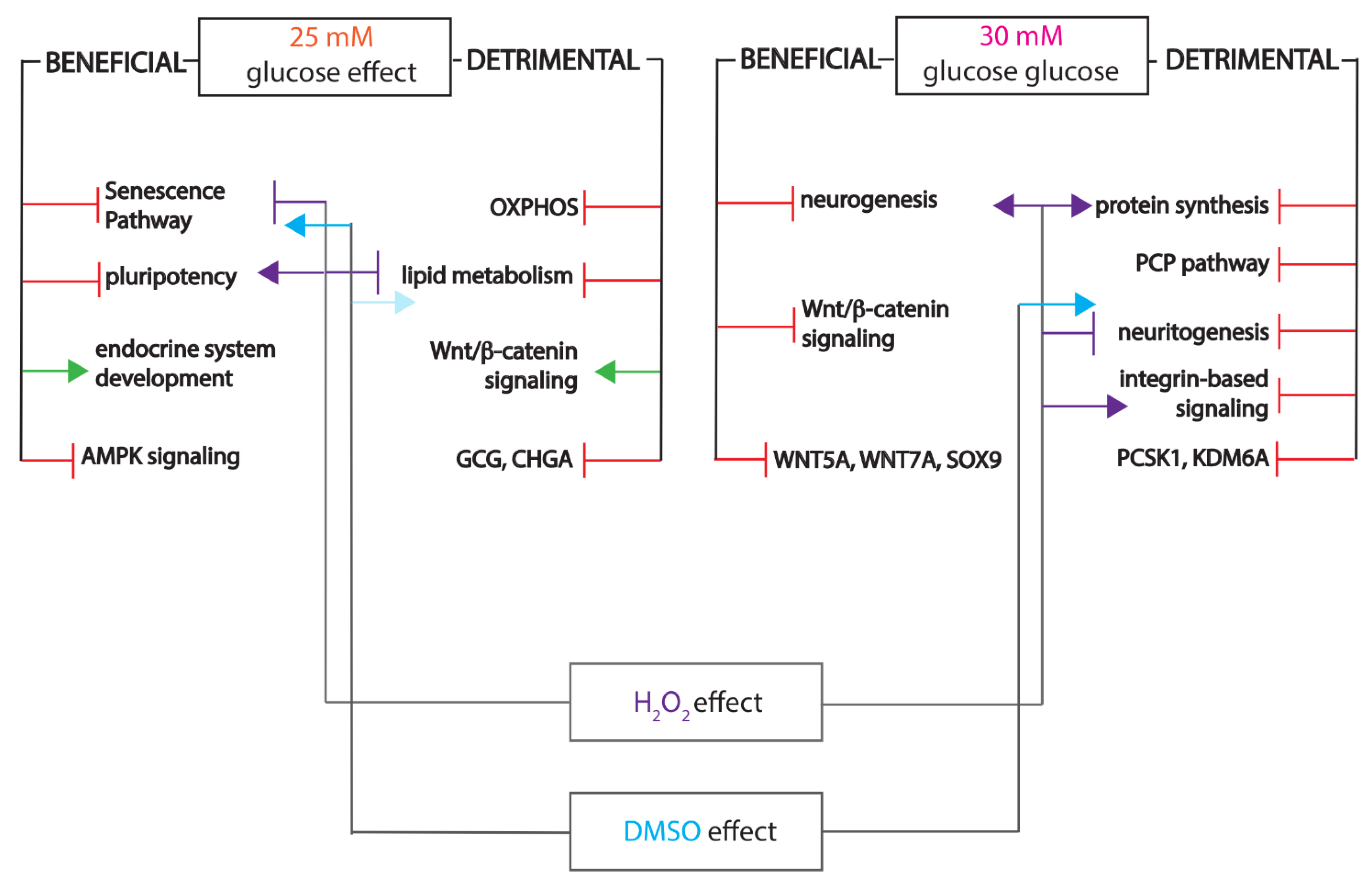
2.3. Mildly Elevated Glucose Level Elicits both Beneficial and Detrimental Effects on the Islet Cell Signature of the Differentiating Cells
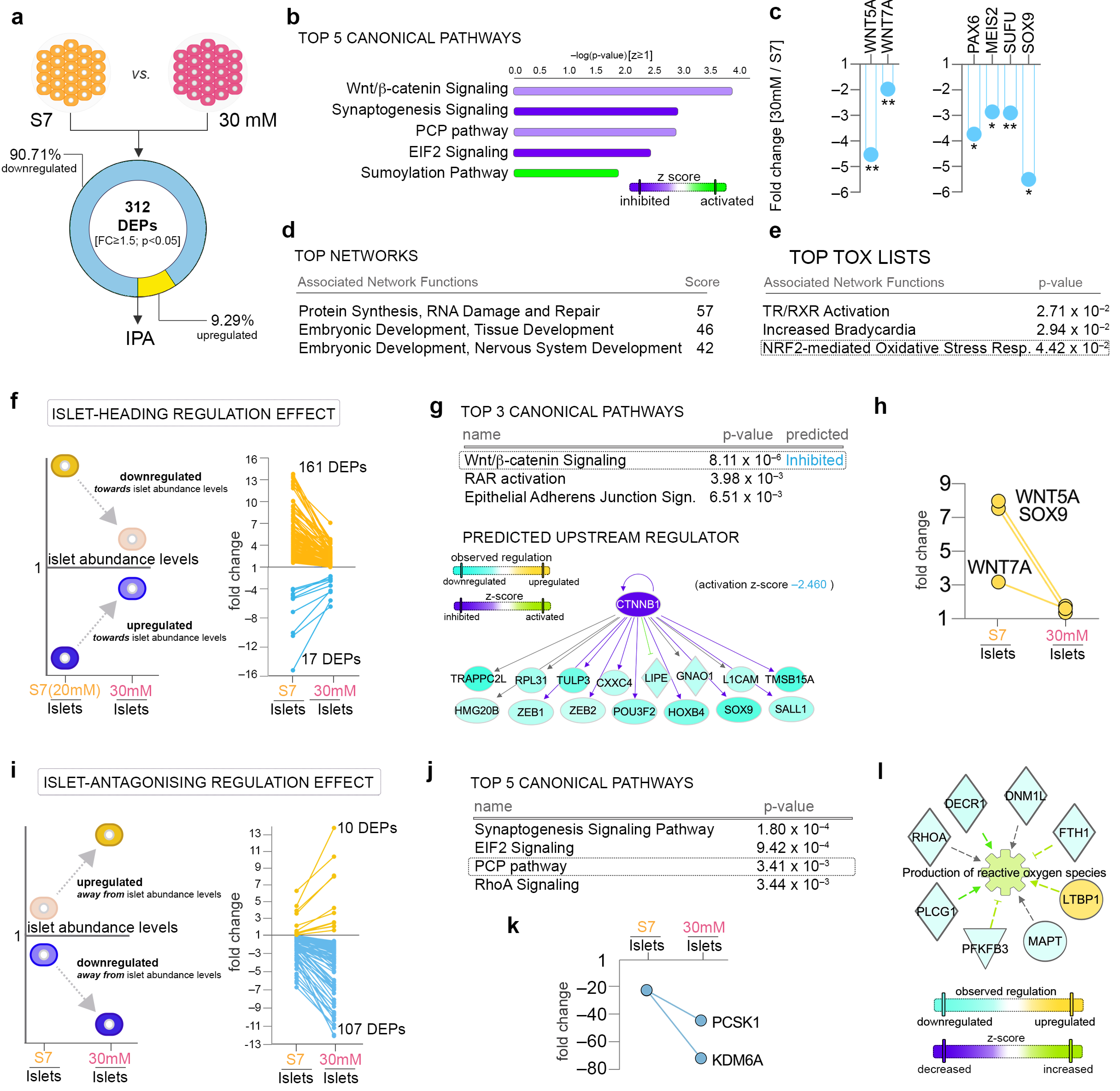
2.4. Pathway Analysis Reveals a Largely Different Protein Regulation in Response to Highly Increased Glucose Levels
2.5. Highly Increased Glucose Level Modulates the Canonical Wnt Signaling towards Native Islet Regulation, while Negatively Impacting Energy Metabolism and Protein Synthesis
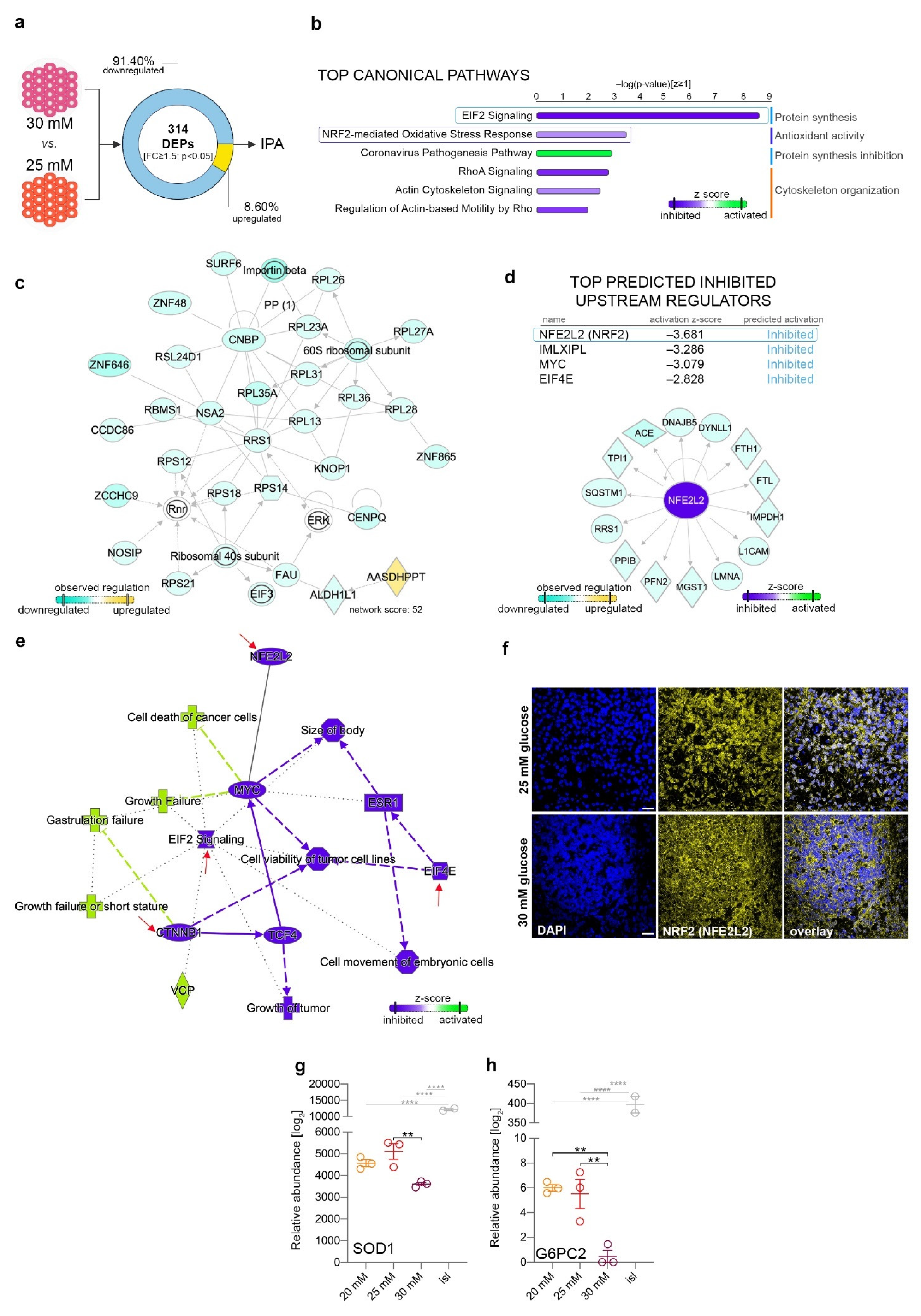
2.6. The Differential Proteome Landscape between Mildly and Highly Increased Glucose Is Characterized by Reduced Protein Synthesis and Low Antioxidant Activity
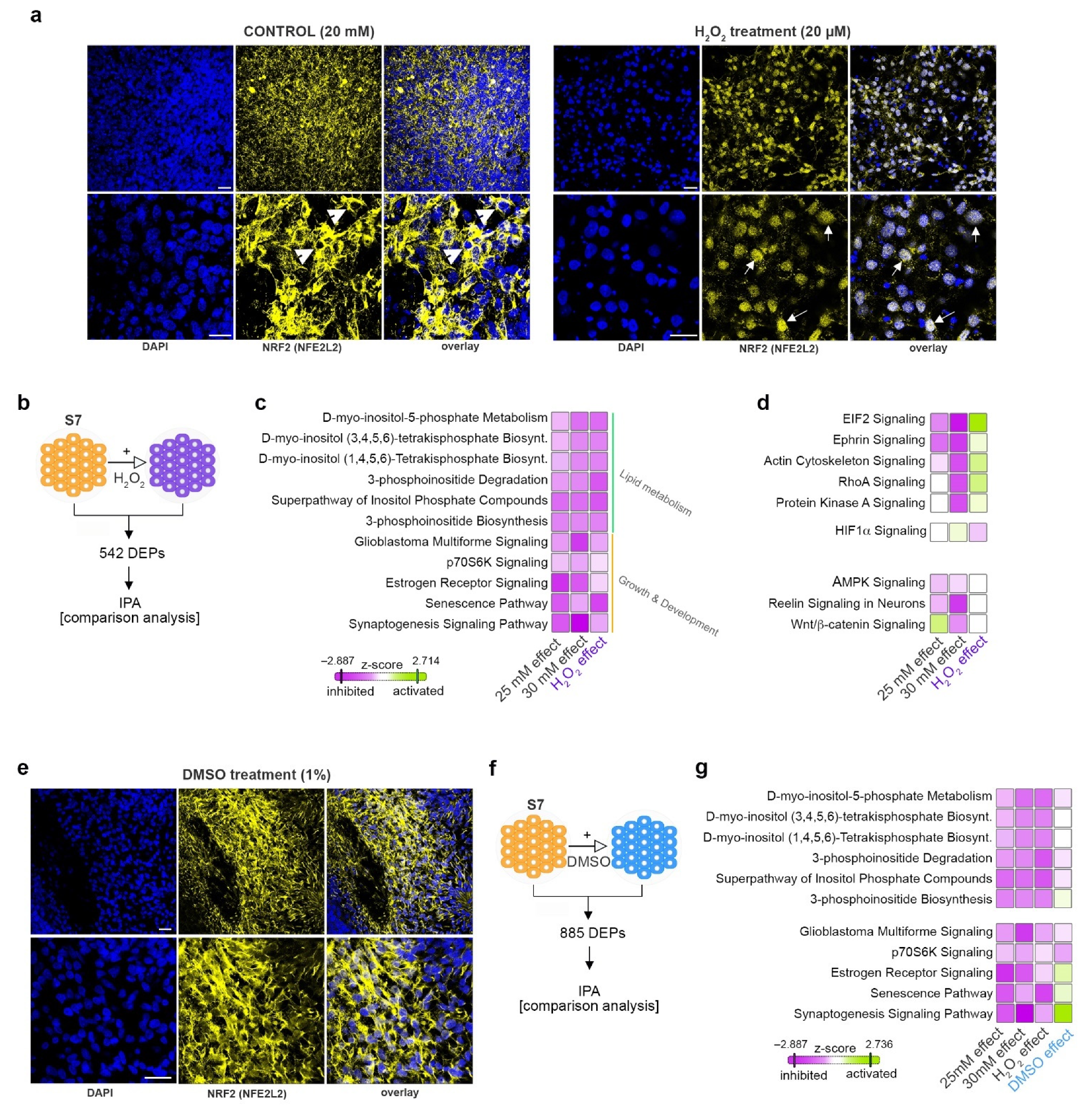
2.7. Elevated Glucose Concentrations and Redox Imbalance Prompt Similar but not Overlapping Responses
3. Discussion
4. Materials and Methods
4.1. Cell Sources and Ethical Statements
4.2. Cell Differentiation
4.3. Cell Counting and Viability Measurements
4.4. H2O2 and DMSO Treatments
4.5. Immunofluorescence Staining
4.6. Global Proteomics Analysis
4.7. Proteomic Data Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cigliola, V.; Ghila, L.; Chera, S.; Herrera, P.L. Tissue repair brakes: A common paradigm in the biology of regeneration. Stem Cells 2020, 38, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; York, N.W.; Nichols, C.G.; Remedi, M.S. Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014, 19, 872–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spijker, H.S.; Song, H.; Ellenbroek, J.H.; Roefs, M.M.; Engelse, M.A.; Bos, E.; Koster, A.J.; Rabelink, T.J.; Hansen, B.C.; Clark, A.; et al. Loss of β-Cell Identity Occurs in Type 2 Diabetes and Is Associated With Islet Amyloid Deposits. Diabetes 2015, 64, 2928–2938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spijker, H.S.; Ravelli, R.B.; Mommaas-Kienhuis, A.M.; van