
Fabry Disease and the Heart: A Comprehensive Review

 , ,
, ,
Abstract
:1. Fabry Disease Overview
2. Cardiac Involvement in FD
2.1. Pathophysiology
2.2. Pathology
3. Cardiac Manifestations of FD
3.1. Hypertrophic Cardiomyopathy
3.1.1. Left Ventricular Hypertrophy
3.1.2. Left Ventricular Storage, Inflammation, and Fibrosis
3.1.3. Left Ventricular Function
3.1.4. Left Ventricular Obstruction
3.1.5. Right Ventricular Involvement
3.1.6. Atrial Involvement
3.1.7. Heart Failure
3.2. Dysrhythmias and Cardiac Conduction Disorders
3.2.1. Bradycardia, Chronotropic Incompetence, and Cardiac Conduction Disorders
3.2.2. Tachydysrhythmias
3.2.3. Cardiac Devices

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cardiac Manifestations | Frequencies | References |
|---|---|---|
| Palpitations |
|
|
| Syncope |
|
|
| Short PR interval |
|
|
| Bradycardia |
|
|
| Bradyarrhythmic events |
|
|
| PR interval > 200 ms |
|
|
| QRS duration > 120 ms |
|
|
| Right bundle branch block | In late-onset phenotype with predominant cardiac involvement
|
|
| Left anterior fascicular block | In late-onset phenotype with predominant cardiac involvement
|
|
| Bifascicular block | In late-onset phenotype with predominant cardiac involvement
|
|
| Complete AV block | In late-onset phenotype with predominant cardiac involvement
|
|
| Atrial Fibrillation |
|
|
|
| |
|
| |
In late-onset phenotype with predominant cardiac involvement
|
| |
| Atrial Flutter | In late-onset phenotype with predominant cardiac involvement
|
|
| Ventricular Arrhythmias |
|
|
|
| |
|
| |
Systematic review of the literature
|
| |
In late-onset phenotype with predominant cardiac involvement
|
| |
| SCD |
|
|
|
| |
Systematic review of the literature
|
| |
| Cardiac device |
|
|
|
| |
| Pacemaker |
|
|
|
| |
In late-onset phenotype with predominant cardiac involvement
|
| |
| ICD |
|
|
In late-onset phenotype with predominant cardiac involvement
|
|
3.3. Coronary Manifestations
3.4. Valvular Disease
3.5. Aortic Dilation
| Cardiac Manifestations | Frequencies | References |
|---|---|---|
| Angina |
|
|
|
| |
| Myocardial infarct |
|
|
|
| |
| Aortic valve dysfunction |
|
|
| Mitral valve dysfunction |
|
|
| Aortic dilation | At the sinuses of Valsalva
|
|
| Aortic aneurysm |
|
|
3.6. Cardiac Events
4. Late-Onset Phenotypes with Predominant Cardiac Involvement (“Cardiac Variants”)
5. Cardiac Treatment in FD
5.1. ERT
5.2. Migalastat
5.3. Supportive Treatment
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Desnick, R.J.; Ioannou, Y.A.; Eng, C.M. Alpha-galactosidase A deficiency: Fabry disease. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Childs, B., Kinzler, K.W., Vogelstein, B., Eds.; McGraw Hill: New York, NY, USA, 2001; pp. 3733–3774. [Google Scholar]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; Biegstraaten, M.; et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J. Am. Soc. Nephrol. 2017, 28, 1631–1641. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, O.; Gal, A.; Faria, R.; Gaspar, P.; Miltenberger-Miltenyi, G.; Gago, M.F.; Dias, F.; Martins, A.; Rodrigues, J.; Reimão, P.; et al. Founder effect of Fabry disease due to p.F113L mutation: Clinical profile of a late-onset phenotype. Mol. Genet. Metab. 2020, 129, 150–160. [Google Scholar] [CrossRef]
- Azevedo, O.; Gago, M.F.; Miltenberger-Miltenyi, G.; Robles, A.R.; Costa, M.A.; Pereira, O.; Vide, A.T.; Branco, G.C.; Simões, S.; Guimarães, M.J.; et al. Natural history of the late-onset phenotype of Fabry disease due to the p.F113L mutation. Mol. Genet. Metab. Rep. 2020, 22, 100565. [Google Scholar] [CrossRef] [PubMed]
- Deegan, P.B.; Baehner, A.F.; Romero, M.-Á.B.; A Hughes, D.; Kampmann, C.; Beck, M. Natural history of Fabry disease in females in the Fabry Outcome Survey. J. Med. Genet. 2005, 43, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Whybra, C.; Kampmann, C.; Willers, I.; Davies, J.; Winchester, B.; Kriegsmann, J.; Brühl, K.; Gal, A.; Bunge, S.; Beck, M. Anderson-Fabry disease: Clinical manifestations of disease in female heterozygotes. J. Inherit. Metab. Dis. 2001, 24, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Elleder, M. Subcellular, cellular, and organ pathology of Fabry disease. In Fabry Disease; Elstein, D., Altarescu, G., Beck, M., Eds.; Springer: Dordrecht, The Netherlands, 2010; pp. 39–79. [Google Scholar]
- Miller, J.J.; Kanack, A.J.; Dahms, N.M. Progress in the understanding and treatment of Fabry disease. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129437. [Google Scholar] [CrossRef] [PubMed]
- Lücke, T.; Höppner, W.; Schmidt, E.; Illsinger, S.; Das, A.M. Fabry disease: Reduced activities of respiratory chain enzymes with decreased levels of energy-rich phosphates in fibroblasts. Mol. Genet. Metab. 2004, 82, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.-S.; Meng, X.-L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, P.N.; Mucci, J.M.; Ceci, R.; Fossati, C.A.; Rozenfeld, P.A. Fabry disease peripheral blood immune cells release inflammatory cytokines: Role of globotriaosylceramide. Mol. Genet. Metab. 2013, 109, 93–99. [Google Scholar] [CrossRef]
- De Francesco, P.N.; Mucci, J.M.; Ceci, R.; Fossati, C.A.; Rozenfeld, P.A. Higher apoptotic state in Fabry disease peripheral blood mononuclear cells: Effect of globotriaosylceramide. Mol. Genet. Metab. 2011, 104, 319–324. [Google Scholar] [CrossRef]
