
Mutations Q93H and E97K in TPM2 Disrupt Ca-Dependent Regulation of Actin Filaments

Abstract
:1. Introduction
2. Results
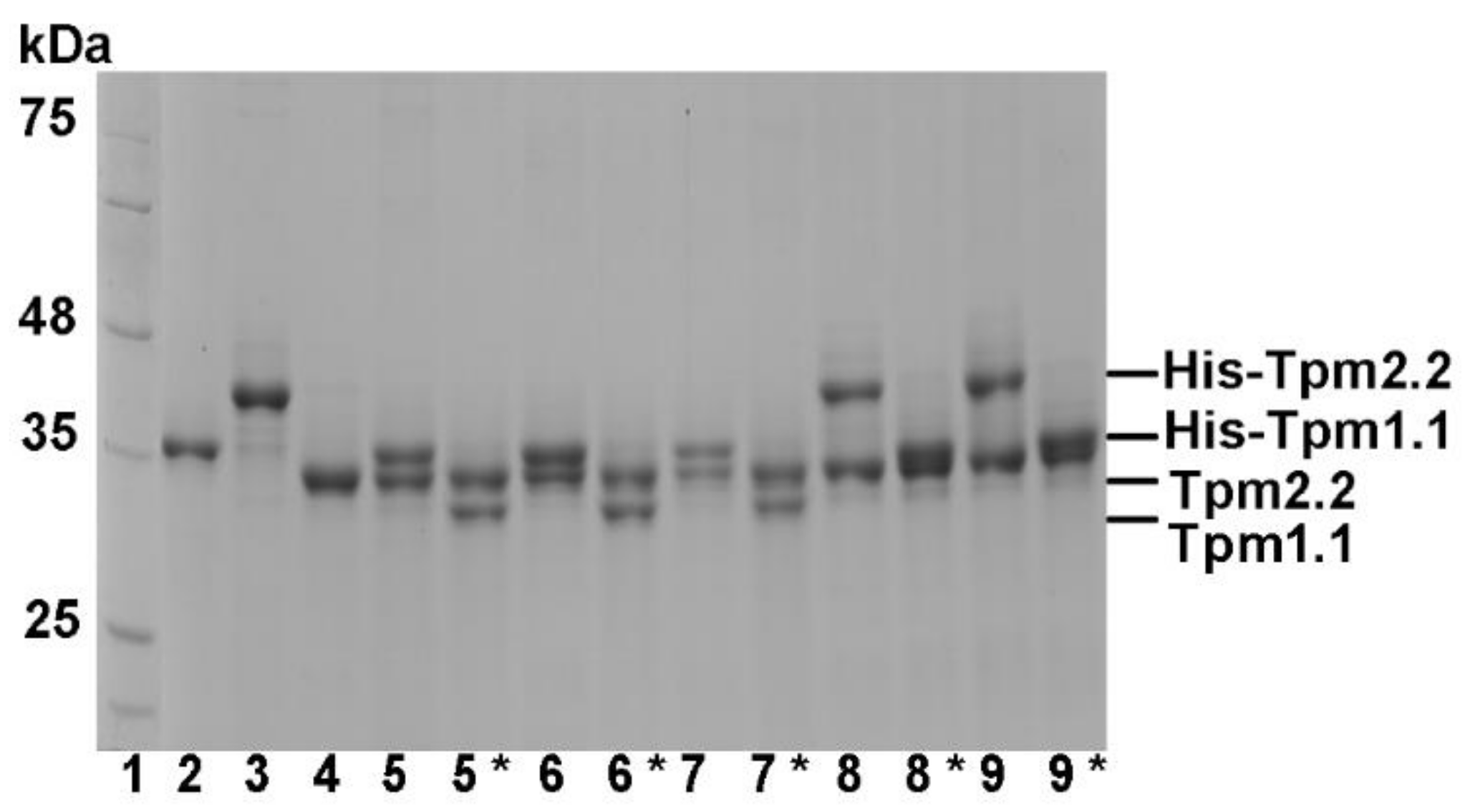
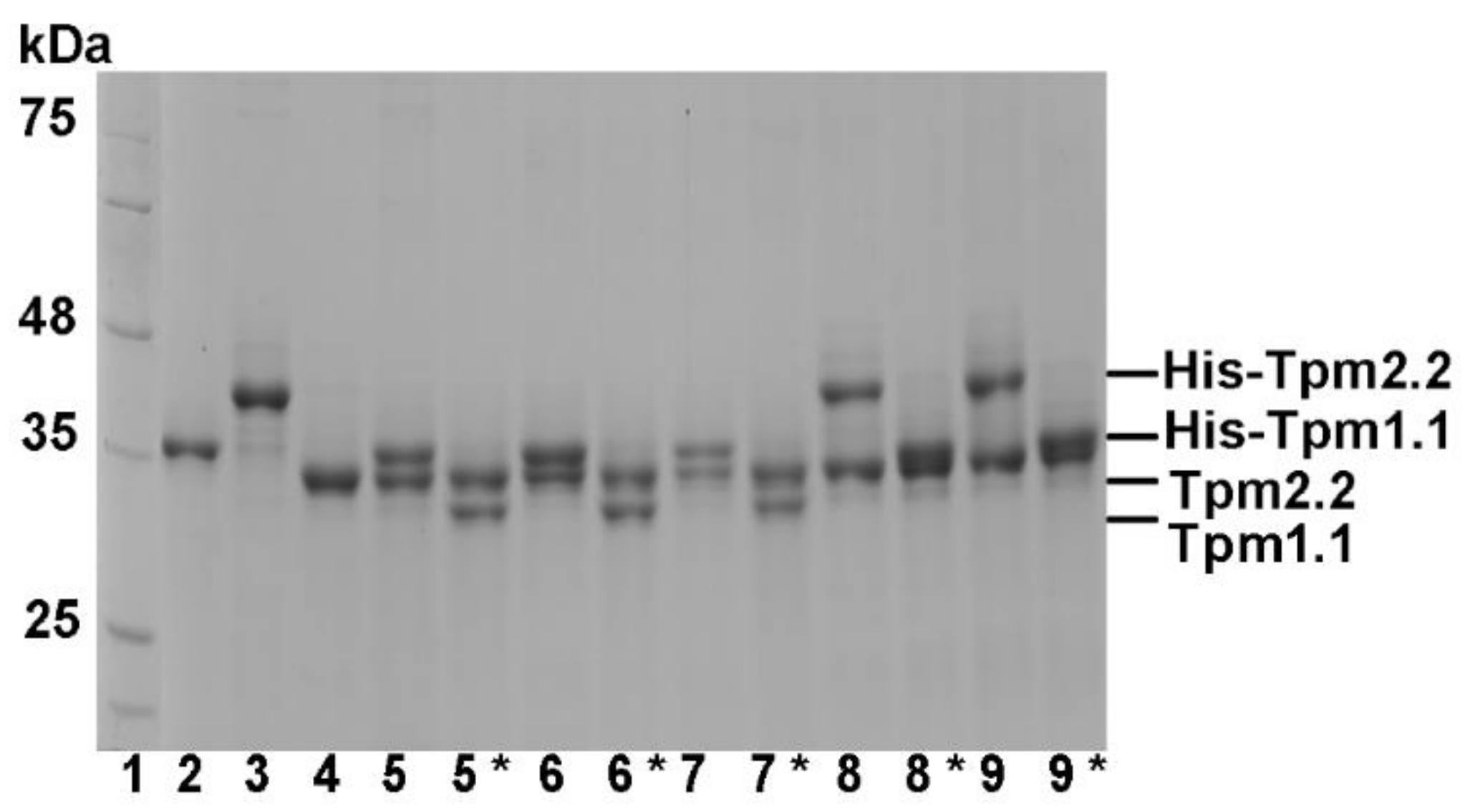
2.1. Formation of Heterodimers by Wild Type and Mutant Tpm2.2
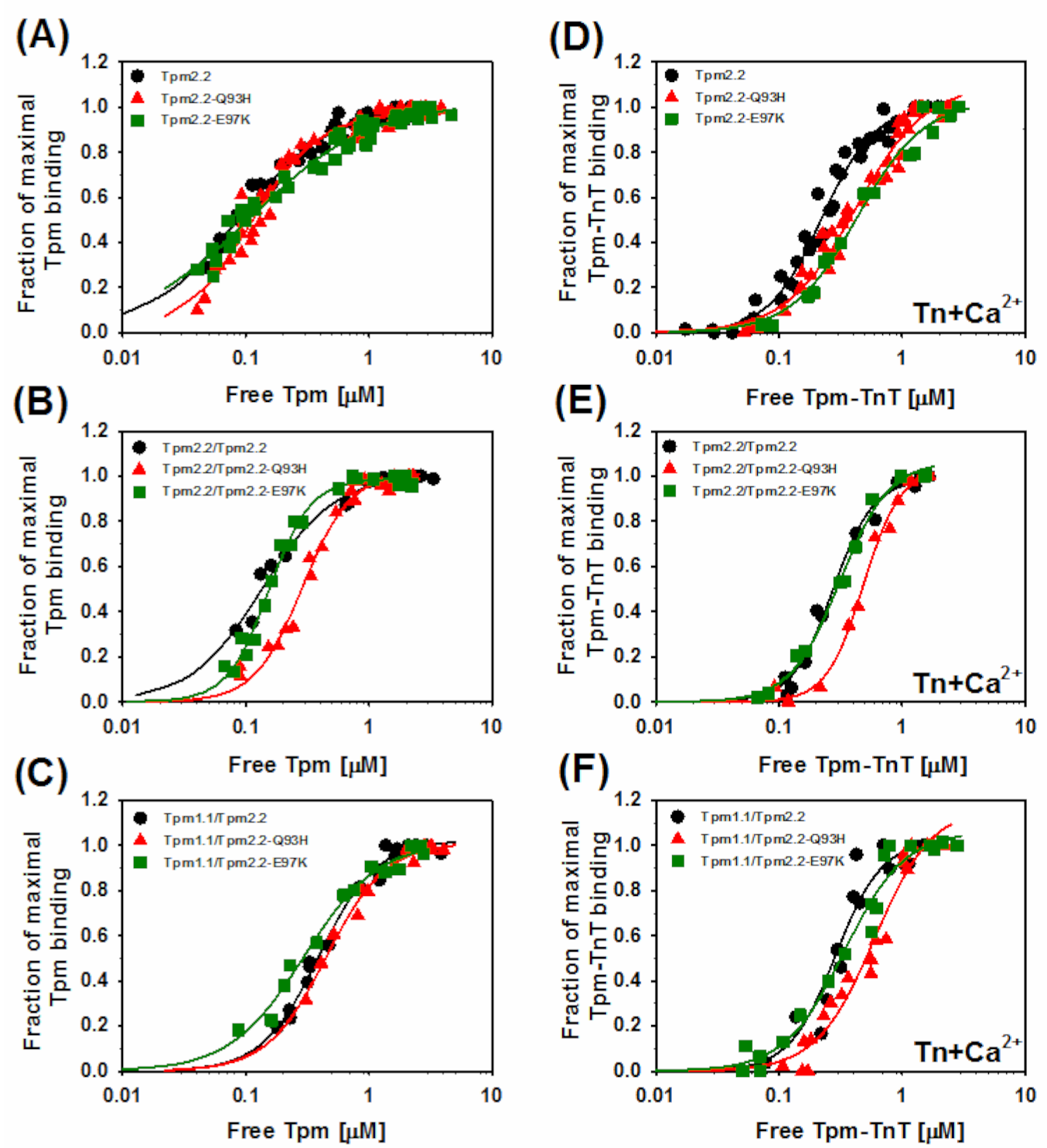
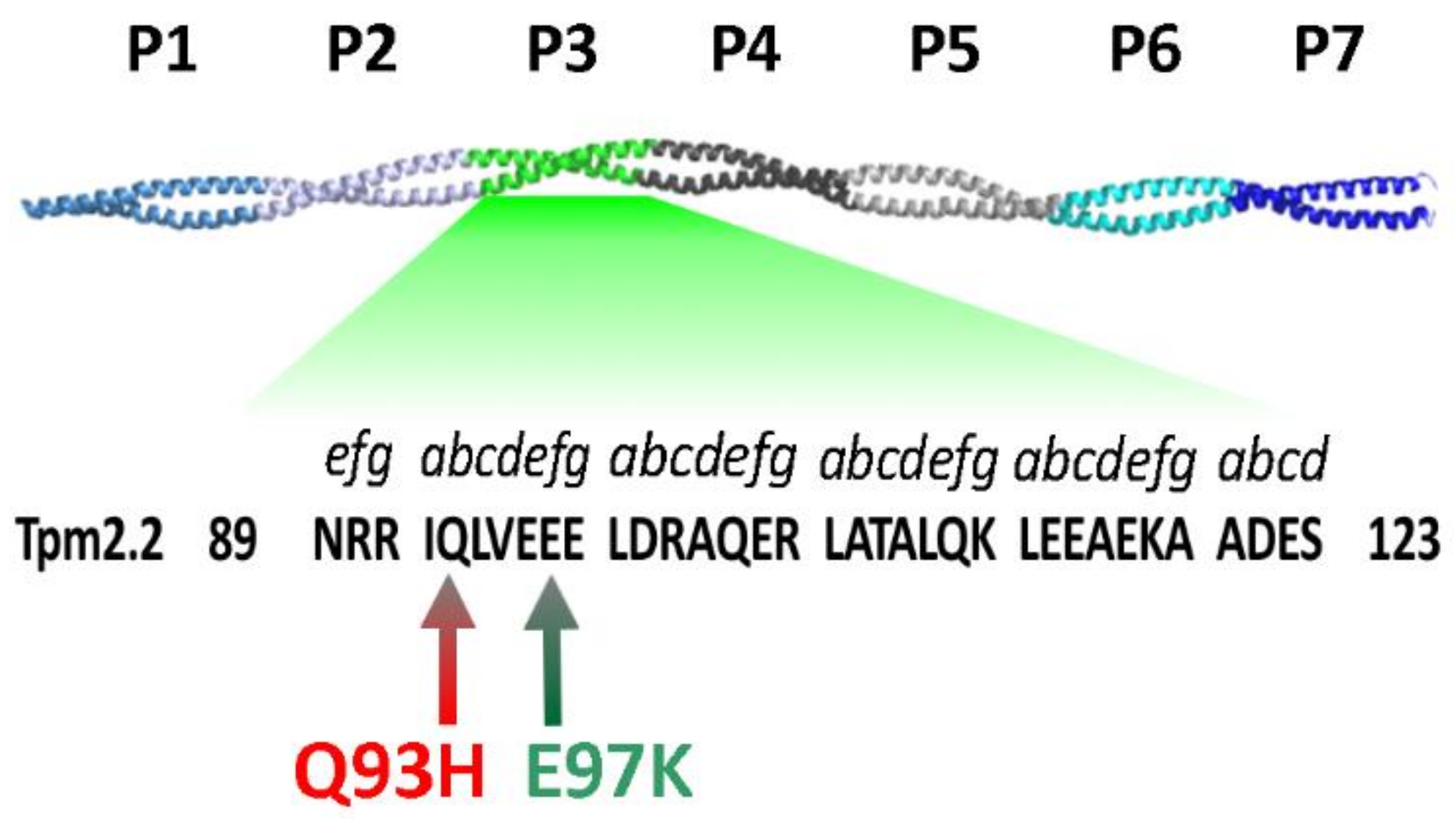
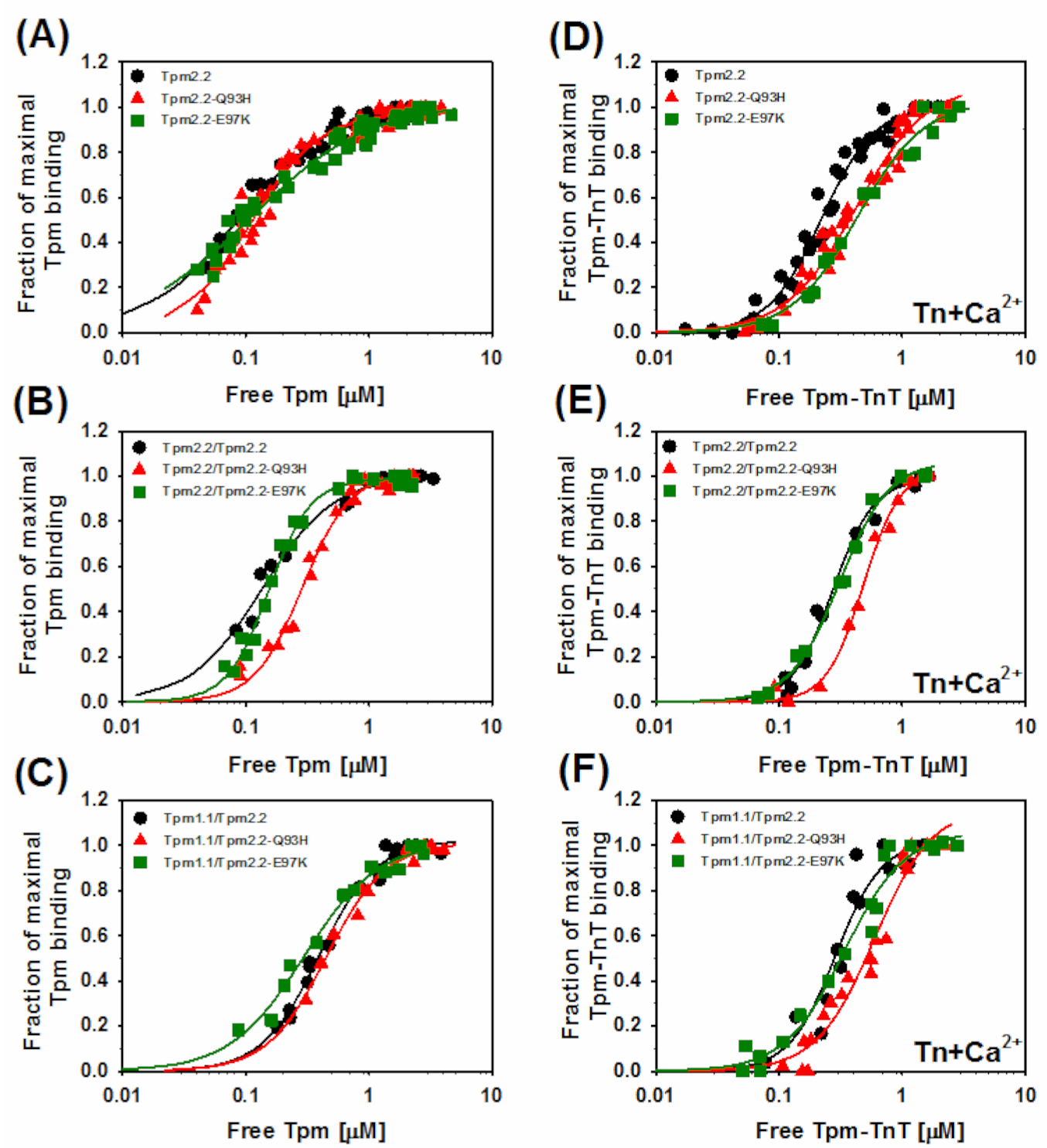
2.2. Effects of the Mutations Q93H and E97K on Interactions of Tpm2.2 Homo- and Heterodimers with Actin Filament
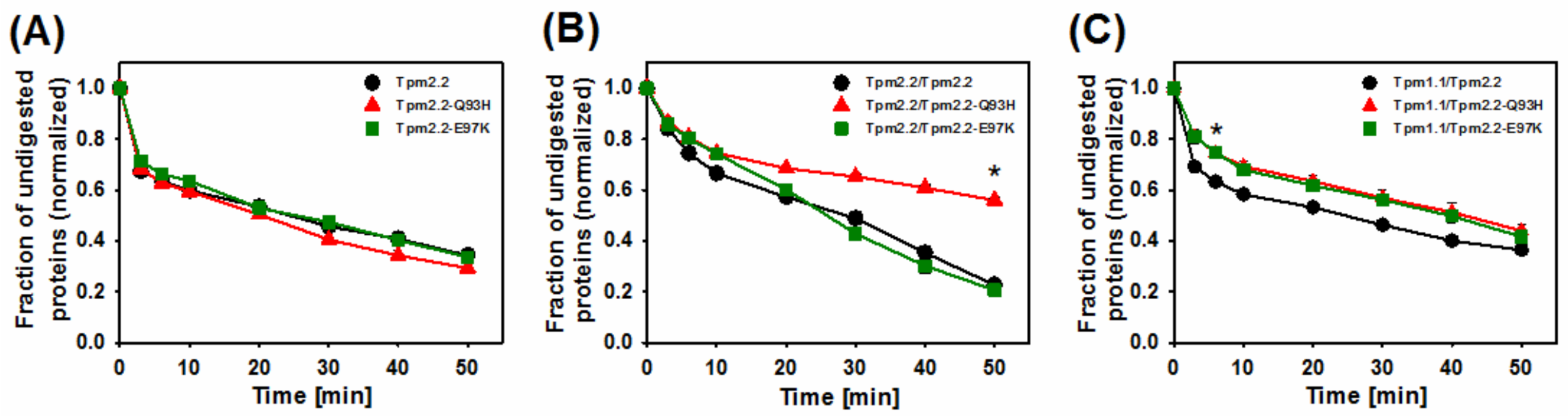
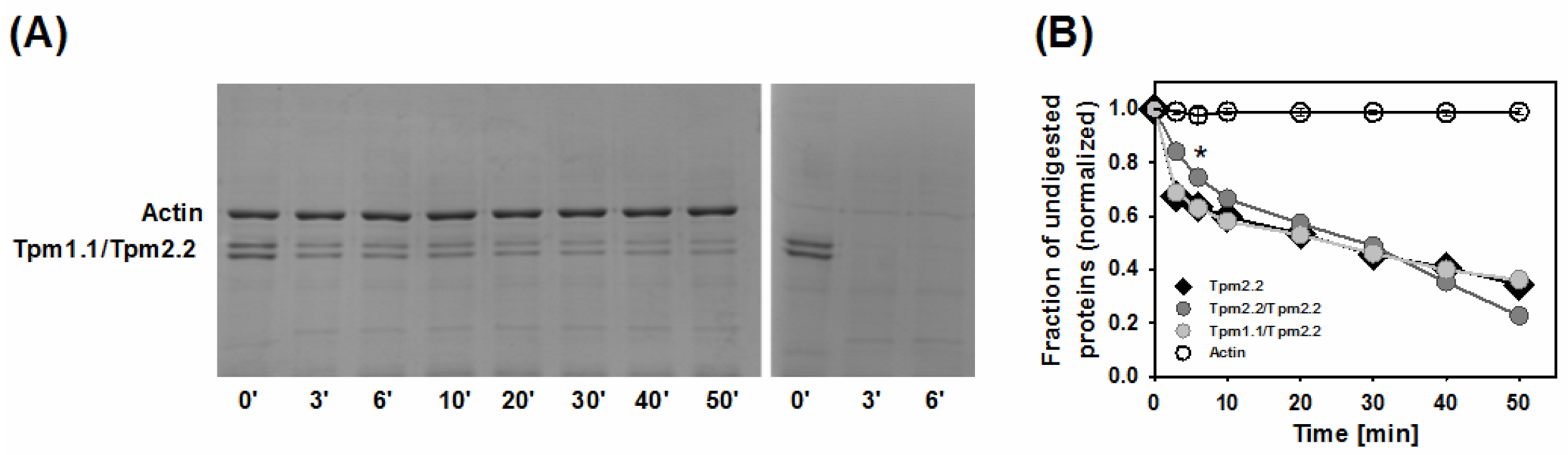
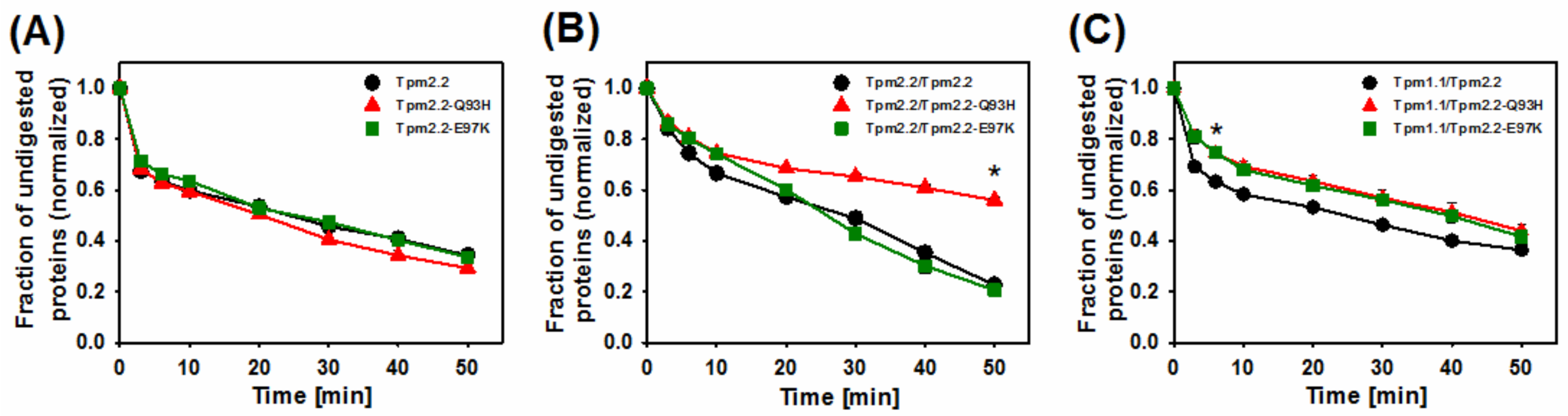
2.3. Effects of Mutations Q93H and E97K on Dynamics of Tropomyosin Interactions with F-actin
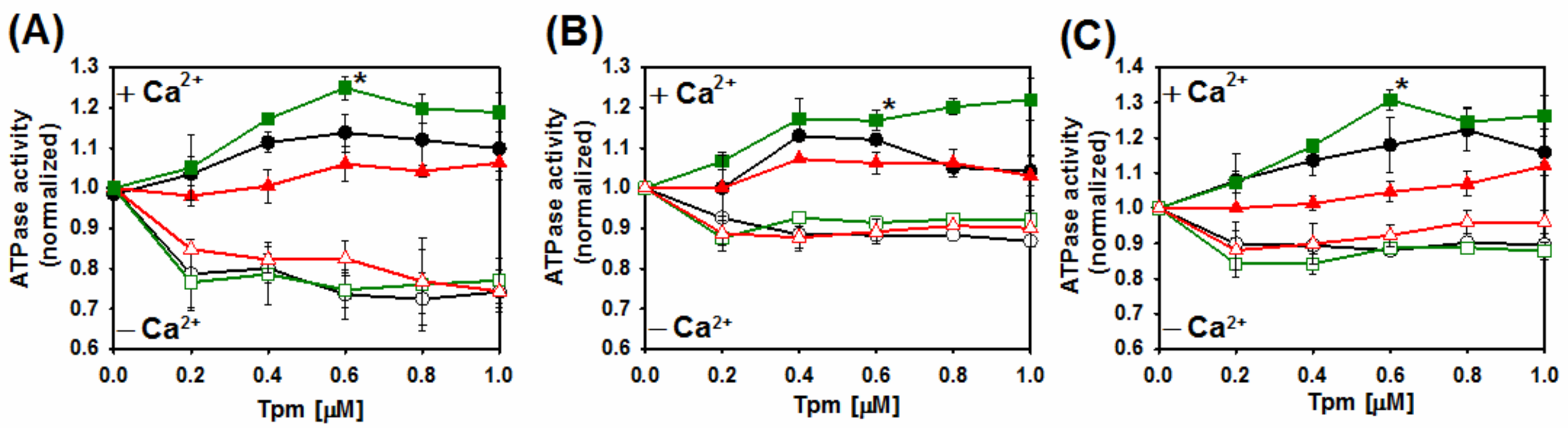
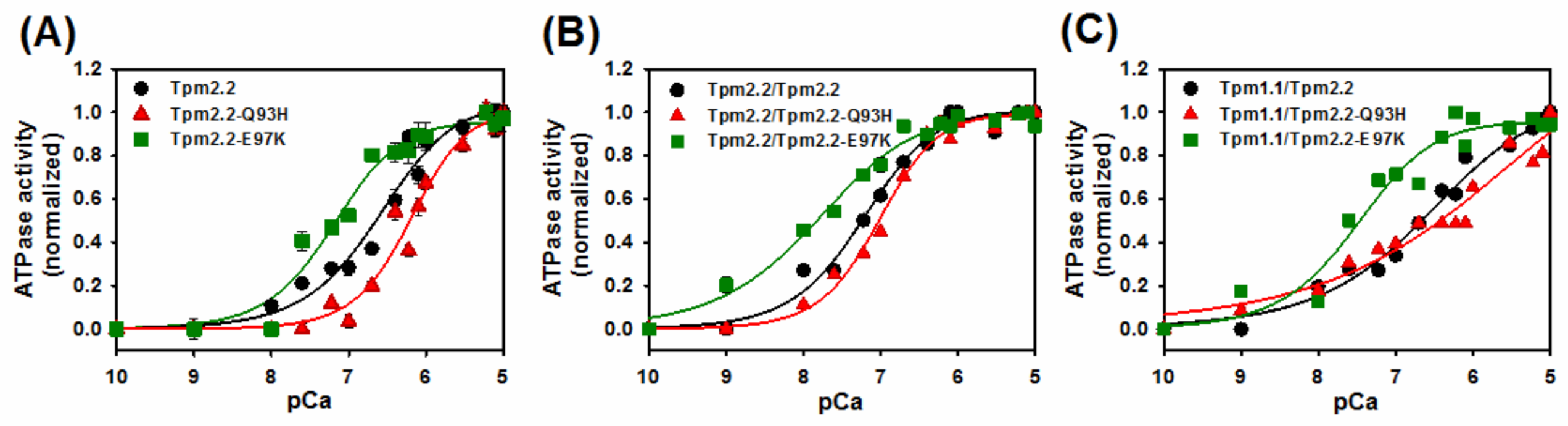
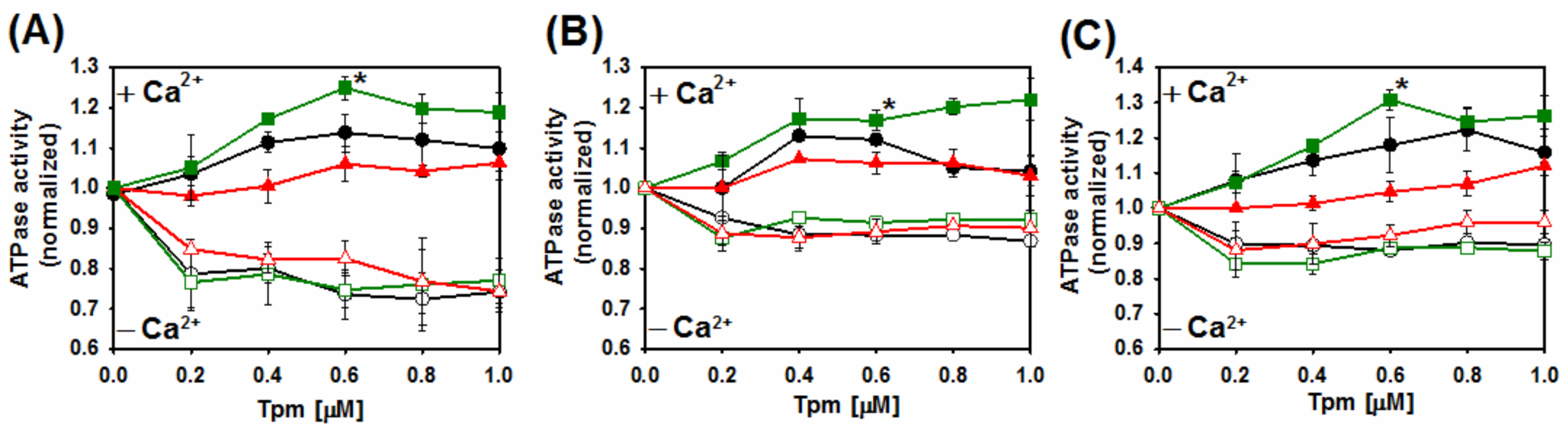
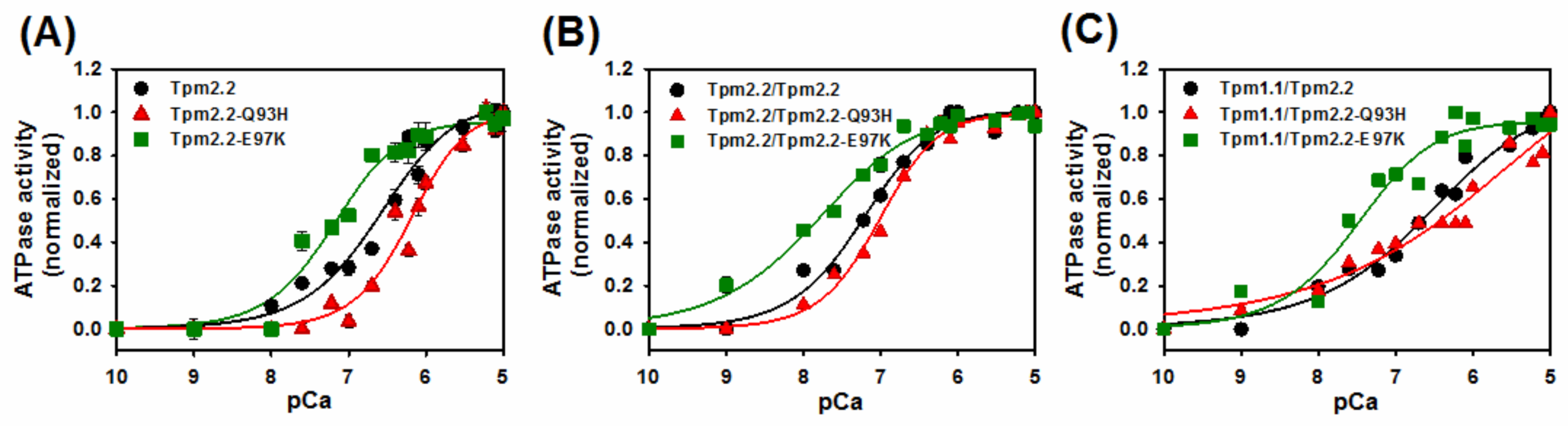
2.4. Ca2+-Dependent Regulation of Actin–Myosin Interactions by Tpm2.2 Mutants
3. Discussion
4. Materials and Methods
4.1. Preparation of Muscle Proteins
4.2. Expression and Purification of Recombinant Wild Type and Mutant Tropomyosin Homo- and Heterodimers
- Q93H: 5′ CTGAACCGTCGTATCCACCTGGTTGAAGAAGAAC 3′
- E97K: 5′ GTATCCAGCTGGTTGAAAAAGAACTGGACCGTGC 3′
- Codons that were mutated are underlined.
4.3. Actin-Binding Assay
4.4. Actomyosin MgATPase Activity
4.5. Tropomyosin Digestion with Trypsin
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Kee, A.J.; Gunning, P.W.; Hardeman, E.C. Diverse roles of the actin cytoskeleton in striated muscle. J. Muscle Res. Cell Motil. 2009, 30, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Gunning, P.W.; Hardeman, E.C.; Lappalainen, P.; Mulvihill, D.P. Tropomyosin—Master regulator of actin filament function in the cytoskeleton. J. Cell Sci. 2015, 128, 2965–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitchcock-DeGregori, S.E.; Singh, A. What makes tropomyosin an actin binding protein? A perspective. J. Struct. Biol. 2010, 170, 319–324. [Google Scholar] [PubMed] [Green Version]