Apeldoorn, A.A.; Engelse, M.A.; Zaldumbide, A.; Bonner-Weir, S.; Rabelink, T.J.; Hoeben, R.C.; Clevers, H.; et al. Conversion of mature human beta-cells into glucagon-producing alpha-cells. Diabetes 2013, 62, 2471–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collombat, P.; Hecksher-Sørensen, J.; Krull, J.; Berger, J.; Riedel, D.; Herrera, P.L.; Serup, P.; Mansouri, A. Embryonic endocrine pancreas and mature beta cells acquire alpha and PP cell phenotypes upon Arx misexpression. J. Clin. Investig. 2007, 117, 961–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.; Zhang, T.; et al. Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metab. 2014, 19, 259–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulsen, R.C.; Knowles, H.J.; Carr, A.J.; Hulley, P.A. Cell differentiation versus cell death: Extracellular glucose is a key determinant of cell fate following oxidative stress exposure. Cell Death Dis. 2014, 5, e1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, S.; Inoguchi, T. The role of oxidative stress in the pathogenesis of diabetic vascular complications. Diabetes Metab. J. 2012, 36, 255–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Deng, R.; Yang, X.; Xu, W.; Liu, Y.; Li, F.; Zhang, J.; Tang, H.; Ji, X.; Bi, Y.; et al. Glucose potentiates beta-cell function by inducing Tph1 expression in rat islets. FASEB J. 2017, 31, 5342–5355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebelato, E.; Santos, L.R.; Carpinelli, A.R.; Rorsman, P.; Abdulkader, F. Short-term high glucose culture potentiates pancreatic beta cell function. Sci. Rep. 2018, 8, 13061. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Jonas, J.C.; Laybutt, D.R. Mechanisms of beta-cell dedifferentiation in diabetes: Recent findings and future research directions. J. Endocrinol. 