
Small GTPases of the Rab and Arf Families: Key Regulators of Intracellular Trafficking in Neurodegeneration



Abstract
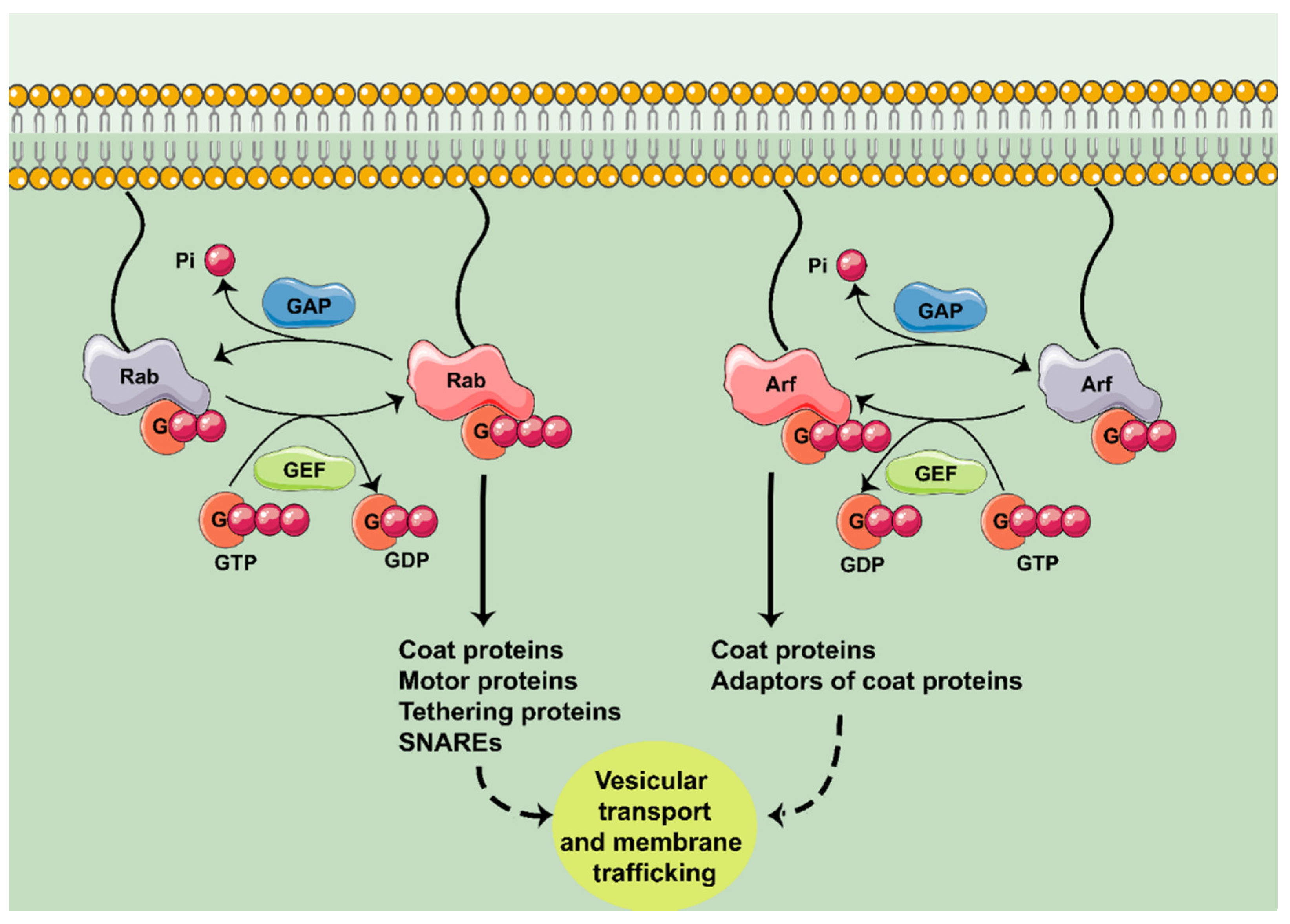
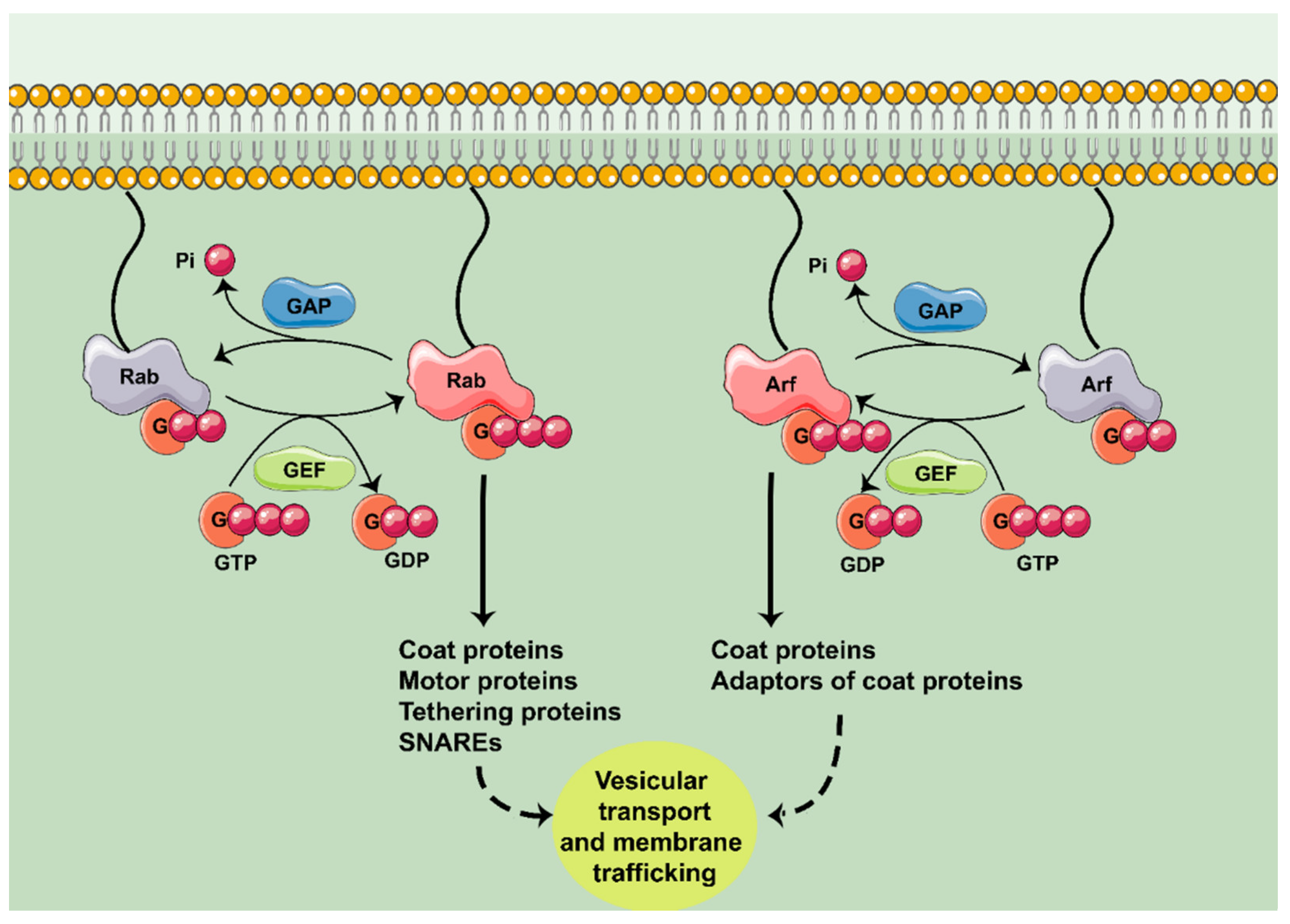
:1. Introduction
2. Rab GTPases in Neurodegeneration
2.1. Rab1
2.1.1. Rab1 and the ER–Golgi Traffic
2.1.2. Rab1 and the Integrity of the GA
2.1.3. Rab1 and the Control of the Autophagosome
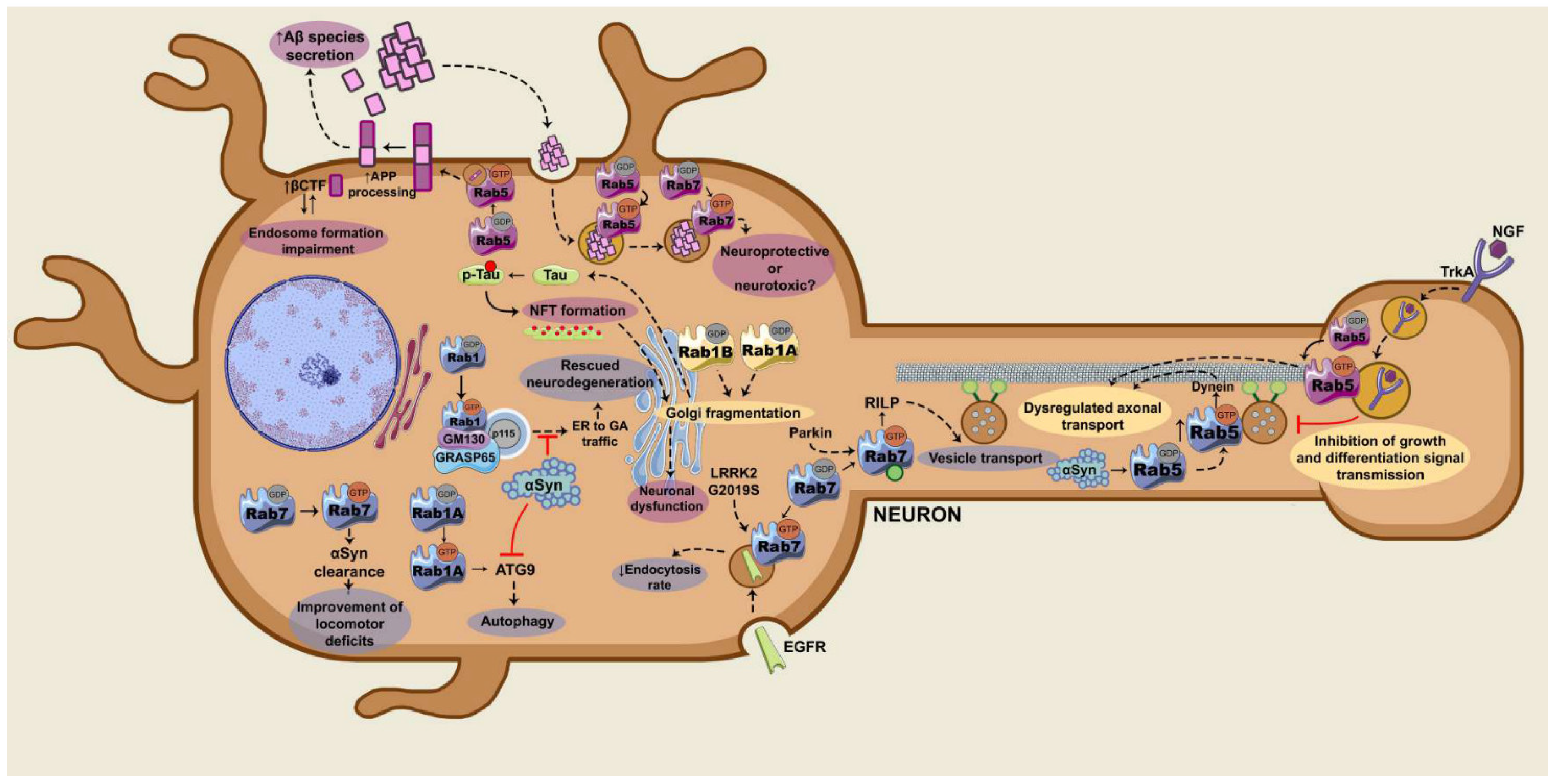
2.2. Rab5
2.2.1. Rab5 and APP Processing
2.2.2. Rab5 and Axonal Transport
2.3. Rab7
2.3.1. Rab7 and Trafficking of Toxic Peptides
2.3.2. Rab7 and Endolysosomal Trafficking of Membrane Receptors
2.3.3. Parkin/Rab7/RILP
2.3.4. Rab7 and Autophagy
3. Arf GTPases in Neurodegeneration
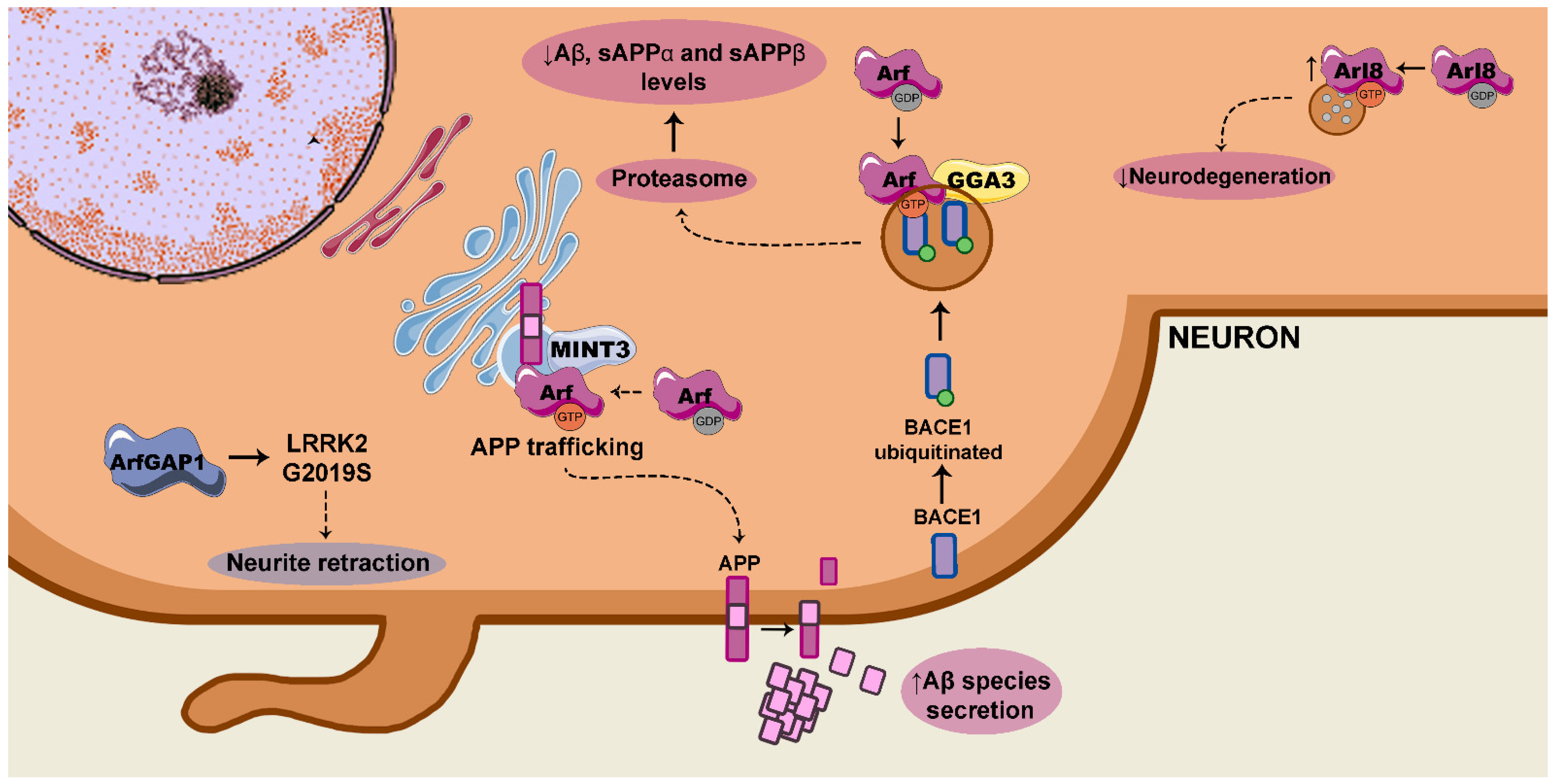
3.1. Arf/MINT and APP Trafficking and Processing
3.2. Arf/GGA/BACE1
3.3. Arl8 and Neuroprotection Against Aβ
3.4. ArfGAP1/LRRK2
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Berridge, M.J. Calcium signalling remodelling and disease. Biochem. Soc. Trans. 2012, 40, 297–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goitre, L.; Trapani, E.; Trabalzini, L.; Retta, S.F. The Ras Superfamily of Small GTPases: The Unlocked Secrets. Methods Mol. Biol. 2014, 1120, 1–18. [Google Scholar]
- Song, S.; Cong, W.; Zhou, S.; Shi, Y.; Dai, W.; Zhang, H.; Wang, X.; He, B.; Zhang, Q. Small GTPases: Structure, biological function and its interaction with nanoparticles. Asian J. Pharm. Sci. 2019, 14, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Toma-Fukai, S.; Shimizu, T. Structural insights into the regulation mechanism of small GTPases by GEFs. Molecules 2019, 24, 3308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrazola Sastre, A.; Luque Montoro, M.; Gálvez-Martín, P.; Lacerda, H.M.; Lucia, A.M.; Llavero, F.; Zugaza, J.L. Small GTPases of the Ras and Rho Families Switch on/off Signaling Pathways in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 6312. [Google Scholar] [CrossRef]
- Peurois, F.; Peyroche, G.; Cherfils, J. Small GTPase peripheral binding to membranes: Molecular determinants and supramolecular organization. Biochem. Soc. Trans. 2018, 47, 13–22. [Google Scholar] [CrossRef]
- Llavero, F.; Arrazola Sastre, A.; Luque Montoro, M.; Martín, M.A.; Arenas, J.; Lucia, A.; Zugaza, J.L. Small GTPases of the Ras superfamily and glycogen phosphorylase regulation in T cells. Small GTPases 2021, 12, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Kiral, F.R.; Kohrs, F.E.; Jin, E.J.; Hiesinger, P.R. Rab GTPases and Membrane Trafficking in Neurodegeneration. Curr. Biol. 2018, 28, R471–R486. [Google Scholar] [CrossRef] [Green Version]
- Sztul, E.; Chen, P.-W.; Casanova, J.E.; Cherfils, J.; Dacks, J.B.; Lambright, D.G.; Lee, F.-J.S.; Randazzo, P.A.; Santy, L.C.; Schürmann, A.; et al. ARF GTPases and their GEFs and GAPs: Concepts and challenges. Mol. Biol. Cell 2019, 30, 1249–1271. [Google Scholar] [CrossRef]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar]
- Xu, W.; Fang, F.; Ding, J.; Wu, C. Dysregulation of Rab5-mediated endocytic pathways in Alzheimer’s disease. Traffic 2018, 19, 253–262. [Google Scholar] [CrossRef]
- Parikh, I.; Fardo, D.W.; Estus, S. Genetics of PICALM expression and Alzheimer’s disease. PLoS ONE 2014, 9, e91242. [Google Scholar]
- Yang, L.; Mao, K.; Yu, H.; Chen, J. Neuroinflammatory Responses and Parkinson’ Disease: Pathogenic Mechanisms and Therapeutic Targets. J. Neuroimmune Pharmacol. 2020, 15, 830–837. [Google Scholar] [CrossRef]
- Guadagno, N.A.; Progida, C. Rab GTPases: Switching to Human Diseases. Cells 2019, 8, 909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, N.-V.; Desjardins, A.; Leclerc, N. Tau secretion is correlated to an increase of Golgi dynamics. PLoS ONE 2017, 12, e0178288. [Google Scholar] [CrossRef]
- Coune, P.G.; Bensadoun, J.C.; Aebischer, P.; Schneider, B.L. Rab1A Over-Expression Prevents Golgi Apparatus Fragmentation and Partially Corrects Motor Deficits in an Alpha-Synuclein Based Rat Model of Parkinson’s Disease. J. Parkinsons Dis. 2011, 1, 373–387. [Google Scholar] [CrossRef]
- Tomás, M.; Martínez-Alonso, E.; Martínez-Martínez, N.; Cara-Esteban, M.; Martínez-Menárguez, J.A. Fragmentation of the Golgi complex of dopaminergic neurons in human substantia nigra: New cytopathological findings in Parkinson’s disease. Histol. Histopathol. 2020, 36, 47–60. [Google Scholar]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [Green Version]
- Winslow, A.R.; Chen, C.-W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soo, K.Y.; Halloran, M.; Sundaramoorthy, V.; Parakh, S.; Toth, R.P.; Southam, K.A.; McLean, C.A.; Lock, P.; King, A.; Farg, M.A.; et al. Rab1-dependent ER-Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP-43 and FUS-associated ALS. Acta Neuropathol. 2015, 130, 679–697. [Google Scholar] [CrossRef] [PubMed]