- Hitchcock-DeGregori, S.E.; Barua, B. Tropomyosin structure, function, and interactions: A dynamic regulator. Fibrous Proteins Struct. Mech. 2017, 82, 253–284. [Google Scholar]
- Barua, B. Periodicities designed in the tropomyosin sequence and structure define its functions. BioArchitecture 2013, 3, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevzorov, I.A.; Levitsky, D.I. Tropomyosin: Double helix from the protein world. Biochemistry 2011, 76, 1507–1527. [Google Scholar] [CrossRef] [PubMed]
- Orzechowski, M.; Li, X.E.; Fischer, S.; Lehman, W. An atomic model of the tropomyosin cable on F-actin. Biophys. J. 2014, 107, 694–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janco, M.; Suphamungmee, W.; Li, X.; Lehman, W.; Lehrer, S.S.; Geeves, M.A. Polymorphism in tropomyosin structure and function. J. Muscle Res. Cell Motil. 2013, 34, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, S. Vertebrate tropomyosin: Distribution, properties and function. J. Muscle Res. Cell Motil. 2001, 22, 5–49. [Google Scholar] [CrossRef] [PubMed]
- Oe, M.; Ohnishi-Kameyama, M.; Nakajima, I.; Muroya, S.; Chikuni, K. Muscle type specific expression of tropomyosin isoforms in bovine skeletal muscles. Meat. Sci. 2007, 75, 558–563. [Google Scholar] [CrossRef]
- Lehrer, S.S. Intramolecular crosslinking of tropomyosin via disulfide bond formation: Evidence for chain register. Proc. Natl. Acad. Sci. USA 1975, 72, 3377–3381. [Google Scholar] [CrossRef] [Green Version]