- Squillaro, T.; Antonucci, I.; Alessio, N.; Esposito, A.; Cipollaro, M.; Melone, M.A.B.; Peluso, G.; Stuppia, L.; Galderisi, U. Impact of lysosomal storage disorders on biology of mesenchymal stem cells: Evidences from in vitro silencing of glucocerebrosidase (GBA) and alpha-galactosidase A (GLA) enzymes. J. Cell. Physiol. 2017, 232, 3454–3467. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Kim, J.A.; Na, H.-Y.; Cho, S.-E.; Park, S.; Jung, S.-C.; Suh, S.H. Globotriaosylceramide Induces Lysosomal Degradation of Endothelial K Ca 3.1 in Fabry Disease. Arter. Thromb. Vasc. Biol. 2014, 34, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Ferraz, M.J.; Marques, A.R.A.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; Van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrans, V.J.; Hibbs, R.G.; Burda, C.D. The heart in Fabry’s disease. Am. J. Cardiol. 1969, 24, 95–110. [Google Scholar] [CrossRef]
- Desnick, R.J.; Blieden, L.C.; Sharp, H.L.; Hofschire, P.J.; Moller, J.H. Cardiac valvular anomalies in Fabry disease. Clinical, morphologic, and biochemical studies. Circulation 1976, 54, 818–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A.E.; Schoorl, R.; Balk, A.G.; van der Heide, R. Cardiac manifestations of Fabry’s disease: Report of a case with mitral insufficiency and electrocardiographic evidence of myocardial infarction. Am. J. Cardiol. 1975, 36, 829–835. [Google Scholar] [CrossRef]
- Sheppard, M.N.; Cane, P.; Florio, R.; Kavantzas, N.; Close, L.; Shah, J.; Lee, P.; Elliott, P. A detailed pathologic examination of heart tissue from three older patients with Anderson–Fabry disease on enzyme replacement therapy. Cardiovasc. Pathol. 2010, 19, 293–301. [Google Scholar] [CrossRef]
- Takenaka, T.; Teraguchi, H.; Yoshida, A.; Taguchi, S.; Ninomiya, K.; Umekita, Y.; Yoshida, H.; Horinouchi, M.; Tabata, K.; Yonezawa, S.; et al. Terminal stage cardiac findings in patients with cardiac Fabry disease: An electrocardiographic, echocardiographic, and autopsy study. J. Cardiol. 2008, 51, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Frustaci, A.; Chimenti, C.; Doheny, D.; Desnick, R.J. Evolution of cardiac pathology in classic Fabry disease: Progressive cardiomyocyte enlargement leads to increased cell death and fibrosis, and correlates with severity of ventricular hypertrophy. Int. J. Cardiol. 2017, 248, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Verardo, R.; Grande, C.; Galea, N.; Piselli, P.; Carbone, I.; Alfarano, M.; Russo, M.A.; Chimenti, C. Immune-Mediated Myocarditis in Fabry Disease Cardiomyopathy. J. Am. Heart Assoc. 2018, 7, e009052. [Google Scholar] [CrossRef] [PubMed]
- Linhart, A.; Elliott, P.M. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart 2007, 93, 528–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Clarke, J.T.R.; Giugliani, R.; Elliott, P.; Linhart, A.; Beck, M.; Sunder-Plassman, G.; on behalf of the FOS Investigators. Natural course of Fabry disease: Changing pattern of causes of death in FOS—Fabry Outcome Survey. J. Med. Genet. 2009, 46, 548–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, C.M.; Fletcher, J.; Wilcox, W.R.; Waldek, S.; Scott, C.R.; Sillence, D.O.; Breunig, F.; Charrow, J.; Germain, D.P.; Nicholls, K.; et al. Fabry disease: Baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J. Inherit. Metab. Dis. 2007, 30, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Waldek, S.; Patel, M.R.; Banikazemi, M.; Lemay, R.; Lee, P. Life expectancy and cause of death in males and females with Fabry disease: Findings from the Fabry Registry. Genet. Med. 2009, 11, 790–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, O.; Marques, N.; Reis, L.; Cruz, I.; Craveiro, N.; Antunes, H.; Lourenço, C.; Gomes, R.; Guerreiro, R.A.; Faria, R.; et al. Predictors of Fabry disease in patients with hypertrophic cardiomyopathy: How to guide the diagnostic strategy? Am. Heart J. 2020, 226, 114–126. [Google Scholar] [CrossRef]
- Doheny, D.; Srinivasan, R.; Pagant, S.; Chen, B.; Yasuda, M.; Desnick, R.J. Fabry Disease: Prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995–2017. J. Med. Genet. 2018, 55, 261–268. [Google Scholar] [CrossRef]
- Mehta, A.; Widmer, U. Natural history of Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; Chapter 18; pp. 183–188. [Google Scholar]
- Kampmann, C.; Linhart, A.; Baehner, F.; Palecek, T.; Wiethoff, C.M.; Miebach, E.; Whybra, C.; Gal, A.; Bultas, J.; Beck, M. Onset and progression of the Anderson–Fabry disease related cardiomyopathy. Int. J. Cardiol. 2008, 130, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Lidove, O.; Barbey, F.; Niu, D.-M.; Brand, E.; Nicholls, K.; Bizjajeva, S.; Hughes, D.A. Fabry in the older patient: Clinical consequences and possibilities for treatment. Mol. Genet. Metab. 2016, 118, 319–325. [Google Scholar] [CrossRef]
- Linhart, A.; Kampmann, C.; Zamorano, J.L.; Sunder-Plassmann, G.; Beck, M.; Mehta, A.; Elliott, P.M.; on behalf of European FOS Investigators. Cardiac manifestations of Anderson-Fabry disease: Results from the international Fabry outcome survey. Eur. Heart J. 2007, 28, 1228–1235. [Google Scholar] [CrossRef] [Green Version]
- Linhart, A.; Paleček, T.; Bultas, J.; Ferguson, J.J.; Hrudová, J.; Karetová, D.; Zeman, J.; Ledvinová, J.; Poupětová, H.; Elleder, M.; et al. New insights in cardiac structural changes in patients with Fabry’s disease. Am. Heart J. 2000, 139, 1101–1108. [Google Scholar] [CrossRef]
- Tower-Rader, A.; Jaber, W.A. Multimodality Imaging Assessment of Fabry Disease. Circ. Cardiovasc. Imaging 2019, 12, e009013. [Google Scholar] [CrossRef]