2018, 236, R109–R143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, G.C.; Aguayo-Mazzucato, C.; Bonner-Weir, S. beta-cell dedifferentiation in diabetes is important, but what is it? Islets 2013, 5, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bensellam, M.; Laybutt, D.R.; Jonas, J.C. The molecular mechanisms of pancreatic beta-cell glucotoxicity: Recent findings and future research directions. Mol. Cell Endocrinol. 2012, 364, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Tsunekawa, S.; Ikeniwa, M.; Izumoto, T.; Iida, A.; Ogata, H.; Uenishi, E.; Seino, Y.; Ozaki, N.; Sugimura, Y.; et al. Long-term pancreatic beta cell exposure to high levels of glucose but not palmitate induces DNA methylation within the insulin gene promoter and represses transcriptional activity. PLoS ONE 2015, 10, e0115350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottosson-Laakso, E.; Krus, U.; Storm, P.; Prasad, R.B.; Oskolkov, N.; Ahlqvist, E.; Fadista, J.; Hansson, O.; Groop, L.; Vikman, P. Glucose-Induced Changes in Gene Expression in Human Pancreatic Islets: Causes or Consequences of Chronic Hyperglycemia. Diabetes 2017, 66, 3013–3028. [Google Scholar] [CrossRef] [Green Version]
- Hall, E.; Dekker Nitert, M.; Volkov, P.; Malmgren, S.; Mulder, H.; Bacos, K.; Ling, C. The effects of high glucose exposure on global gene expression and DNA methylation in human pancreatic islets. Mol. Cell Endocrinol. 2018, 472, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, C.; Anazawa, T.; Cross, S.E.; Labriola, L.; Meier, R.P.H.; Redfield, R.R.; Scholz, H.; Stock, P.G.; Zammit, N.W.; Committee, I.Y.Y.I. β Cell Replacement Therapy: The Next 10 Years. Transplantation 2018, 102, 215–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, G.G.; Tzanakakis, E.S.; Hebrok, M. Emerging routes to the generation of functional beta-cells for diabetes mellitus cell therapy. Nat. Rev. Endocrinol. 2020, 16, 506–518. [Google Scholar] [CrossRef]