- Janco, M.; Kalyva, A.; Scellini, B.; Piroddi, N.; Tesi, C.; Poggesi, C.; Geeves, M.A. α-Tropomyosin with a D175N or E180G mutation in only one chain differs from tropomyosin with mutations in both chains. Biochemie 2012, 51, 9880–9890. [Google Scholar] [CrossRef]
- Kalyva, A.; Schmidtmann, A.; Geeves, M.A. In vitro formation and characterization of the skeletal muscle alpha.beta tropomyosin heterodimers. Biochemistry 2012, 51, 6388–6399. [Google Scholar] [CrossRef] [Green Version]
- Matyushenko, A.M.; Shchepkin, D.V.; Kopylova, G.V.; Bershitsky, S.Y.; Levitsky, D.I. Unique functional properties of slow skeletal muscle tropomyosin. Biochimie 2020, 174, 1–8. [Google Scholar] [CrossRef]
- Śliwińska, M.; Robaszkiewicz, K.; Czajkowska, M.; Zheng, W.; Moraczewska, J. Functional effects of substitutions I92T and V95A in actin-binding period 3 of tropomyosin. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2018, 1866, 558–568. [Google Scholar] [CrossRef]
- McKillop, D.F.; Geeves, M.A. Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys. J. 1993, 65, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Vibert, P.; Craig, R.; Lehman, W. Steric-model for activation of muscle thin filaments 1 1 Edited by P.E. Wright. J. Mol. Biol. 1997, 266, 8–14. [Google Scholar] [CrossRef]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of contraction in striated muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef]
- Lehman, W. Thin filament structure and the steric blocking model. Compr. Physiol. 2016, 6, 1043–1069. [Google Scholar] [CrossRef]
- Redwood, C.; Robinson, P. Alpha-tropomyosin mutations in inherited cardiomyopathies. J. Muscle. Res. Cell. Motil. 2013, 34, 285–294. [Google Scholar] [CrossRef]