- Hazari, H.; Belenkie, I.; Kryski, A.; White, J.A.; Oudit, G.Y.; Thompson, R.; Fung, T.; Dehar, N.; Khan, A. Comparison of Cardiac Magnetic Resonance Imaging and Echocardiography in Assessment of Left Ventricular Hypertrophy in Fabry Disease. Can. J. Cardiol. 2018, 34, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Kozor, R.; Grieve, S.M.; Tchan, M.C.; Callaghan, F.; Hamilton-Craig, C.; Denaro, C.; Moon, J.C.; A Figtree, G. Cardiac involvement in genotype-positive Fabry disease patients assessed by cardiovascular MR. Heart 2016, 102, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Satriano, A.; Afzal, Y.; Afzal, M.S.; Hassanabad, A.F.; Wu, C.; Dykstra, S.; Flewitt, J.; Feuchter, P.; Sandonato, R.; Heydari, B.; et al. Neural-Network-Based Diagnosis Using 3-Dimensional Myocardial Architecture and Deformation: Demonstration for the Differentiation of Hypertrophic Cardiomyopathy. Front. Cardiovasc. Med. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Adachi, K.; Yamashita, Y.; Toshima, H.; Koga, Y. Four cases of Fabry’s disease mimicking hypertrophic cardio-myopathy. J. Cardiol. 1988, 18, 705–718. [Google Scholar] [PubMed]
- Cianciulli, T.F.; Saccheri, M.C.; Fernández, S.P.; Fernández, C.C.; Rozenfeld, P.A.; Kisinovsky, I. Apical Left Ventricular Hypertrophy and Mid-Ventricular Obstruction in Fabry Disease. Echocardiography 2015, 32, 860–863. [Google Scholar] [CrossRef]
- Pieroni, M.; Chimenti, C.; De Cobelli, F.; Morgante, E.; Del Maschio, A.; Gaudio, C.; Russo, M.A.; Frustaci, A. Fabry’s Disease Cardiomyopathy: Echocardiographic detection of endomyocardial glycosphingolipid compartmentalization. J. Am. Coll. Cardiol. 2006, 47, 1663–1671. [Google Scholar] [CrossRef] [Green Version]
- Mundigler, G.; Gaggl, M.; Heinze, G.; Graf, S.; Zehetgruber, M.; Lajic, N.; Voigtländer, T.; Mannhalter, C.; Sunder-Plassmann, R.; Paschke, E.; et al. The endocardial binary appearance (’binary sign’) is an unreliable marker for echocardiographic detection of Fabry disease in patients with left ventricular hypertrophy. Eur. J. Echocardiogr. 2011, 12. [Google Scholar] [CrossRef] [Green Version]
- Niemann, M.; Liu, D.; Hu, K.; Herrmann, S.; Breunig, F.; Strotmann, J.; Störk, S.; Voelker, W.; Ertl, G.; Wanner, C.; et al. Prominent Papillary Muscles in Fabry Disease: A Diagnostic Marker? Ultrasound Med. Biol. 2011, 37, 37–43. [Google Scholar] [CrossRef]
- Hoigné, P.; Jost, C.A.; Duru, F.; Oechslin, E.; Seifert, B.; Widmer, U.; Frischknecht, B.; Jenni, R. Simple criteria for differentiation of Fabry disease from amyloid heart disease and other causes of left ventricular hypertrophy. Int. J. Cardiol. 2006, 111, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Kozor, R.; Callaghan, F.; Tchan, M.; Hamilton-Craig, C.; A Figtree, G.; Grieve, S.M. A disproportionate contribution of papillary muscles and trabeculations to total left ventricular mass makes choice of cardiovascular magnetic resonance analysis technique critical in Fabry disease. J. Cardiovasc. Magn. Reson. 2015, 17, 22. [Google Scholar] [CrossRef] [Green Version]
- Al-Arnawoot, A.; O’Brien, C.; Karur, G.R.; Nguyen, E.T.; Wasim, S.; Iwanochko, R.M.; Morel, C.F.; Hanneman, K. Clinical Significance of Papillary Muscles on Left Ventricular Mass Quantification Using Cardiac Magnetic Resonance Imaging: Reproducibility and Prognostic Value in Fabry Disease. J. Thorac. Imaging 2020. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.C.; Sachdev, B.; Elkington, A.G.; McKenna, W.J.; Mehta, A.; Pennell, D.J.; Leed, P.J.; Elliott, P.M. Gadolinium enhanced car-diovascular magnetic resonance in Anderson-Fabry disease. Evidence for a disease specific abnormality of the myocardi-al interstitium. Eur. Heart J. 2003, 24, 2151–2155. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.C.; Sheppard, M.; Reed, E.; Lee, P.; Elliott, P.M.; Pennell, D.J. The Histological Basis of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in a Patient with Anderson-Fabry Disease. J. Cardiovasc. Magn. Reson. 2006, 8, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Deva, D.P.; Hanneman, K.; Li, Q.; Ng, M.Y.; Wasim, S.; Morel, C.; Iwanochko, R.M.; Thavendiranathan, P.; Crean, A.M. Cardiovascular magnetic resonance demonstration of the spectrum of morphological phenotypes and patterns of myocardial scarring in Anderson-Fabry disease. J. Cardiovasc. Magn. Reson. 2016, 18, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Authors/Task Force Members; Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: The Task Force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef]
- Park, J.-H.; Kwon, D.H.; Starling, R.C.; Marwick, T.H. Role of Imaging in the Detection of Reversible Cardiomyopathy. J. Cardiovasc. Ultrasound 2013, 21, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Niemann, M.; Herrmann, S.; Hu, K.; Breunig, F.; Strotmann, J.; Beer, M.; Machann, W.; Voelker, W.; Ertl, G.; Wanner, C.; et al. Differences in Fabry Cardiomyopathy Between Female and Male Patients: Consequences for diagnostic assessment. JACC Cardiovasc. Imaging 2011, 4, 592–601. [Google Scholar] [CrossRef] [Green Version]
- Moonen, A.; Lal, S.; Ingles, J.; Yeates, L.; Semsarian, C.; Puranik, R. Prevalence of Anderson-Fabry disease in a cohort with unexplained late gadolinium enhancement on cardiac MRI. Int. J. Cardiol. 2020, 304, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Niemann, M.; Herrmann, S.; Kung, M.; Störk, S.; Waller, C.; Beer, M.; Breunig, F.; Wanner, C.; Voelker, W.; et al. A new echocardiographic approach for the detection of non-ischaemic fibrosis in hypertrophic myocardium. Eur. Heart J. 2007, 28, 3020–3026. [Google Scholar] [CrossRef] [Green Version]
- Krämer, J.; Niemann, M.; Liu, D.; Hu, K.; Machann, W.; Beer, M.; Wanner, C.; Ertl, G.; Weidemann, F. Two-dimensional speckle tracking as a non-invasive tool for identification of myocardial fibrosis in Fabry disease. Eur. Heart J. 2013, 34, 1587–1596. [Google Scholar] [CrossRef] [Green Version]