- Castro-Gutierrez, R.; Michels, A.W.; Russ, H.A. β Cell replacement: Improving on the design. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 251–257. [Google Scholar] [CrossRef]
- Chera, S.; Herrera, P.L. Regeneration of pancreatic insulin-producing cells by in situ adaptive cell conversion. Curr. Opin. Genet. Dev. 2016, 40, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balboa, D.; Saarimaki-Vire, J.; Otonkoski, T. Concise Review: Human Pluripotent Stem Cells for the Modeling of Pancreatic beta-Cell Pathology. Stem Cells 2019, 37, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, G.G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.L.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived beta cells. Nat. Cell Biol. 2019, 21, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Millman, J.R.; Gurtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, S.; Manenti, F.; Chimienti, R.; Nano, R.; Ottoboni, L.; Ruffini, F.; Martino, G.; Ravassard, P.; Piemonti, L.; Sordi, V. Differentiation of Sendai Virus-Reprogrammed iPSC into beta Cells, Compared with Human Pancreatic Islets and Immortalized beta Cell Line. Cell Transplant. 2018, 27, 1548–1560. [Google Scholar] [CrossRef]
- Zhang, D.; Jiang, W.; Liu, M.; Sui, X.; Yin, X.; Chen, S.; Shi, Y.; Deng, H. Highly efficient differentiation of human ES cells and iPS cells into mature pancreatic insulin-producing cells. Cell Res. 2009, 19, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Lithovius, V.; Saarimaki-Vire, J.; Balboa, D.; Ibrahim, H.; Montaser, H.; Barsby, T.; Otonkoski, T. SUR1-mutant iPS cell-derived islets recapitulate the pathophysiology of congenital hyperinsulinism. Diabetologia 2021. [Google Scholar] [CrossRef] [PubMed]