- Moraczewska, J. Thin filament dysfunctions caused by mutations in tropomyosin Tpm3.12 and Tpm1. J. Muscle Res. Cell Motil. 2020, 41, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Marttila, M.; Lehtokari, V.-L.; Marston, S.; Nyman, T.A.; Barnerias, C.; Beggs, A.H.; Bertini, E.; Ceyhan-Birsoy, Ö.; Cintas, P.; Gerard, M.; et al. Mutation update and genotype-phenotype correlations of novel and previously described mutations in TPM2 and TPM3 causing congenital myopathies. Hum. Mutat. 2014, 35, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.E.; McMillin, M.J.; Gildersleeve, H.I.S.; Kezele, P.R.; Shively, K.M.B.; Carey, J.C.; Regnier, M.; Bamshad, M.J. Spectrum of mutations that cause distal arthrogryposis types 1 and 2B. Am. J. Med. Genet. Part A 2013, 161, 550–555. [Google Scholar] [CrossRef] [Green Version]
- Tajsharghi, H.; Ohlsson, M.; Palm, L.; Oldfors, A. Myopathies associated with beta-tropomyosin mutations. Neuromuscul. Disord. 2012, 22, 923–933. [Google Scholar] [CrossRef]
- Matyushenko, A.M.; Levitsky, D.I. Molecular mechanisms of pathologies of skeletal and cardiac muscles caused by point mutations in the tropomyosin genes. Biochemistry 2020, 85, S20–S33. [Google Scholar] [CrossRef]
- Kawai, M.; Lu, X.; Hitchcock-DeGregori, S.E.; Stanton, K.J.; Wandling, M.W. Tropomyosin period 3 is essential for enhancement of isometric tension in thin filament-reconstituted bovine myocardium. J. Biophys. 2009, 2009, 380967. [Google Scholar] [CrossRef] [Green Version]
- Oguchi, Y.; Ishizuka, J.; Hitchcock-DeGregori, S.E.; Ishiwata, S.; Kawai, M. The role of tropomyosin domains in cooperative activation of the actin-myosin interaction. J. Mol. Biol. 2011, 414, 667–680. [Google Scholar] [CrossRef] [Green Version]
- Barua, B.; Winkelmann, D.A.; White, H.D.; Hitchcock-DeGregori, S.E. Regulation of actin-myosin interaction by conserved periodic sites of tropomyosin. Proc. Natl. Acad. Sci. USA 2012, 109, 18425–18430. [Google Scholar] [CrossRef] [Green Version]