- Niemann, M.; Hartmann, T.; Namdar, M.; Breunig, F.; Beer, M.; Machann, W.; Herrmann, S.; Ertl, G.; Wanner, C.; Weidemann, F. Cross-sectional baseline analysis of electrocardiography in a large cohort of patients with untreated Fabry disease. J. Inherit. Metab. Dis. 2012, 36, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Nordin, S.; Kozor, R.; Bulluck, H.; Castelletti, S.; Rosmini, S.; Abdel-Gadir, A.; Baig, S.; Mehta, A.; Hughes, D.; Moon, J.C. Cardiac Fabry Disease With Late Gadolinium Enhancement Is a Chronic Inflammatory Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 68, 1707–1708. [Google Scholar] [CrossRef] [PubMed]
- Seydelmann, N.; Liu, D.; Krämer, J.; Drechsler, C.; Hu, K.; Nordbeck, P.; Schneider, A.; Störk, S.; Bijnens, B.; Ertl, G.; et al. High-Sensitivity Troponin: A Clinical Blood Biomarker for Staging Cardiomyopathy in Fabry Disease. J. Am. Heart Assoc. 2016, 5, e002839, Erratum in: J. Am. Heart Assoc. 2016, 5, e002114, doi:10.1161/JAHA.116.002114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yogasundaram, H.; Nikhanj, A.; Putko, B.N.; Boutin, M.; Jain-Ghai, S.; Khan, A.; Auray-Blais, C.; West, M.L.; Oudit, G.Y. Elevated Inflammatory Plasma Biomarkers in Patients With Fabry Disease: A Critical Link to Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2018, 7, e009098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augusto, J.B.; Nordin, S.; Vijapurapu, R.; Baig, S.; Bulluck, H.; Castelletti, S.; Alfarih, M.; Knott, K.; Captur, G.; Kotecha, T.; et al. Myocardial Edema, Myocyte Injury, and Disease Severity in Fabry Disease. Circ. Cardiovasc. Imaging 2020, 13, e010171. [Google Scholar] [CrossRef]
- Nappi, C.; Altiero, M.; Imbriaco, M.; Nicolai, E.; Giudice, C.A.; Aiello, M.; Diomiaiuti, C.T.; Pisani, A.; Spinelli, L.; Cuocolo, A. First experience of simultaneous PET/MRI for the early detection of cardiac involvement in patients with Anderson-Fabry disease. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 1025–1031. [Google Scholar] [CrossRef]
- Imbriaco, M.; Nappi, C.; Ponsiglione, A.; Pisani, A.; Dell’Aversana, S.; Nicolai, E.; Spinelli, L.; Aiello, M.; Diomiaiuti, C.T.; Riccio, E.; et al. Hybrid positron emission tomography-magnetic resonance imaging for assessing different stages of cardiac impairment in patients with Anderson–Fabry disease: AFFINITY study group. Eur. Heart J. Cardiovasc. Imaging 2019, 20, 1004–1011. [Google Scholar] [CrossRef]
- Spinelli, L.; Imbriaco, M.; Nappi, C.; Nicolai, E.; Giugliano, G.; Ponsiglione, A.; Diomiaiuti, T.C.; Riccio, E.; Duro, G.; Pisani, A.; et al. Early Cardiac Involvement Affects Left Ventricular Longitudinal Function in Females Carrying α-Galactosidase A Mutation: Role of Hybrid Positron Emission Tomography and Magnetic Resonance Imaging and Speck-le-Tracking Echocardiography. Circ. Cardiovasc. Imaging 2018, 11, e007019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidemann, F.; Niemann, M.; Breunig, F.; Herrmann, S.; Beer, M.; Störk, S.; Voelker, W.; Ertl, G.; Wanner, C.; Strotmann, J. Long-Term Effects of Enzyme Replacement Therapy on Fabry Cardiomyopathy: Evidence for a better outcome with early treatment. Circulation 2009, 119, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krämer, J.; Niemann, M.; Störk, S.; Frantz, S.; Beer, M.; Ertl, G.; Wanner, C.; Weidemann, F. Relation of Burden of Myocardial Fibrosis to Malignant Ventricular Arrhythmias and Outcomes in Fabry Disease. Am. J. Cardiol. 2014, 114, 895–900. [Google Scholar] [CrossRef] [Green Version]
- Baig, S.; Edward, N.C.; Kotecha, D.; Liu, B.; Nordin, S.; Kozor, R.; Moon, J.C.; Geberhiwot, T.; Steeds, R.P. Ventricular arrhythmia and sudden cardiac death in Fabry disease: A systematic review of risk factors in clinical practice. Europace 2017, 20, f153–f161. [Google Scholar] [CrossRef]
- Sado, D.M.; White, S.K.; Piechnik, S.K.; Banypersad, S.M.; Treibel, T.; Captur, G.; Fontana, M.; Maestrini, V.; Flett, A.S.; Robson, M.D.; et al. Identification and Assessment of Anderson-Fabry Disease by Cardiovascular Magnetic Resonance Noncontrast Myocardial T1 Mapping. Circ. Cardiovasc. Imaging 2013, 6, 392–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordin, S.; Kozor, R.; Baig, S.; Abdel-Gadir, A.; Medina-Menacho, K.; Rosmini, S.; Captur, G.; Tchan, M.; Geberhiwot, T.; Murphy, E.; et al. Cardiac Phenotype of Prehypertrophic Fabry Disease. Circ. Cardiovasc. Imaging 2018, 11, e007168. [Google Scholar] [CrossRef] [Green Version]
- Camporeale, A.; Pieroni, M.; Pieruzzi, F.; Lusardi, P.; Pica, S.; Spada, M.; Mignani, R.; Burlina, A.; Bandera, F.; Guazzi, M.; et al. Predictors of Clinical Evolution in Prehypertrophic Fabry Disease. Circ. Cardiovasc. Imaging 2019, 12, e008424. [Google Scholar] [CrossRef]
- Pica, S.; Sado, D.M.; Maestrini, V.; Fontana, M.; White, S.K.; Treibel, T.; Captur, G.; Anderson, S.; Piechnik, S.K.; Robson, M.D.; et al. Reproducibility of native myocardial T1 mapping in the assessment of Fabry disease and its role in early detection of cardiac involvement by cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2014, 16, 1–9. [Google Scholar] [CrossRef]
- Vijapurapu, R.; Nordin, S.; Baig, S.; Liu, B.; Rosmini, S.; Augusto, J.; Tchan, M.; A Hughes, D.; Geberhiwot, T.; Moon, J.C.; et al. Global longitudinal strain, myocardial storage and hypertrophy in Fabry disease. Heart 2018, 105, 470–476. [Google Scholar] [CrossRef]
- Messroghli, D.R.; Moon, J.C.; Ferreira, V.M.; Grosse-Wortmann, L.; He, T.; Kellman, P.; Mascherbauer, J.; Nezafat, R.; Salerno, M.; Schelbert, E.B.; et al. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: A consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J. Cardiovasc. Magn. Reson. 2017, 19, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Pagano, J.J.; Chow, K.; Khan, A.; Michelakis, E.; Paterson, I.; Oudit, G.Y.; Thompson, R.B. Reduced Right Ventricular Native Myocardial T1 in Anderson-Fabry Disease: Comparison to Pulmonary Hypertension and Healthy Controls. PLoS ONE 2016, 11, e0157565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordin, S.; Kozor, R.; Medina-Menacho, K.; Abdel-Gadir, A.; Baig, S.; Sado, D.M.; Lobascio, I.; Murphy, E.; Lachmann, R.H.; Mehta, A.; et al. Proposed Stages of Myocardial Phenotype Development in Fabry Disease. JACC: Cardiovasc. Imaging 2019, 12, 1673–1683. [Google Scholar] [CrossRef]