- Legoy, T.A.; Vethe, H.; Abadpour, S.; Strand, B.L.; Scholz, H.; Paulo, J.A.; Raeder, H.; Ghila, L.; Chera, S. Encapsulation boosts islet-cell signature in differentiating human induced pluripotent stem cells via integrin signalling. Sci. Rep. 2020, 10, 414. [Google Scholar] [CrossRef] [Green Version]
- Legoy, T.A.; Mathisen, A.F.; Salim, Z.; Vethe, H.; Bjorlykke, Y.; Abadpour, S.; Paulo, J.A.; Scholz, H.; Raeder, H.; Ghila, L.; et al. In vivo Environment Swiftly Restricts Human Pancreatic Progenitors Toward Mono-Hormonal Identity via a HNF1A/HNF4A Mechanism. Front. Cell Dev. Biol. 2020, 8, 109. [Google Scholar] [CrossRef]
- Legoy, T.A.; Ghila, L.; Vethe, H.; Abadpour, S.; Mathisen, A.F.; Paulo, J.A.; Scholz, H.; Raeder, H.; Chera, S. In vivo hyperglycaemia exposure elicits distinct period-dependent effects on human pancreatic progenitor differentiation, conveyed by oxidative stress. Acta Physiol. 2020, 228, e13433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vethe, H.; Bjørlykke, Y.; Ghila, L.M.; Paulo, J.A.; Scholz, H.; Gygi, S.P.; Chera, S.; Ræder, H. Probing the missing mature β-cell proteomic landscape in differentiating patient iPSC-derived cells. Sci. Rep. 2017, 7, 4780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghila, L.; Bjorlykke, Y.; Legoy, T.A.; Vethe, H.; Furuyama, K.; Chera, S.; Raeder, H. Bioinformatic Analyses of miRNA-mRNA Signature during hiPSC Differentiation towards Insulin-Producing Cells upon HNF4alpha Mutation. Biomedicines 2020, 8, 179. [Google Scholar] [CrossRef] [PubMed]
- Regazzi, R.; Wollheim, C.B.; Lang, J.; Theler, J.M.; Rossetto, O.; Montecucco, C.; Sadoul, K.; Weller, U.; Palmer, M.; Thorens, B. VAMP-2 and cellubrevin are expressed in pancreatic beta-cells and are essential for Ca(2+)-but not for GTP gamma S-induced insulin secretion. EMBO J. 1995, 14, 2723–2730. [Google Scholar] [CrossRef]