- Pavadai, E.; Lehman, W.; Rynkiewicz, M.J. Protein-protein docking reveals dynamic interactions of tropomyosin on actin filaments. Biophys. J. 2020, 119, 75–86. [Google Scholar] [CrossRef]
- Li, X.E.; Tobacman, L.S.; Mun, J.Y.; Craig, R.; Fischer, S.; Lehman, W. Tropomyosin position on f-actin revealed by em reconstruction and computational chemistry. Biophys. J. 2011, 100, 1005–1013. [Google Scholar] [CrossRef] [Green Version]
- Barua, B.; Pamula, M.C.; Hitchcock-DeGregori, S.E. Evolutionarily conserved surface residues constitute actin binding sites of tropomyosin. Proc. Natl. Acad. Sci. USA 2011, 108, 10150–10155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, E.; Kielley, W.W. Troponin-tropomyosin complex—Column chromatographic-separation and activity of 3 active troponin components with and without tropomyosin present. J. Biol. Chem. 1974, 249, 4742–4748. [Google Scholar] [CrossRef]
- Khaitlina, S.; Fitz, H.; Hinssen, H. The interaction of gelsolin with tropomyosin modulates actin dynamics. FEBS J. 2013, 280, 4600–4611. [Google Scholar] [CrossRef] [PubMed]
- Robaszkiewicz, K.; Śliwinska, M.; Moraczewska, J. Regulation of actin filament length by muscle isoforms of tropomyosin and cofilin. Int. J. Mol. Sci. 2020, 21, 4285. [Google Scholar] [CrossRef]
- Kee, A.J.; Hardeman, E.C. Tropomyosins in skeletal muscle diseases. Adv. Exp. Med. Biol. 2008, 644, 143–157. [Google Scholar]
- Yamada, Y.; Namba, K.; Fujii, T. Cardiac muscle thin filament structures reveal calcium regulatory mechanism. Nat. Commun. 2020, 11, 153. [Google Scholar] [CrossRef]