- Kawano, M.; Takenaka, T.; Otsuji, Y.; Teraguchi, H.; Yoshifuku, S.; Yuasa, T.; Yu, B.; Miyata, M.; Hamasaki, S.; Minagoe, S.; et al. Significance of Asymmetric Basal Posterior Wall Thinning in Patients With Cardiac Fabry’s Disease. Am. J. Cardiol. 2007, 99, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Niemann, M.; Breunig, F.; Beer, M.; Hu, K.; Liu, D.; Emmert, A.; Herrmann, S.; Ertl, G.; Wanner, C.; Takenaka, T.; et al. Tei Index in Fabry Disease. J. Am. Soc. Echocardiogr. 2011, 24, 1026–1032. [Google Scholar] [CrossRef]
- Liu, D.; Oder, D.; Salinger, T.; Hu, K.; Müntze, J.; Weidemann, F.; Herrmann, S.; Ertl, G.; Wanner, C.; Frantz, S.; et al. Association and diagnostic utility of diastolic dysfunction and myocardial fibrosis in patients with Fabry disease. Open Heart 2018, 5, e000803. [Google Scholar] [CrossRef]
- Torralba-Cabeza, M.-Á.; Olivera, S.; Hughes, D.A.; Pastores, G.M.; Mateo, R.N.; Pérez-Calvo, J.-I. Cystatin C and NT-proBNP as prognostic biomarkers in Fabry disease. Mol. Genet. Metab. 2011, 104, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.; Ho, C.Y.; Skali, H.; Abichandani, R.; Wilcox, W.R.; Banikazemi, M.; Packman, S.; Sims, K.; Solomon, S.D. Cardiovascular manifestations of Fabry disease: Relationships between left ventricular hypertrophy, disease severity, and alpha-galactosidase A activity. Eur. Heart J. 2010, 31, 1088–1097. [Google Scholar] [CrossRef] [Green Version]
- Pieroni, M.; Chimenti, C.; Ricci, R.; Sale, P.; Russo, M.A.; Frustaci, A. Early Detection of Fabry Cardiomyopathy by Tissue Doppler Imaging. Circulation 2003, 107, 1978–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saccheri, M.C.; Cianciulli, T.F.; Lax, J.A.; Gagliardi, J.A.; Cáceres, G.L.; Quarin, A.E.; Kisinovsky, I.; Rozenfeld, P.A.; Reisin, R.C. Aadelfa Two-Dimensional Speckle Tracking Echocardiography for Early Detection of Myocardial Damage in Young Patients with Fabry Disease. Echocardiography 2013, 30. [Google Scholar] [CrossRef]
- Spinelli, L.; Giugliano, G.; Imbriaco, M.; Esposito, G.; Nappi, C.; Riccio, E.; Ponsiglione, A.; Pisani, A.; Cuocolo, A.; Trimarco, B. Left ventricular radial strain impairment precedes hypertrophy in Anderson–Fabry disease. Int. J. Cardiovasc. Imaging 2020, 36, 1465–1476. [Google Scholar] [CrossRef]
- Zada, M.; Lo, Q.; Boyd, A.C.; Bradley, S.; Devine, K.; Denaro, C.P.; Sadick, N.; Richards, D.A.; Tchan, M.C.; Thomas, L. Basal Segmental Longitudinal Strain: A Marker of Subclinical Myocardial Involvement in Anderson-Fabry Disease. J. Am. Soc. Echocardiogr. 2020, 24. [Google Scholar] [CrossRef]
- Shanks, M.; Thompson, R.B.; Paterson, I.D.; Putko, B.; Khan, A.; Chan, A.; Becher, H.; Oudit, G.Y. Systolic and Diastolic Function Assessment in Fabry Disease Patients Using Speckle-Tracking Imaging and Comparison with Conventional Echocardiographic Measurements. J. Am. Soc. Echocardiogr. 2013, 26, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Cianciulli, T.F.; Saccheri, M.C.; Rísolo, M.A.; Lax, J.A.; Méndez, R.J.; Morita, L.A.; Beck, M.A.; Kazelián, L.R. Mechanical dispersion in Fabry disease assessed with speckle tracking echocardiography. Echocardiography 2020, 37, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Réant, P.; Testet, E.; Reynaud, A.; Bourque, C.; Michaud, M.; Rooryck, C.; Goizet, C.; Lacombe, D.; De-Précigout, V.; Peyrou, J.; et al. Characterization of Fabry Disease cardiac involvement according to longitudinal strain, cardiometabolic exercise test, and T1 mapping. Int. J. Cardiovasc. Imaging 2020, 36, 1333–1342. [Google Scholar] [CrossRef] [PubMed]
- Gruner, C.; Verocai, F.; Carasso, S.; Vannan, M.A.; Jamorski, M.; Clarke, J.T.; Care, M.; Iwanochko, R.M.; Rakowski, H. Systolic Myocardial Mechanics in Patients with Anderson-Fabry Disease with and without Left Ventricular Hypertrophy and in Comparison to Nonobstructive Hypertrophic Cardiomyopathy. Echocardiography 2012, 29, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Graziani, F.; Lillo, R.; Panaioli, E.; Spagnoletti, G.; Pieroni, M.; Ferrazzi, P.; Camporeale, A.; Verrecchia, E.; Sicignano, L.L.; Manna, R.; et al. Evidence of evolution towards left midventricular obstruction in severe Anderson–Fabry cardiomyopathy. ESC Heart Fail. 2021, 8, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Calcagnino, M.; O’Mahony, C.; Coats, C.; Cardona, M.; Garcia, A.; Janagarajan, K.; Mehta, A.; Hughes, D.; Murphy, E.; Lachmann, R.; et al. Exercise-Induced Left Ventricular Outflow Tract Obstruction in Symptomatic Patients with Anderson-Fabry Disease. J. Am. Coll. Cardiol. 2011, 58, 88–89. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, L.; Nicolai, E.; Acampa, W.; Imbriaco, M.; Pisani, A.; Rao, M.A.E.; Scopacasa, F.; Cianciaruso, B.; De Luca, N.; Cuocolo, A. Cardiac performance during exercise in patients with Fabry’s disease. Eur. J. Clin. Investig. 2008, 38, 910–917. [Google Scholar] [CrossRef]
- Niemann, M.; Breunig, F.; Beer, M.; Herrmann, S.; Strotmann, J.; Hu, K.; Emmert, A.; Voelker, W.; Ertl, G.; Wanner, C.; et al. The right ventricle in Fabry disease: Natural history and impact of enzyme replacement therapy. Heart 2010, 96, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Graziani, F.; Laurito, M.; Pieroni, M.; Pennestrì, F.; Lanza, G.A.; Coluccia, V.; Camporeale, A.; Pedicino, D.; Verrecchia, E.; Manna, R.; et al. Right Ventricular Hypertrophy, Systolic Function, and Disease Severity in Anderson-Fabry Disease: An Echocardiographic Study. J. Am. Soc. Echocardiogr. 2017, 30, 282–291. [Google Scholar] [CrossRef]