- Grbovic, O.M.; Mathews, P.M.; Jiang, Y.; Schmidt, S.D.; Dinakar, R.; Summers-Terio, N.B.; Ceresa, B.P.; Nixon, R.A.; Cataldo, A.M. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Abeta production. J. Biol. Chem. 2003, 278, 31261–31268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Kanekiyo, T.; Shinohara, M.; Zhang, Y.; LaDu, M.J.; Xu, H.; Bu, G. Differential Regulation of Amyloid-β Endocytic Trafficking and Lysosomal Degradation by Apolipoprotein E Isoforms. J. Biol. Chem. 2012, 287, 44593–44601. [Google Scholar] [CrossRef] [Green Version]
- Song, M.S.; Baker, G.B.; Todd, K.G.; Kar, S. Inhibition of β-amyloid1-42 internalization attenuates neuronal death by stabilizing the endosomal-lysosomal system in rat cortical cultured neurons. Neuroscience 2011, 178, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Gillooly, D.J.; Raiborg, C.; Stenmark, H. Phosphatidylinositol 3-phosphate is found in microdomains of early endosomes. Histochem. Cell Biol. 2003, 120, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Bucci, C. Role of the RAB7 Protein in Tumor Progression and Cisplatin Chemoresistance. Cancers 2019, 11, 1096. [Google Scholar] [CrossRef] [Green Version]
- Nordmann, M.; Cabrera, M.; Perz, A.; Bröcker, C.; Ostrowicz, C.; Engelbrecht-Vandré, S.; Ungermann, C. The Mon1-Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr. Biol. 2010, 20, 1654–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafar, S.; Younas, N.; Correia, S.; Shafiq, M.; Tahir, W.; Schmitz, M.; Ferrer, I.; Andréoletti, O.; Zerr, I. Strain-Specific Altered Regulatory Response of Rab7a and Tau in Creutzfeldt-Jakob Disease and Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 697–709. [Google Scholar] [CrossRef]
- Rodriguez, L.; Mohamed, N.; Desjardins, A.; Lippé, R.; Fon, E.A.; Leclerc, N. Rab7A regulates tau secretion. J. Neurochem. 2017, 141, 592–605. [Google Scholar] [CrossRef]
- Dinter, E.; Saridaki, T.; Nippold, M.; Plum, S.; Diederichs, L.; Komnig, D.; Fensky, L.; May, C.; Marcus, K.; Voigt, A.; et al. Rab7 induces clearance of α-synuclein aggregates. J. Neurochem. 2016, 138, 758–774. [Google Scholar] [CrossRef]
- Gómez-Suaga, P.; Rivero-Ríos, P.; Fdez, E.; Blanca Ramírez, M.; Ferrer, I.; Aiastui, A.; López De Munain, A.; Hilfiker, S. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum. Mol. Genet. 2014, 23, 6779–6796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, P.; Trajkovic, K.; Tsunemi, T.; Krainc, D. Parkin Modulates Endosomal Organization and Function of the Endo-Lysosomal Pathway. J. Neurosci. 2016, 36, 2425–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaver, E.J.; van der Pouw Kraan, T.C.T.M.; Laan, L.C.; Kringel, H.; Cummings, R.D.; Bouma, G.; Kraal, G.; van Die, I. Trichuris suis soluble products induce Rab7b expression and limit TLR4 responses in human dendritic cells. Genes Immun. 2015, 16, 378–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colecchia, D.; Stasi, M.; Leonardi, M.; Manganelli, F.; Nolano, M.; Veneziani, B.M.; Santoro, L.; Eskelinen, E.-L.; Chiariello, M.; Bucci, C. Alterations of autophagy in the peripheral neuropathy Charcot-Marie-Tooth type 2B. Autophagy 2018, 14, 930–941. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.; Li, Y.; Bennett, M.; McKay, M.; Zhu, X.; Shern, J.; Torre, E.; Lah, J.J.; Levey, A.I.; Kahn, R.A. Munc18 Interacting Proteins: ADP-ribosylation factor-dependent coat proteins that regulate the traffic of β-Alzheimer’s precursor protein. J. Biol. Chem. 2003, 278, 36032–36040. [Google Scholar] [CrossRef] [Green Version]
- Bansal, A.; Kirschner, M.; Zu, L.; Cai, D.; Zhang, L. Coconut oil decreases expression of amyloid precursor protein (APP) and secretion of amyloid peptides through inhibition of ADP-ribosylation factor 1 (ARF1). Brain Res. 2019, 1704, 78–84. [Google Scholar] [CrossRef]
- Griffin, E.F.; Yan, X.; Caldwell, K.A.; Caldwell, G.A. Distinct functional roles of Vps41-mediated neuroprotection in Alzheimer’s and Parkinson’s disease models of neurodegeneration. Hum. Mol. Genet. 2018, 27, 4176–4193. [Google Scholar] [CrossRef] [PubMed]
- Goud, B.; Liu, S.; Storrie, B. Rab proteins as major determinants of the Golgi complex structure. Small GTPases 2018, 9, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homma, Y.; Hiragi, S.; Fukuda, M. Rab family of small GTPases: An updated view on their regulation and functions. FEBS J. 2021, 288, 36–55. [Google Scholar] [CrossRef]