- Behrmann, E.; Müller, M.; Penczek, P.A.; Mannherz, H.G.; Manstein, D.J.; Raunser, S. Structure of the rigor actin-tropomyosin-myosin complex. Cell 2012, 150, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Doran, M.H.; Pavadai, E.; Rynkiewicz, M.J.; Walklate, J.; Bullitt, E.; Moore, J.R.; Regnier, M.; Geeves, M.A.; Lehman, W. Cryo-em and molecular docking shows myosin loop 4 contacts actin and tropomyosin on thin filaments. Biophys. J. 2020, 119, 821–830. [Google Scholar] [CrossRef]
- Robaszkiewicz, K.; Dudek, E.; Kasprzak, A.A.; Moraczewska, J. Functional effects of congenital myopathy-related mutations in gamma-tropomyosin gene. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2012, 1822, 1562–1569. [Google Scholar] [CrossRef] [Green Version]
- Hitchcock-DeGregori, S.E.; Song, Y.; Greenfield, N.J. Functions of tropomyosin’s periodic repeats. Biochemistry 2002, 41, 15036–15044. [Google Scholar] [CrossRef]
- Bershitsky, S.Y.; Logvinova, D.S.; Shchepkin, D.V.; Kopylova, G.V.; Matyushenko, A.M. Myopathic mutations in the beta-chain of tropomyosin differently affect the structural and functional properties of betabeta- and alphabeta-dimers. FASEB J. 2019, 33, 1963–1971. [Google Scholar] [CrossRef] [Green Version]
- Rynkiewicz, M.J.; Fischer, S.; Lehman, W. The propensity for tropomyosin twisting in the presence and absence of F-actin. Arch. Biochem. Biophys. 2016, 609, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Ochala, J. Thin filament proteins mutations associated with skeletal myopathies: Defective regulation of muscle contraction. J. Mol. Med. 2008, 86, 1197–1204. [Google Scholar] [CrossRef]