- Morris, D.A.; Blaschke, D.; Canaan-Kühl, S.; Krebs, A.; Knobloch, G.; Walter, T.C.; Haverkamp, W. Global cardiac alterations detected by speckle-tracking echocardiography in Fabry disease: Left ventricular, right ventricular, and left atrial dysfunction are common and linked to worse symptomatic status. Int. J. Cardiovasc. Imaging 2014, 31, 301–313. [Google Scholar] [CrossRef]
- Graziani, F.; Lillo, R.; Panaioli, E.; Pieroni, M.; Camporeale, A.; Verrecchia, E.; Sicignano, L.L.; Manna, R.; Lombardo, A.; Lanza, G.A.; et al. Prognostic significance of right ventricular hypertrophy and systolic function in Anderson–Fabry disease. ESC Heart Fail. 2020, 7, 1605–1614. [Google Scholar] [CrossRef]
- Boyd, A.C.; Lo, Q.; Devine, K.; Tchan, M.C.; Sillence, D.O.; Sadick, N.; Richards, D.A.; Thomas, L. Left Atrial Enlargement and Reduced Atrial Compliance Occurs Early in Fabry Cardiomyopathy. J. Am. Soc. Echocardiogr. 2013, 26, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Esposito, R.; Russo, C.; Santoro, C.; Cocozza, S.; Riccio, E.; Sorrentino, R.; Pontillo, G.; Luciano, F.; Imbriaco, M.; Brunetti, A.; et al. Association between Left Atrial Deformation and Brain Involvement in Patients with Anderson-Fabry Disease at Diagnosis. J. Clin. Med. 2020, 9, 2741. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.S.; Hughes, D.A.; Sachdev, B.; Tome, M.; Ward, D.; Lee, P.; Mehta, A.B.; Elliott, P.M. Prevalence and Clinical Significance of Cardiac Arrhythmia in Anderson-Fabry Disease. Am. J. Cardiol. 2005, 96, 842–846. [Google Scholar] [CrossRef]
- Coats, C.J.; Parisi, V.; Ramos, M.; Janagarajan, K.; O’Mahony, C.; Dawnay, A.; Lachmann, R.H.; Murphy, E.; Mehta, A.; Hughes, D.; et al. Role of Serum N-Terminal Pro-Brain Natriuretic Peptide Measurement in Diagnosis of Cardiac Involvement in Patients With Anderson-Fabry Disease. Am. J. Cardiol. 2013, 111, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.; O’Mahony, C.; Hughes, D.; Rahman, M.S.; Coats, C.; Murphy, E.; Lachmann, R.; Mehta, A.; Elliott, P.M. Clinical and genetic predictors of major cardiac events in patients with Anderson–Fabry Disease. Heart 2015, 101, 961–966. [Google Scholar] [CrossRef]
- Kampmann, C.; Wiethoff, C.M.; Whybra, C.; Baehner, F.A.; Mengel, E.; Beck, M. Cardiac manifestations of Anderson-Fabry disease in children and adolescents. Acta Paediatr. 2008, 97, 463–469. [Google Scholar] [CrossRef]
- Frustaci, A.; Morgante, E.; Russo, M.A.; Scopelliti, F.; Grande, C.; Verardo, R.; Franciosa, P.; Chimenti, C. Pathology and Function of Conduction Tissue in Fabry Disease Cardiomyopathy. Circ. Arrhythmia Electrophysiol. 2015, 8, 799–805. [Google Scholar] [CrossRef] [Green Version]
- Roudebush, C.P.; Foerster, J.M.; Bing, O.H.L. The Abbreviated Pr Interval of Fabry’s Disease. N. Engl. J. Med. 1973, 289, 357–358. [Google Scholar] [CrossRef]
- Omar, A.R.; Harris, L.; Cameron, D.A.; Chauhan, V.S. WPW and Fabry’s disease: Evidence for atrioventricular and atriohisian acces-sory pathway conduction. HeartRhythm 2006, 3, S214. [Google Scholar]
- Jastrzebski, M.; Bacior, B.; Dimitrow, P.P.; Kawecka-Jaszcz, K. Electrophysiological study in a patient with Fabry disease and a short PQ interval. Europace 2006, 8, 1045–1047. [Google Scholar] [CrossRef]
- Waldek, S. PR Interval and the Response to Enzyme-Replacement Therapy for Fabry’s Disease. N. Engl. J. Med. 2003, 348, 1186–1187. [Google Scholar] [CrossRef]
- Wolf, C.M.; Arad, M.; Ahmad, F.; Sanbe, A.; Bernstein, S.A.; Toka, O.; Konno, T.; Morley, G.; Robbins, J.; Seidman, J.; et al. Reversibility of PRKAG2 Glycogen-Storage Cardiomyopathy and Electrophysiological Manifestations. Circulation 2008, 117, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Namdar, M.; Kampmann, C.; Steffel, J.; Walder, D.; Holzmeister, J.; Lüscher, T.F.; Jenni, R.; Duru, F. PQ Interval in Patients With Fabry Disease. Am. J. Cardiol. 2010, 105, 753–756. [Google Scholar] [CrossRef]
- Mehta, J.; Tuna, N.; Moller, J.H.; Desnick, R.J. Electrocardiographic and Vectorcardiographic Observations in Fabrys Disease. Adv. Cardiol. 1977, 21, 220–222. [Google Scholar] [CrossRef]
- Ikari, Y.; Kuwako, K.; Yamaguchi, T. Fabry’s disease with complete atrioventricular block: Histological evidence of involvement of the conduction system. Heart 1992, 68, 323–325. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, C.; Coats, C.; Cardona, M.; Garcia, A.; Calcagnino, M.; Murphy, E.; Lachmann, R.; Mehta, A.; Hughes, D.; Elliott, P.M. Incidence and predictors of anti-bradycardia pacing in patients with Anderson-Fabry disease. Europace 2011, 13, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Lobo, T.; Morgan, J.; Bjorksten, A.; Nicholls, K.; Grigg, L.; Centra, E.; Becker, G. Cardiovascular testing in Fabry disease: Exercise capacity reduction, chronotropic incompetence and improved anaerobic threshold after enzyme replacement. Intern. Med. J. 2008, 38, 407–414. [Google Scholar] [CrossRef]
- Powell, A.W.; Jefferies, J.L.; Hopkin, R.J.; Mays, W.A.; Goa, Z.; Chin, C. Cardiopulmonary fitness assessment on maximal and submaximal exercise testing in patients with Fabry disease. Am. J. Med. Genet. Part A 2018, 176, 1852–1857. [Google Scholar] [CrossRef]
- Di, L.Z.; Pichette, M.; Nadeau, R.; Bichet, D.G.; Poulin, F. Severe bradyarrhythmia linked to left atrial dysfunction in Fabry disease-A cross-sectional study. Clin. Cardiol. 2018, 41, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Chimenti, C.; Russo, M.A.; Frustaci, A. Atrial biopsy evidence of Fabry disease causing lone atrial fibrillation. Heart 2010, 96, 1782–1783. [Google Scholar] [CrossRef]