- Marat, A.L.; Dokainish, H.; McPherson, P.S. DENN Domain Proteins: Regulators of Rab GTPases. J. Biol. Chem. 2011, 286, 13791–13800. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.P.; Goody, R.S. Molecular control of Rab activity by GEFs, GAPs and GDI. Small GTPases 2018, 9, 5–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, D.; Rai, A.; Ali, I.; Bleimling, N.; Friese, T.; Brockmeyer, A.; Janning, P.; Goud, B.; Itzen, A.; Müller, M.P.; et al. A pull-down procedure for the identification of unknown GEFs for small GTPases. Small GTPases 2016, 7, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 2016, 5, e12813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madero-Pérez, J.; Fdez, E.; Fernández, B.; Ordóñez, A.J.L.; Ramírez, M.B.; Gómez-Suaga, P.; Waschbüsch, D.; Lobbestael, E.; Baekelandt, V.; Nairn, A.C.; et al. Parkinson disease-associated mutations in LRRK2 cause centrosomal defects via Rab8a phosphorylation. Mol. Neurodegener. 2018, 13, 3. [Google Scholar] [CrossRef] [Green Version]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [Green Version]
- Carroll, K.S.; Hanna, J.; Simon, I.; Krise, J.; Barbero, P.; Pfeffer, S.R. Role of Rab9 GTPase in facilitating receptor recruitment by TIP47. Science 2001, 292, 1373–1376. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-T.; Gomez, T.S.; Sackey, B.K.; Billadeau, D.D.; Burd, C.G. Rab GTPase regulation of retromer-mediated cargo export during endosome maturation. Mol. Biol. Cell 2012, 23, 2505–2515. [Google Scholar] [CrossRef]
- Horgan, C.P.; Mccaffrey, M.W. Rab GTPases and microtubule motors. Biochem. Soc. Trans. 2011, 39, 1202–1206. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, A.J.; Jollivet, F.; Horgan, C.P.; Khan, A.R.; Raposo, G.; McCaffrey, M.W.; Goud, B. Identification and characterization of multiple novel Rab-myosin Va interactions. Mol. Biol. Cell 2013, 24, 3420–3434. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, K.; Torii, S.; Yi, Z.; Igarashi, M.; Okamoto, K.; Takeuchi, T.; Izumi, T. Melanophilin directly links Rab27a and myosin Va through its distinct coiled-coil regions. FEBS Lett. 2002, 517, 233–238. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Linstedt, A.D. Binding of the vesicle docking protein p115 to the GTPase Rab1b regulates membrane recruitment of the COPI vesicle coat. Cell. Logist. 2013, 3, e27687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, N. Emerging new roles of GM130, a cis-Golgi matrix protein, in higher order cell functions. J. Pharmacol. Sci. 2010, 112, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, E.; Christoforidis, S.; Uttenweiler-Joseph, S.; Miaczynska, M.; Dewitte, F.; Wilm, M.; Hoflack, B.; Zerial, M. Rabenosyn-5, a novel Rab5 effector, is complexed with hVPS45 and recruited to endosomes through a FYVE finger domain. J. Cell Biol. 2000, 151, 601–612. [Google Scholar] [CrossRef] [Green Version]
- Rahajeng, J.; Caplan, S.; Naslavsky, N. Common and distinct roles for the binding partners Rabenosyn-5 and Vps45 in the regulation of endocytic trafficking in mammalian cells. Exp. Cell Res. 2010, 316, 859–874. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Huang, T.Y.; Yancey, J.; Luo, H.; Zhang, Y.-W. Role of Rab GTPases in Alzheimer’s Disease. ACS Chem. Neurosci. 2019, 10, 828–838. [Google Scholar] [CrossRef]
- Shi, M.; Shi, C.; Xu, Y. Rab GTPases: The Key Players in the Molecular Pathway of Parkinson’s Disease. Front. Cell. Neurosci. 2017, 11, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, G.R.; Jang, E.-H.; Bae, J.R.; Jun, S.; Kang, H.C.; Park, C.-H.; Shin, J.-H.; Yamamoto, Y.; Tanaka-Yamamoto, K.; Dawson, V.L.; et al. Dysregulated phosphorylation of Rab GTPases by LRRK2 induces neurodegeneration. Mol. Neurodegener. 2018, 13, 8. [Google Scholar] [CrossRef]
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, S.R.; et al. Systematic proteomic analysis of LRRK2-mediated rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 2017, 6, e31012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Storrie, B. How Rab proteins determine Golgi structure. Int. Rev. Cell Mol. Biol. 2015, 315, 1–22. [Google Scholar] [PubMed] [Green Version]
- Ishida, M.; Oguchi, M.E.; Fukuda, M. Multiple Types of Guanine Nucleotide Exchange Factors (GEFs) for Rab Small GTPases. Cell Struct. Funct. 2016, 41, 61–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, M. TBC proteins: GAPs for mammalian small GTPase Rab? Biosci. Rep. 2011, 31, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Sztul, E.; Lupashin, V. Role of tethering factors in secretory membrane traffic. Am. J. Physiol. Cell Physiol. 2006, 290, C11–C26. [Google Scholar] [CrossRef]
- Sztul, E.; Lupashin, V. Role of vesicle tethering factors in the ER–Golgi membrane traffic. FEBS Lett. 2009, 583, 3770–3783. [Google Scholar] [CrossRef] [Green Version]