- Orzechowski, M.; Fischer, S.; Moore, J.R.; Lehman, W.; Farman, G.P. Energy landscapes reveal the myopathic effects of tropomyosin mutations. Arch. Biochem. Biophys. 2014, 564, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Moraczewska, J.; Robaszkiewicz, K.; Śliwinska, M.; Czajkowska, M.; Ly, T.; Kostyukova, A.; Wen, H.; Zheng, W. Congenital myopathy-related mutations in tropomyosin disrupt regulatory function through altered actin affinity and tropomodulin binding. FEBS J. 2019, 286, 1877–1893. [Google Scholar] [CrossRef]
- Lehman, W.; Rynkiewicz, M.J. An atomicmodel of the thin filament describing troponin-i -tropomyosin binding at low-calcium. Biophys. J. 2021, 120, 251a. [Google Scholar] [CrossRef]
- Vinogradova, M.V.; Stone, D.B.; Malanina, G.G.; Karatzaferi, C.; Cooke, R.; Mendelson, R.A.; Fletterick, R.J. Ca2+-regulated structural changes in troponin. Proc. Natl. Acad. Sci. USA 2005, 102, 5038–5043. [Google Scholar] [CrossRef] [Green Version]
- Spudich, J.A.; Watt, S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 1971, 246, 4866–4871. [Google Scholar] [CrossRef]
- Margossian, S.S.; Lowey, S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzymol. 1982, 85, 55–71. [Google Scholar]
- Potter, J.D. Preparation of troponin and its subunits. Methods Enzymol. 1982, 85 Pt B, 241–263. [Google Scholar]
- Moraczewska, J.; Hitchcock-DeGregori, S.E. Independent functions for the N- and C-termini in the overlap region of tropomyosin. Biochemistry 2000, 39, 6891–6897. [Google Scholar] [CrossRef]
- White, H.D. Special instrumentation and techniques for kinetic studies of contractile systems. Methods Enzymol. 1982, 85, 698–708. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tropomyosin Variants | −Tn | +Tn (Ca2+) | ||
|---|---|---|---|---|
| Kapp [µM−1] | αH | Kapp [µM−1] | αH | |
| Tpm2.2 | 11.6 ± 1.9 | 1.2 ± 0.3 | 4.5 ± 0.2 | 2.0 ± 0.2 |
| Tpm2.2-Q93H | 9.1 ± 0.7 | 1.6 ± 0.3 | 2.4 ± 0.2 * | 1.5 ± 0.1 |
| Tpm2.2-E97K | 9.1 ± 0.9 | 0.9 ± 0.1 | 2.3 ± 0.2 * | 1.6 ± 0.2 |
| Tpm2.2/Tpm2.2 | 7.4 ± 0.5 | 1.5 ± 0.2 | 3.7 ± 0.2 | 2.6 ± 0.3 |