- Zhong, W.; Li, Z.; Zhou, M.; Xu, T.; Wang, Y. DDX1 regulates alternative splicing and insulin secretion in pancreatic beta cells. Biochem. Biophys. Res. Commun. 2018, 500, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhu, D.; Feng, C.; Chen, G.; Mao, X.; Wang, Q.; Wang, J.; Liu, C. Inhibition of peptidyl-prolyl cis-trans isomerase B mediates cyclosporin A-induced apoptosis of islet beta cells. Exp. Ther. Med. 2018, 16, 3959–3964. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Korbutt, G.S.; Hellerstrom, C. Prolonged exposure of human pancreatic islets to high glucose concentrations in vitro impairs the beta-cell function. J. Clin. Investig. 1992, 90, 1263–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glynn, E.; Thompson, B.; Vadrevu, S.; Lu, S.; Kennedy, R.T.; Ha, J.; Sherman, A.; Satin, L.S. Chronic Glucose Exposure Systematically Shifts the Oscillatory Threshold of Mouse Islets: Experimental Evidence for an Early Intrinsic Mechanism of Compensation for Hyperglycemia. Endocrinology 2016, 157, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Olofsson, C.S.; Collins, S.; Bengtsson, M.; Eliasson, L.; Salehi, A.; Shimomura, K.; Tarasov, A.; Holm, C.; Ashcroft, F.; Rorsman, P. Long-term exposure to glucose and lipids inhibits glucose-induced insulin secretion downstream of granule fusion with plasma membrane. Diabetes 2007, 56, 1888–1897. [Google Scholar] [CrossRef] [Green Version]
- Khaldi, M.Z.; Guiot, Y.; Gilon, P.; Henquin, J.C.; Jonas, J.C. Increased glucose sensitivity of both triggering and amplifying pathways of insulin secretion in rat islets cultured for 1 wk in high glucose. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E207–E217. [Google Scholar] [CrossRef] [PubMed]