- Grosshans, B.L.; Ortiz, D.; Novick, P. Rabs and their effectors: Achieving specificity in membrane traffic. Proc. Natl. Acad. Sci. USA 2006, 103, 11821–11827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabski, R.; Hay, J.; Sztul, E. Tethering factor P115: A new model for tether-SNARE interactions. Bioarchitecture 2012, 2, 175–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, F.; Shi, X.; Li, B.; Huang, X.; Morelli, X.; Shi, N. Structural basis for the interaction between the Golgi reassembly-stacking protein GRASP65 and the Golgi matrix protein GM130. J. Biol. Chem. 2015, 290, 26373–26382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wang, Y. GRASPs in Golgi Structure and Function. Front. Cell Dev. Biol. 2015, 3, 84. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, C.; Garcia-Mata, R.; Brandon, E.; Sztul, E. COPI Recruitment Is Modulated by a Rab1b-dependent Mechanism. Mol. Biol. Cell 2003, 14, 2116–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monetta, P.; Slavin, I.; Romero, N.; Alvarez, C. Rab1b interacts with GBF1 and modulates both ARF1 dynamics and COPI association. Mol. Biol. Cell 2007, 18, 2400–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Menárguez, J.Á.; Tomás, M.; Martínez-Martínez, N.; Martínez-Alonso, E. Golgi Fragmentation in Neurodegenerative Diseases: Is There a Common Cause? Cells 2019, 8, 748. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Wang, L.; Guan, Y.; Xu, H.; Niu, Y.; Han, L.; Wei, Y.-P.; Lin, L.; Chu, J.; Wang, Q.; et al. Golgin-84-associated Golgi fragmentation triggers tau hyperphosphorylation by activation of cyclin-dependent kinase-5 and extracellular signal-regulated kinase. Neurobiol. Aging 2014, 35, 1352–1363. [Google Scholar] [CrossRef]
- Antón-Fernández, A.; Aparicio-Torres, G.; Tapia, S.; DeFelipe, J.; Muñoz, A. Morphometric alterations of Golgi apparatus in Alzheimer’s disease are related to tau hyperphosphorylation. Neurobiol. Dis. 2017, 97, 11–23. [Google Scholar] [CrossRef]
- Ao, X.; Zou, L.; Wu, Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014, 21, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Kakuta, S.; Yamamoto, H.; Negishi, L.; Kondo-Kakuta, C.; Hayashi, N.; Ohsumi, Y. Atg9 vesicles recruit vesicle-tethering proteins Trs85 and Ypt1 to the autophagosome formation site. J. Biol. Chem. 2012, 287, 44261–44269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Klionsky, D.J. Autophagic membrane delivery through ATG9. Cell Res. 2017, 27, 161–162. [Google Scholar] [CrossRef]
- Yuan, W.; Song, C. The Emerging Role of Rab5 in Membrane Receptor Trafficking and Signaling Pathways. Biochem. Res. Int. 2020, 2020, 4186308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajiho, H.; Sakurai, K.; Minoda, T.; Yoshikawa, M.; Nakagawa, S.; Fukushima, S.; Kontani, K.; Katada, T. Characterization of RIN3 as a guanine nucleotide exchange factor for the Rab5 subfamily GTPase Rab31. J. Biol. Chem. 2011, 286, 24364–24373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauer, J.; Segeletz, S.; Cezanne, A.; Guaitoli, G.; Raimondi, F.; Gentzel, M.; Alva, V.; Habeck, M.; Kalaidzidis, Y.; Ueffing, M.; et al. Auto-regulation of Rab5 GEF activity in Rabex5 by allosteric structural changes, catalytic core dynamics and ubiquitin binding. Elife 2019, 8, e46302. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H.; Vitale, G.; Ullrich, O.; Zerial, M. Rabaptin-5 is a direct effector of the small GTPase Rab5 in endocytic membrane fusion. Cell 1995, 83, 423–432. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Tsai, Y.C.; Mattera, R.; Smith, W.J.; Kostelansky, M.S.; Weissman, A.M.; Bonifacino, J.S.; Hurley, J.H. Structural basis for ubiquitin recognition and autoubiquitination by Rabex-5. Nat. Struct. Mol. Biol. 2006, 13, 264–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattera, R.; Tsai, Y.C.; Weissman, A.M.; Bonifacino, J.S. The Rab5 Guanine Nucleotide Exchange Factor Rabex-5 Binds Ubiquitin (Ub) and Functions as a Ub Ligase through an Atypical Ub-interacting Motif and a Zinc Finger Domain. J. Biol. Chem. 2006, 281, 6874–6883. [Google Scholar] [CrossRef] [Green Version]
- Christoforidis, S.; Miaczynska, M.; Ashman, K.; Wilm, M.; Zhao, L.; Yip, S.-C.; Waterfield, M.D.; Backer, J.M.; Zerial, M. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nat. Cell Biol. 1999, 1, 249–252. [Google Scholar] [CrossRef]
- Murray, J.T.; Panaretou, C.; Stenmark, H.; Miaczynska, M.; Backer, J.M. Role of Rab5 in the Recruitment of hVps34/p150 to the Early Endosome. Traffic 2002, 3, 416–427. [Google Scholar] [CrossRef]