- Pinderski, L.J.; Strotmann, J. Congestive heart failure in Fabry cardiomyopathy: Natural history experience in an interna-tional cohort of 1448 patients. J. Heart Lung Transpl. 2006, 25, S70. [Google Scholar] [CrossRef]
- Hanneman, K.; Karur, G.R.; Wasim, S.; Wald, R.M.; Iwanochko, R.M.; Morel, C.F. Left Ventricular Hypertrophy and Late Gadolinium Enhancement at Cardiac MRI Are Associated with Adverse Cardiac Events in Fabry Disease. Radiology 2020, 294, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Chimenti, C. Images in cardiovascular medicine. Cryptogenic Ventricular Arrhythmias and Sudden Death by Fabry Disease: Prominent Infiltration of Cardiac Conduction Tissue. Circulation 2007, 116, e350–e351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, H.; Yamagata, K.; Noda, T.; Satomi, K. Endocardial and epicardial substrates of ventricular tachycardia in a patient with Fabry disease. Heart Rhythm. 2011, 8, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Niemann, M.; Störk, S.; Breunig, F.; Beer, M.; Sommer, C.; Herrmann, S.; Ertl, G.; Wanner, C. Long-term outcome of enzyme-replacement therapy in advanced F abry disease: Evidence for disease progression towards serious complications. J. Intern. Med. 2013, 274, 331–341. [Google Scholar] [CrossRef]
- Imbriaco, M.; Pellegrino, T.; Piscopo, V.; Petretta, M.; Ponsiglione, A.; Nappi, C.; Puglia, M.; Dell’Aversana, S.; Riccio, E.; Spinelli, L.; et al. Cardiac sympathetic neuronal damage precedes myocardial fibrosis in patients with Anderson-Fabry disease. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 2266–2273. [Google Scholar] [CrossRef] [Green Version]
- Weidemann, F.; Maier, S.K.; Störk, S.; Brunner, T.; Liu, D.; Hu, K.; Seydelmann, N.; Schneider, A.; Becher, J.; Canan-Kühl, S.; et al. Usefulness of an Implantable Loop Recorder to Detect Clinically Relevant Arrhythmias in Patients with Advanced Fabry Cardiomyopathy. Am. J. Cardiol. 2016, 118, 264–274. [Google Scholar] [CrossRef] [Green Version]
- Sené, T.; Lidove, O.; Sebbah, J.; Darondel, J.-M.; Picard, H.; Aaron, L.; Fain, O.; Zenone, T.; Joly, D.; Charron, P.; et al. Cardiac device implantation in Fabry disease: A retrospective monocentric study. Medicine 2016, 95, e4996. [Google Scholar] [CrossRef]
- Vijapurapu, R.; Geberhiwot, T.; Jovanovic, A.; Baig, S.; Nordin, S.; Kozor, R.; Leyva, F.; Kotecha, D.; Wheeldon, N.; Deegan, P.; et al. Study of indications for cardiac device implantation and utilisation in Fabry cardiomyopathy. Heart 2019, 105, 1825–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, D.; Robertson, P.; Kay, G.N.; Jackson, L.; Warnock, D.G.; Plumb, V.J.; Tallaj, J.A. Arrhythmias in Fabry Cardiomyopathy. Clin. Cardiol. 2012, 35, 738–740. [Google Scholar] [CrossRef]
- Chimenti, C.; Morgante, E.; Tanzilli, G.; Mangieri, E.; Critelli, G.; Gaudio, C.; Russo, M.A.; Frustaci, A. Angina in Fabry Disease Reflects Coronary Small Vessel Disease. Circ. Heart Fail. 2008, 1, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Kalliokoski, R.J.; Kalliokoski, K.K.; Sundell, J.; Engblom, E.; Penttinen, M.; Kantola, I.; Raitakari, O.T.; Knuuti, J.; Nuutila, P. Impaired myocardial perfusion reserve but preserved peripheral endothelial function in patients with Fabry disease. J. Inherit. Metab. Dis. 2005, 28, 563–573. [Google Scholar] [CrossRef]
- Elliott, P.M.; Kindler, H.; Shah, J.S.; Sachdev, B.; E Rimoldi, O.; Thaman, R.; Tome, M.T.; McKenna, W.J.; Lee, P.; Camici, P.G. Coronary microvascular dysfunction in male patients with Anderson-Fabry disease and the effect of treatment with alphagalactosidase A. Heart 2005, 92, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Tomberli, B.; Cecchi, F.; Sciagrà, R.; Berti, V.; Lisi, F.; Torricelli, F.; Morrone, A.; Castelli, G.; Yacoub, M.H.; Olivotto, I. Coronary microvascular dysfunction is an early feature of cardiac involvement in patients with Anderson-Fabry disease. Eur. J. Heart Fail. 2013, 15, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Knott, K.D.; Augusto, J.B.; Nordin, S.; Kozor, R.; Camaioni, C.; Xue, H.; Hughes, R.K.; Manisty, C.; Brown, L.A.; Kellman, P.; et al. Quantitative Myocardial Perfusion in Fabry Disease. Circ. Cardiovasc. Imaging 2019, 12, e008872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitani, Y.; Nakagawa, N.; Sakamoto, N.; Takeuchi, T.; Takahashi, F.; Momosaki, K.; Nakamura, K.; Endo, F.; Maruyama, H.; Hasebe, N. Unexpectedly High Prevalence of Coronary Spastic Angina in Patients with Anderson-Fabry Disease. Circ. J. 2019, 83, 481–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, T.; Kawai, M.; Matsui, T.; Seo, A.; Aizawa, O.; Hongo, K.; Shibata, T.; Yoshida, S.; Okamura, T.; Nishikawa, T.; et al. Vasospastic Angina in a Patient with Fabry’s Disease Who Showed Normal Coronary Angiographic Findings. Jpn. Circ. J. 1996, 60, 315–318. [Google Scholar] [CrossRef] [Green Version]
- Kodama, K.; Ozawa, T.; Dochi, K.; Ueno, Y. Ventricular fibrillation associated with vasospastic angina pectoris in Fabry disease: A case report. Eur. Heart J. Case Rep. 2019, 3, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Linhart, A. The heart in Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; Chapter 18; pp. 189–201. [Google Scholar]