- Scheibner, K.; Bakhti, M.; Bastidas-Ponce, A.; Lickert, H. Wnt signaling: Implications in endoderm development and pancreas organogenesis. Curr. Opin. Cell Biol. 2019, 61, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Sharon, N.; Vanderhooft, J.; Straubhaar, J.; Mueller, J.; Chawla, R.; Zhou, Q.; Engquist, E.N.; Trapnell, C.; Gifford, D.K.; Melton, D.A. Wnt Signaling Separates the Progenitor and Endocrine Compartments during Pancreas Development. Cell Rep. 2019, 27, 2281–2291.e2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, E.; Migliorini, A.; Gegg, M.; Moruzzi, N.; Gerdes, J.; Roscioni, S.S.; Bakhti, M.; Brandl, E.; Irmler, M.; Beckers, J.; et al. Identification of proliferative and mature β-cells in the islets of Langerhans. Nature 2016, 535, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Rains, J.L.; Jain, S.K. Oxidative stress, insulin signaling, and diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Bjørlykke, Y.; Søviknes, A.M.; Hoareau, L.; Vethe, H.; Mathisen, A.F.; Chera, S.; Vaudel, M.; Ghila, L.; Ræder, H. Reprogrammed cells display distinct proteomic signatures associated with colony morphology variability. Stem Cells Int. 2019, 2019, 8036035. [Google Scholar] [CrossRef]
- Friberg, A.S.; Stahle, M.; Brandhorst, H.; Korsgren, O.; Brandhorst, D. Human islet separation utilizing a closed automated purification system. Cell Transplant. 2008, 17, 1305–1313. [Google Scholar] [CrossRef]
- Paulo, J.A.; Gygi, S.P. A comprehensive proteomic and phosphoproteomic analysis of yeast deletion mutants of 14-3-3 orthologs and associated effects of rapamycin. Proteomics 2015, 15, 474–486. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Islet Preparation | 1 | 2 | 3 |
|---|---|---|---|
| Mandatory Information | |||
| Donor age (years) | 60 | 35 | 57 |
| Donor sex (male: M/female: F) | M | M | F |
| Donor BMI (kg/m2) | 27.8 | 25.2 | 24.2 |
| Donor HbA1c or other measure of blood glucose control (mmol/mol) | 44 | not assessed | not assessed |
| Origin/source of islets | ECIT a | EDIT | ECIT |
| Islet isolation centre | Oslo | Oslo | Oslo |
| Donor history of diabetes? | No | No | No |
| Recommended Information | |||