- Wilson, J.M.; de Hoop, M.; Zorzi, N.; Toh, B.H.; Dotti, C.G.; Parton, R.G. EEA1, a tethering protein of the early sorting endosome, shows a polarized distribution in hippocampal neurons, epithelial cells, and fibroblasts. Mol. Biol. Cell 2000, 11, 2657–2671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, F.; Rocheleau, C.E. Vps34 and the Armus/TBC-2 Rab GAPs: Putting the brakes on the endosomal Rab5 and Rab7 GTPases. Cell. Logist. 2017, 7, e1403530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laifenfeld, D.; Patzek, L.J.; McPhie, D.L.; Chen, Y.; Levites, Y.; Cataldo, A.M.; Neve, R.L. Rab5 mediates an amyloid precursor protein signaling pathway that leads to apoptosis. J. Neurosci. 2007, 27, 7141–7153. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Mufson, E.J.; Counts, S.E.; Wuu, J.; Alldred, M.J.; Nixon, R.A.; Che, S. Regional selectivity of rab5 and rab7 protein upregulation in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers Dis. 2010, 22, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginsberg, S.D.; Alldred, M.J.; Counts, S.E.; Cataldo, A.M.; Neve, R.L.; Jiang, Y.; Wuu, J.; Chao, M.V.; Mufson, E.J.; Nixon, R.A.; et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol. Psychiatry 2010, 68, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Ginsberg, S.D.; Mufson, E.J.; Alldred, M.J.; Counts, S.E.; Wuu, J.; Nixon, R.A.; Che, S. Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J. Chem. Neuroanat. 2011, 42, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Weissmiller, A.M.; White, J.A.; Fang, F.; Wang, X.; Wu, Y.; Pearn, M.L.; Zhao, X.; Sawa, M.; Chen, S.; et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Investig. 2016, 126, 1815–1833. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Sato, Y.; Mohan, P.S.; Peterhoff, C.; Pensalfini, A.; Rigoglioso, A.; Jiang, Y.; Nixon, R.A. Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease. Mol. Psychiatry 2016, 21, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Spencer, B.; Desplats, P.A.; Overk, C.R.; Valera-Martin, E.; Rissman, R.A.; Wu, C.; Mante, M.; Adame, A.; Florio, J.; Rockenstein, E.; et al. Reducing Endogenous α-Synuclein Mitigates the Degeneration of Selective Neuronal Populations in an Alzheimer’s Disease Transgenic Mouse Model. J. Neurosci. 2016, 36, 7971–7984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, F.; Yang, W.; Florio, J.B.; Rockenstein, E.; Spencer, B.; Orain, X.M.; Dong, S.X.; Li, H.; Chen, X.; Sung, K.; et al. Synuclein impairs trafficking and signaling of BDNF in a mouse model of Parkinson’s disease. Sci. Rep. 2017, 7, 3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, A.; Severin, F.; Lommer, B.; Shevchenko, A.; Zerial, M. Huntingtin-HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington’s disease. J. Cell Biol. 2006, 172, 605–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer’s disease and down syndrome: Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Jiang, Y.; Mullaney, K.A.; Peterhoff, C.M.; Che, S.; Schmidt, S.D.; Boyer-Boiteau, A.; Ginsberg, S.D.; Cataldo, A.M.; Mathews, P.M.; Nixon, R.A. Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 1630–1635. [Google Scholar] [CrossRef] [Green Version]
- Kajiho, H.; Saito, K.; Tsujita, K.; Kontani, K.; Araki, Y.; Kurosu, H.; Katada, T. RIN3: A novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway. J. Cell Sci. 2003, 116, 4159–4168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.-C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Guerra, F.; Bucci, C. Multiple Roles of the Small GTPase Rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef]
- Langemeyer, L.; Fröhlich, F.; Ungermann, C. Rab GTPase Function in Endosome and Lysosome Biogenesis. Trends Cell Biol. 2018, 28, 957–970. [Google Scholar] [CrossRef]
- Kuchitsu, Y.; Fukuda, M. Revisiting Rab7 Functions in Mammalian Autophagy: Rab7 Knockout Studies. Cells 2018, 7, 215. [Google Scholar] [CrossRef] [Green Version]
- Wen, H.; Zhan, L.; Chen, S.; Long, L.; Xu, E. Rab7 may be a novel therapeutic target for neurologic diseases as a key regulator in autophagy. J. Neurosci. Res. 2017, 95, 1993–2004. [Google Scholar] [CrossRef]
- Poteryaev, D.; Datta, S.; Ackema, K.; Zerial, M.; Spang, A. Identification of the switch in early-to-late endosome transition. Cell 2010, 141, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Stroupe, C. This Is the End: Regulation of Rab7 Nucleotide Binding in Endolysosomal Trafficking and Autophagy. Front. Cell Dev. Biol. 2018, 6, 129. [Google Scholar] [CrossRef] [Green Version]
- Langemeyer, L.; Borchers, A.-C.; Herrmann, E.; Füllbrunn, N.; Han, Y.; Perz, A.; Auffarth, K.; Kümmel, D.; Ungermann, C. A conserved and regulated mechanism drives endosomal Rab transition. Elife 2020, 9, e56090. [Google Scholar] [CrossRef]
- Cantalupo, G.; Alifano, P.; Roberti, V.; Bruni, C.B.; Bucci, C. Rab-interacting lysosomal protein (RILP): The Rab7 effector required for transport to lysosomes. EMBO J. 2001, 20, 683–693. [Google Scholar] [CrossRef]