| Tpm2.2/Tpm2.2-Q93H | 3.5 ± 0.1 * | 2.2 ± 0.2 | 2.1 ± 0.1 * | 2.9 ± 0.3 |
| Tpm2.2/Tpm2.2-E97K | 6.5 ± 0.2 * | 2.6 ± 0.2 | 3.2 ± 0.1 | 2.2 ± 0.2 |
| Tpm1.1/Tpm2.2 | 2.5 ± 0.1 | 1.9 ± 0.2 | 3.5 ± 0.3 | 2.3 ± 0.5 |
| Tpm1.1/Tpm2.2-Q93H | 2.2 ± 0.4 | 1.8 ± 0.7 | 1.7 ± 0.2 * | 1.8 ± 0.3 |
| Tpm1.1/Tpm2.2-E97K | 3.4 ± 0.3 | 1.4 ± 0.2 | 2.9 ± 0.2 | 1.8 ± 0.2 |
| Tropomyosin Variants | pCa50 [M] |
|---|---|
| Tpm2.2 | 6.6 ± 0.01 |
| Tpm2.2-Q93H | 6.2 ± 0.1 * |
| Tpm2.2-E97K | 7.2 ± 0.1 * |
| Tpm2.2/Tpm2.2 | 7.2 ± 0.1 |
| Tpm2.2/Tpm2.2-Q93H | 7.0 ± 0.1 |
| Tpm2.2/Tpm2.2-E97K | 7.8 ± 0.1 * |
| Tpm1.1/Tpm2.2 | 6.6 ± 0.1 |
| Tpm1.1/Tpm2.2-Q93H | 6.0 ± 0.1 * |
| Tpm1.1/Tpm2.2-E97K | 7.5 ± 0.1 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Śliwinska, M.; Robaszkiewicz, K.; Wasąg, P.; Moraczewska, J. Mutations Q93H and E97K in TPM2 Disrupt Ca-Dependent Regulation of Actin Filaments. Int. J. Mol. Sci. 2021, 22, 4036. https://doi.org/10.3390/ijms22084036
Śliwinska M, Robaszkiewicz K, Wasąg P, Moraczewska J. Mutations Q93H and E97K in TPM2 Disrupt Ca-Dependent Regulation of Actin Filaments. International Journal of Molecular Sciences. 2021; 22(8):4036. https://doi.org/10.3390/ijms22084036
Chicago/Turabian StyleŚliwinska, Małgorzata, Katarzyna Robaszkiewicz, Piotr Wasąg, and Joanna Moraczewska. 2021. "Mutations Q93H and E97K in TPM2 Disrupt Ca-Dependent Regulation of Actin Filaments" International Journal of Molecular Sciences 22, no. 8: 4036. https://doi.org/10.3390/ijms22084036
APA StyleŚliwinska, M., Robaszkiewicz, K., Wasąg, P., & Moraczewska, J. (2021). Mutations Q93H and E97K in TPM2 Disrupt Ca-Dependent Regulation of Actin Filaments. International Journal of Molecular Sciences, 22(8), 4036. https://doi.org/10.3390/ijms22084036