| Donor cause of death | DBD b | DBD | DBD |
| Warm ischemia time (h) | 02:00 | 03:00 | 02:25 |
| Cold ischemia time (h) | 05:03 | 09:48 | 07:07 |
| Estimated purity (%) | 53 | 50 | 70 |
| Estimated viability (%) | 90 | 95 | 90 |
| Total culture time (h) c | 72 | 72 | 72 |
| Glucose-stimulated insulin secretion | 2.3 | 2.2 | 4.3 |
| Handpicked to purity? | Yes | Yes | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghila, L.; Legøy, T.A.; Mathisen, A.F.; Abadpour, S.; Paulo, J.A.; Scholz, H.; Ræder, H.; Chera, S. Chronically Elevated Exogenous Glucose Elicits Antipodal Effects on the Proteome Signature of Differentiating Human iPSC-Derived Pancreatic Progenitors. Int. J. Mol. Sci. 2021, 22, 3698. https://doi.org/10.3390/ijms22073698
Ghila L, Legøy TA, Mathisen AF, Abadpour S, Paulo JA, Scholz H, Ræder H, Chera S. Chronically Elevated Exogenous Glucose Elicits Antipodal Effects on the Proteome Signature of Differentiating Human iPSC-Derived Pancreatic Progenitors. International Journal of Molecular Sciences. 2021; 22(7):3698. https://doi.org/10.3390/ijms22073698
Chicago/Turabian StyleGhila, Luiza, Thomas Aga Legøy, Andreas Frøslev Mathisen, Shadab Abadpour, Joao A. Paulo, Hanne Scholz, Helge Ræder, and Simona Chera. 2021. "Chronically Elevated Exogenous Glucose Elicits Antipodal Effects on the Proteome Signature of Differentiating Human iPSC-Derived Pancreatic Progenitors" International Journal of Molecular Sciences 22, no. 7: 3698. https://doi.org/10.3390/ijms22073698
APA StyleGhila, L., Legøy, T. A., Mathisen, A. F., Abadpour, S., Paulo, J. A., Scholz, H., Ræder, H., & Chera, S. (2021). Chronically Elevated Exogenous Glucose Elicits Antipodal Effects on the Proteome Signature of Differentiating Human iPSC-Derived Pancreatic Progenitors. International Journal of Molecular Sciences, 22(7), 3698. https://doi.org/10.3390/ijms22073698