- Jordens, I.; Fernandez-Borja, M.; Marsman, M.; Dusseljee, S.; Janssen, L.; Calafat, J.; Janssen, H.; Wubbolts, R.; Neefjes, J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 2001, 11, 1680–1685. [Google Scholar] [CrossRef] [Green Version]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.-A.; Lamark, T.; Overvatn, A.; Bjørkøy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-K.; Lee, J.-A. Role of the mammalian ATG8/LC3 family in autophagy: Differential and compensatory roles in the spatiotemporal regulation of autophagy. BMB Rep. 2016, 49, 424–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, N.; Ganesh, S. Emerging nexus between RAB GTPases, autophagy and neurodegeneration. Autophagy 2016, 12, 900–904. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunello, C.A.; Merezhko, M.; Uronen, R.-L.; Huttunen, H.J. Mechanisms of secretion and spreading of pathological tau protein. Cell. Mol. Life Sci. 2020, 77, 1721–1744. [Google Scholar] [CrossRef] [Green Version]
- Vidyadhara, D.J.; Lee, J.E.; Chandra, S.S. Role of the endolysosomal system in Parkinson’s disease. J. Neurochem. 2019, 150, 487–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, M.W.; Zhang, T.; Jiang, C.; Chen, S.; Guo, M. Roles of the Drosophila LRRK2 homolog in Rab7-dependent lysosomal positioning. Hum. Mol. Genet. 2012, 21, 1350–1363. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C. Recycling Endosomes and TLR Signaling—The Rab11 GTPase Leads the Way. Immunity 2010, 33, 578–580. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.S.; Tang, B.L. The Evi5 family in cellular physiology and pathology. FEBS Lett. 2013, 587, 1703–1710. [Google Scholar] [CrossRef] [Green Version]
- Mazdeh, M.; Ghafouri-Fard, S.; Noroozi, R.; Sayad, A.; Khani, M.; Taheri, M.; Davood Omrani, M. Ecotropic Viral Integration Site 5 (EVI5) variants are associated with multiple sclerosis in Iranian population. Mult. Scler. Relat. Disord. 2017, 18, 15–19. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Taheri, M.; Omrani, M.D.; Daaee, A.; Mohammad-Rahimi, H. Application of Artificial Neural Network for Prediction of Risk of Multiple Sclerosis Based on Single Nucleotide Polymorphism Genotypes. J. Mol. Neurosci. 2020, 70, 1081–1087. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Parkin Plays a Role in Sporadic Parkinson’s Disease. Neurodegener. Dis. 2013, 13, 69–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Underwood, R.; Wang, B.; Carico, C.; Whitaker, R.H.; Placzek, W.J.; Yacoubian, T. Rab27b regulates the release, autophagic clearance, and toxicity of alpha-synuclein. J. Biol. Chem. 2020, 295, 8005–8016. [Google Scholar] [CrossRef]
- Wilson, G.R.; Sim, J.C.H.; McLean, C.; Giannandrea, M.; Galea, C.A.; Riseley, J.R.; Stephenson, S.E.M.; Fitzpatrick, E.; Haas, S.A.; Pope, K.; et al. Mutations in RAB39B cause X-linked intellectual disability and early-onset parkinson disease with α-synuclein pathology. Am. J. Hum. Genet. 2014, 95, 729–735. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Bras, J.; Cormier-Dequaire, F.; Condroyer, C.; Nicolas, A.; Darwent, L.; Guerreiro, R.; Majounie, E.; Federoff, M.; Heutink, P.; et al. Loss-of-function mutations in RAB39B are associated with typical early-onset Parkinson disease. Neurol. Genet. 2015, 1, e9. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.-C.; Yeh, T.-H.; Lai, S.-C.; Weng, Y.-H.; Huang, Y.-C.; Cheng, Y.-C.; Chen, R.-S.; Huang, Y.-Z.; Hung, J.; Chen, C.-C.; et al. Increased Rab35 expression is a potential biomarker and implicated in the pathogenesis of Parkinson’s disease. Oncotarget 2016, 7, 54215–54227. [Google Scholar] [CrossRef] [Green Version]
- Kahn, R.A.; Cherfils, J.; Elias, M.; Lovering, R.C.; Munro, S.; Schurmann, A. Nomenclature for the human Arf family of GTP-binding proteins: ARF, ARL, and SAR proteins. J. Cell Biol. 2006, 172, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.L.; Bouvet, S. Arfs at a glance. J. Cell Sci. 2014, 127, 4103–4109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mossessova, E.; Gulbis, J.M.; Goldberg, J. Structure of the guanine nucleotide exchange factor Sec7 domain of human arno and analysis of the interaction with ARF GTPase. Cell 1998, 92, 415–423. [Google Scholar] [CrossRef] [Green Version]
- Cox, R.; Mason-Gamer, R.J.; Jackson, C.L.; Segev, N. Phylogenetic analysis of Sec7-domain-containing Arf nucleotide exchangers. Mol. Biol. Cell 2004, 15, 1487–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randazzo, P.A.; Hirsch, D.S. Arf GAPs: Multifunctional proteins that regulate membrane traffic and actin remodelling. Cell. Signal. 2004, 16, 401–413. [Google Scholar] [CrossRef]
- Inoue, H.; Randazzo, P.A. Arf GAPs and their interacting proteins. Traffic 2007, 8, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Spang, A.; Shiba, Y.; Randazzo, P.A. Arf GAPs: Gatekeepers of vesicle generation. FEBS Lett. 2010, 584, 2646–2651. [Google Scholar] [CrossRef] [Green Version]
- Bowzard, J.B.; Cheng, D.; Peng, J.; Kahn, R.A. ELMOD2 is an Arl2 GTPase-activating protein that also acts on Arfs. J. Biol. Chem. 2007, 282, 17568–17580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- East, M.P.; Bowzard, J.B.; Dacks, J.B.; Kahn, R.A. ELMO domains, evolutionary and functional characterization of a novel GTPase-activating protein (GAP) domain for Arf protein family GTPases. J. Biol. Chem. 2012, 287, 39538–39553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, A.A.; East, M.P.; Yi, S.L.; Kahn, R.A. Characterization of recombinant ELMOD (cell engulfment and motility domain) proteins as GTPase-activating proteins (GAPs) for ARF family GTPases. J. Biol. Chem. 2014, 289, 11111–11121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, Y.; Hongu, T.; Yamauchi, Y.; Funakoshi, Y.; Katagiri, N.; Ohbayashi, N.; Kanaho, Y. ACAP3 regulates neurite outgrowth through its GAP activity specific to Arf6 in mouse hippocampal neurons. Biochem. J. 2016, 473, 2591–2602. [Google Scholar] [CrossRef]
- Inaba, Y.; Tian, Q.B.; Okano, A.; Zhang, J.; Sakagami, H.; Miyazawa, S.; Li, W.; Komiyama, A.; Inokuchi, K.; Kondo, H.; et al. Brain-specific potential guanine nucleotide exchange factor for Arf, synArfGEF (Po), is localized to postsynaptic density. J. Neurochem. 2004, 89, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Mignot, C.; McMahon, A.C.; Bar, C.; Campeau, P.M.; Davidson, C.; Buratti, J.; Nava, C.; Jacquemont, M.-L.; Tallot, M.; Milh, M.; et al. IQSEC2-related encephalopathy in males and females: A comparative study including 37 novel patients. Genet. Med. 2019, 21, 837–849. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Torii, T.; Tago, K.; Tanoue, A.; Takashima, S.; Yamauchi, J. BIG1/Arfgef1 and Arf1 regulate the initiation of myelination by Schwann cells in mice. Sci. Adv. 2018, 4, eaar4471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherfils, J. Arf GTPases and their effectors: Assembling multivalent membrane-binding platforms. Curr. Opin. Struct. Biol. 2014, 29, 67–76. [Google Scholar] [CrossRef]
- Jackson, L.P. Structure and mechanism of COPI vesicle biogenesis. Curr. Opin. Cell Biol. 2014, 29, 67–73. [Google Scholar] [CrossRef]
- Okamoto, M.; Südhof, T.C. Mints, Munc18-interacting proteins in synaptic vesicle exocytosis. J. Biol. Chem. 1997, 272, 31459–31464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, L.; Pan, C.; He, S.M.; Lang, B.; Gao, G.D.; Wang, X.L.; Wang, Y. The ras superfamily of small gtpases in non-neoplastic cerebral diseases. Front. Mol. Neurosci. 2019, 12, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Einem, B.; Wahler, A.; Schips, T.; Serrano-Pozo, A.; Proepper, C.; Boeckers, T.M.; Rueck, A.; Wirth, T.; Hyman, B.T.; Danzer, K.M.; et al. The Golgi-Localized γ-Ear-Containing ARF-Binding (GGA) Proteins Alter Amyloid-β Precursor Protein (APP) Processing through Interaction of Their GAE Domain with the Beta-Site APP Cleaving Enzyme 1 (BACE1). PLoS ONE 2015, 10, e0129047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, M.; Südhof, T.C. Mint 3: A ubiquitous mint isoform that does not bind to munc18-1 or -2. Eur. J. Cell Biol. 1998, 77, 161–165. [Google Scholar] [CrossRef]
- Shrivastava-Ranjan, P.; Faundez, V.; Fang, G.; Rees, H.; Lah, J.J.; Levey, A.I.; Kahn, R.A. Mint3/X11gamma is an ADP-ribosylation factor-dependent adaptor that regulates the traffic of the Alzheimer’s Precursor protein from the trans-Golgi network. Mol. Biol. Cell 2008, 19, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, P.; Kornfeld, S. The GGA proteins: Key players in protein sorting at the trans-Golgi network. Eur. J. Cell Biol. 2004, 83, 257–262. [Google Scholar] [CrossRef]
- Puertollano, R.; Bonifacino, J.S. Interactions of GGA3 with the ubiquitin sorting machinery. Nat. Cell Biol. 2004, 6, 244–251. [Google Scholar] [CrossRef]
- Scott, P.M.; Bilodeau, P.S.; Zhdankina, O.; Winistorfer, S.C.; Hauglund, M.J.; Allaman, M.M.; Kearney, W.R.; Robertson, A.D.; Boman, A.L.; Piper, R.C. GGA proteins bind ubiquitin to facilitate sorting at the trans-Golgi network. Nat. Cell Biol. 2004, 6, 252–259. [Google Scholar] [CrossRef]
- Ren, X.; Hurley, J.H. VHS domains of ESCRT-0 cooperate in high-avidity binding to polyubiquitinated cargo. EMBO J. 2010, 29, 1045–1054. [Google Scholar] [CrossRef]
- Kang, E.L.; Cameron, A.N.; Piazza, F.; Walker, K.R.; Tesco, G. Ubiquitin regulates GGA3-mediated degradation of BACE1. J. Biol. Chem. 2010, 285, 24108–24119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Arnim, C.A.F.; Spoelgen, R.; Peltan, I.D.; Deng, M.; Courchesne, S.; Koker, M.; Matsui, T.; Kowa, H.; Lichtenthaler, S.F.; Irizarry, M.C.; et al. GGA1 Acts as a Spatial Switch Altering Amyloid Precursor Protein Trafficking and Processing. J. Neurosci. 2006, 26, 9913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, S.; Sharma, M.; Ung, C.; Tuli, A.; Barral, D.C.; Hava, D.L.; Veerapen, N.; Besra, G.S.; Hacohen, N.; Brenner, M.B. Lysosomal Trafficking, Antigen Presentation, and Microbial Killing Are Controlled by the Arf-like GTPase Arl8b. Immunity 2011, 35, 182–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatter, D.; Sindhwani, A.; Sharma, M. Arf-like GTPase Arl8: Moving from the periphery to the center of lysosomal biology. Cell. Logist. 2015, 5, e1086501. [Google Scholar] [CrossRef] [Green Version]
- Rosa-Ferreira, C.; Sweeney, S.T.; Munro, S. The small G protein Arl8 contributes to lysosomal function and long-range axonal transport in Drosophila. Biol. Open 2018, 7, bio035964. [Google Scholar] [CrossRef] [Green Version]
- Balderhaar, H.J.K.; Ungermann, C. CORVET and HOPS tethering complexes–coordinators of endosome and lysosome fusion. J. Cell Sci. 2013, 126, 1307–1316. [Google Scholar] [CrossRef] [Green Version]
- Khatter, D.; Raina, V.B.; Dwivedi, D.; Sindhwani, A.; Bahl, S.; Sharma, M. The small GTPase Arl8b regulates assembly of the mammalian HOPS complex on lysosomes. J. Cell Sci. 2015, 128, 1746–1761. [Google Scholar] [CrossRef] [Green Version]
- Rosa-Ferreira, C.; Munro, S. Arl8 and SKIP act together to link lysosomes to kinesin-1. Dev. Cell 2011, 21, 1171–1178. [Google Scholar] [CrossRef] [Green Version]
- Griffin, E.F.; Caldwell, K.A.; Caldwell, G.A. Vacuolar protein sorting protein 41 (VPS41) at an intersection of endosomal traffic in neurodegenerative disease. Neural Regen. Res. 2019, 14, 1210–1212. [Google Scholar]
- Nguyen, A.P.T.; Moore, D.J. Understanding the GTPase activity of LRRK2: Regulation, function, and neurotoxicity. Adv. Neurobiol. 2017, 14, 71–88. [Google Scholar]
- Stafa, K.; Trancikova, A.; Webber, P.J.; Glauser, L.; West, A.B.; Moore, D.J. GTPase activity and neuronal toxicity of Parkinson’s disease-associated LRRK2 is regulated by ArfGAP1. PLoS Genet. 2012, 8, e1002526. [Google Scholar] [CrossRef]
- Nazam, F.; Shaikh, S.; Nazam, N.; Alshahrani, A.S.; Hasan, G.M.; Hassan, M.I. Mechanistic insights into the pathogenesis of neurodegenerative diseases: Towards the development of effective therapy. Mol. Cell. Biochem. 2021. [Google Scholar] [CrossRef]
- Paintlia, A.S.; Paintlia, M.K.; Singh, A.K.; Singh, I. Inhibition of Rho family functions by lovastatin promotes myelin repair in ameliorating experimental autoimmune encephalomyelitis. Mol. Pharmacol. 2008, 73, 1381–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Vergallo, A.; Caraci, F.; Cuello, A.C.; Lemercier, P.; Vellas, B.; Giudici, K.V.; Baldacci, F.; Hänisch, B.; Haberkamp, M.; et al. Future avenues for Alzheimer’s disease detection and therapy: Liquid biopsy, intracellular signaling modulation, systems pharmacology drug discovery. Neuropharmacology 2021, 185, 108081. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; O’Bryant, S.E.; Molinuevo, J.L.; Zetterberg, H.; Masters, C.L.; Lista, S.; Kiddle, S.J.; Batrla, R.; Blennow, K. Blood-based biomarkers for Alzheimer disease: Mapping the road to the clinic. Nat. Rev. Neurol. 2018, 14, 639–652. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| GTPases | Pathology | Role | Model Used | References |
|---|---|---|---|---|
| Rab1 | AD |
|
| [16] |
| PD |
|
| [17] | |
| PD |
|
| [18] | |
| PD |
|
| [19] | |
| PD |
|
| [20] | |
| ALS |
|
| [21] | |
| Rab5 | AD |
|
| [22] |
| AD |
|
| [23,24] | |
| AD, PD |
|
| [12] | |
| HD |
|
| [25] | |
| Rab7 | AD |
|
| [26,27] |
| AD |
|
| [28] | |
| AD |
|
| [29] | |
| PD |
|
| [30] | |
| PD |
|
| [31] | |
| PD |
|
| [32] | |
| MS |
|
| [33] | |
| CMT2B |
|
| [34] | |
| Arf | AD |
|
| [35] |
| AD |
|
| [36] | |
| Arl8 | AD |
|
| [37] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrazola Sastre, A.; Luque Montoro, M.; Lacerda, H.M.; Llavero, F.; Zugaza, J.L. Small GTPases of the Rab and Arf Families: Key Regulators of Intracellular Trafficking in Neurodegeneration. Int. J. Mol. Sci. 2021, 22, 4425. https://doi.org/10.3390/ijms22094425
Arrazola Sastre A, Luque Montoro M, Lacerda HM, Llavero F, Zugaza JL. Small GTPases of the Rab and Arf Families: Key Regulators of Intracellular Trafficking in Neurodegeneration. International Journal of Molecular Sciences. 2021; 22(9):4425. https://doi.org/10.3390/ijms22094425
Chicago/Turabian StyleArrazola Sastre, Alazne, Miriam Luque Montoro, Hadriano M. Lacerda, Francisco Llavero, and José L. Zugaza. 2021. "Small GTPases of the Rab and Arf Families: Key Regulators of Intracellular Trafficking in Neurodegeneration" International Journal of Molecular Sciences 22, no. 9: 4425. https://doi.org/10.3390/ijms22094425
APA StyleArrazola Sastre, A., Luque Montoro, M., Lacerda, H. M., Llavero, F., & Zugaza, J. L. (2021). Small GTPases of the Rab and Arf Families: Key Regulators of Intracellular Trafficking in Neurodegeneration. International Journal of Molecular Sciences, 22(9), 4425. https://doi.org/10.3390/ijms22094425