- Graziani, F.; Lillo, R.; Panaioli, E.; Spagnoletti, G.; Bruno, I.; Leccisotti, L.; Marano, R.; Manna, R.; Crea, F. Massive Coronary Microvascular Dysfunction in Severe Anderson-Fabry Disease Cardiomyopathy. Circ. Cardiovasc. Imaging 2019, 12, e009104. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Rapkiewicz, A.; Abu-Asab, M.; Ries, M.; Askari, H.; Tsokos, M.; Quezado, M. Pathological findings in a patient with Fabry disease who died after 2.5 years of enzyme replacement. Virchows Arch. 2005, 448, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Frustaci, A.; Russo, M.A.; Francone, M.; Chimenti, C. Microvascular Angina as Prehypertrophic Presentation of Fabry Disease Cardiomyopathy. Circulation 2014, 130, 1530–1531. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.R.; Cecchi, F.; Cizmarik, M.; Kantola, I.; Linhart, A.; Nicholls, K.; Strotmann, J.; Tallaj, J.; Tran, T.C.; West, M.L.; et al. Cardiovascular Events in Patients With Fabry Disease: Natural history data from the fabry registry. J. Am. Coll. Cardiol. 2011, 57, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Hernández, A.; Diez-López, C.; Azevedo, O.; Palomino-Doza, J.; Alfonso, F.; Fuentes-Cañamero, M.E.; Jiménez, M.V.M.; Rodríguez, C.C.; Ruz, A.; Tirón, C.; et al. Screening of Fabry Disease in Patients with Chest Pain Without Obstructive Coronary Artery Disease. J. Cardiovasc. Transl. Res. 2021, 1–3. [Google Scholar] [CrossRef]
- Sakuraba, H.; Yanagawa, Y.; Igarashi, T.; Suzuki, Y.; Suzuki, T.; Watanabe, K.; Ieki, K.; Shimoda, K.; Yamanaka, T. Cardiovascular manifestations in Fabry’s disease. A high incidence of mitral valve prolapse in hemizygotes and heterozygotes. Clin. Genet. 1986, 29, 276–283. [Google Scholar] [CrossRef]
- Barbey, F.; Qanadli, S.D.; Juli, C.; Brakch, N.; Palaček, T.; Rizzo, E.; Jeanrenaud, X.; Eckhardt, B.; Linhart, A. Aortic remodelling in Fabry disease. Eur. Heart J. 2009, 31, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, L.; Giugliano, G.; Pisani, A.; Imbriaco, M.; Riccio, E.; Russo, C.; Cuocolo, A.; Trimarco, B.; Esposito, G. Does left ventricular function predict cardiac outcome in Anderson–Fabry disease? Int. J. Cardiovasc. Imaging 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Talbot, A.S.; Lewis, N.T.; Nicholls, K.M. Cardiovascular outcomes in Fabry disease are linked to severity of chronic kidney disease. Heart 2014, 101, 287–293. [Google Scholar] [CrossRef]
- Rosmini, S.; Biagini, E.; O’Mahony, C.; Bulluck, H.; Ruozi, N.; Lopes, L.R.; Guttmann, O.; Reant, P.; Quarta, C.C.; Pantazis, A.; et al. Relationship between aetiology and left ventricular systolic dysfunction in hypertrophic cardiomyopathy. Heart 2016, 103, 300–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linhart, A.; Germain, D.P.; Olivotto, I.; Akhtar, M.M.; Anastasakis, A.; Hughes, D.; Namdar, M.; Pieroni, M.; Hagège, A.; Cecchi, F.; et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur. J. Heart Fail. 2020, 22, 1076–1096. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.M.; Ashley, G.A.; Burgert, T.S.; Enriquez, A.L.; D’Souza, M.; Desnick, R.J. Fabry disease: Thirty-five mutations in the alpha-galactosidase A gene in patients with classic and variant phenotypes. Mol. Med. 1997, 3, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Bishop, D.F.; Grabowski, G.A.; Desnick, R.J. Fabry disease: An asymptomatic hemizygote with significant residual a-galactosidase activity. Am. J. Hum. Genet. 1981, 33, 71A. [Google Scholar]
- Eng, C.M.; Resnick-Silverman, L.A.; Niehaus, D.J.; Astrin, K.H.; Desnick, R.J. Nature and frequency of mutations in the alpha-galactosidase A gene that cause Fabry disease. Am. J. Hum. Genet. 1993, 53, 1186–1197. [Google Scholar] [PubMed]
- Germain, D.P.; Brand, E.; Burlina, A.; Cecchi, F.; Garman, S.C.; Kempf, J.; Laney, D.A.; Linhart, A.; Maródi, L.; Nicholls, K.; et al. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: A multicenter Fabry Registry study. Mol. Genet. Genom. Med. 2018, 6, 492–503. [Google Scholar] [CrossRef] [Green Version]
- LaValle, L.; Thomas, A.S.; Beaton, B.; Ebrahim, H.; Reed, M.; Ramaswami, U.; Elliott, P.; Mehta, A.B.; Hughes, D.A. Phenotype and biochemical heterogeneity in late onset Fabry disease defined by N215S mutation. PLoS ONE 2018, 13, e0193550. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Nakao, S.; Minamikawa-Tachino, R.; Desnick, R.J.; Fan, J.-Q. Alternative Splicing in the alpha-Galactosidase A Gene: Increased Exon Inclusion Results in the Fabry Cardiac Phenotype. Am. J. Hum. Genet. 2002, 70, 994–1002. [Google Scholar] [CrossRef] [Green Version]
- Nakao, S.; Takenaka, T.; Maeda, M.; Kodama, C.; Tanaka, A.; Tahara, M.; Yoshida, A.; Kuriyama, M.; Hayashibe, H.; Sakuraba, H.; et al. An Atypical Variant of Fabry’s Disease in Men with Left Ventricular Hypertrophy. N. Engl. J. Med. 1995, 333, 288–293. [Google Scholar] [CrossRef]
- Visoiu, I.-S.; Ciobanu, A.O.; Nicula, A.I.; Iascone, M.; Jurcut, R.; Vinereanu, D.; Rimbas, R.C. Severe Late-Onset Fabry Cardiomyopathy Unmasked by a Multimodality Imaging Approach. Circ. Cardiovasc. Imaging 2019, 12, e009709. [Google Scholar] [CrossRef]
- Adalsteinsdottir, B.; Palsson, R.; Desnick, R.J.; Gardarsdottir, M.; Teekakirikul, P.; Maron, M.; Appelbaum, E.; Neisius, U.; Maron, B.J.; Burke, M.A.; et al. Fabry Disease in Families with Hypertrophic Cardiomyopathy: Clinical Manifestations in the Clas-sic and Later-Onset Phenotypes. Circ. Cardiovasc. Genet. 2017, 10, e001639, Erratum in: Circ. Cardiovasc. Genet. 2017, 10, e000039. [Google Scholar] [CrossRef] [Green Version]
- Csányi, B.; Hategan, L.; Nagy, V.; Obál, I.; Varga, E.T.; Borbás, J.; Tringer, A.; Eichler, S.; Forster, T.; Rolfs, A.; et al. Identification of a Novel GLA Gene Mutation, p.Ile239Met, in Fabry Disease With a Predominant Cardiac Phenotype. Int. Heart J. 2017, 58, 454–458. [Google Scholar] [CrossRef] [Green Version]
- Ishii, S.; Kase, R.; Sakuraba, H.; Suzuki, Y. Characterization of a Mutant alpha-Galactosidase Gene Product for the Late-Onset Cardiac Form of Fabry Disease. Biochem. Biophys. Res. Commun. 1993, 197, 1585–1589. [Google Scholar] [CrossRef]
- Kase, R.; Bierfreund, U.; Klein, A.; Kolter, T.; Utsumi, K.; Itoh, K.; Sandhoff, K.; Sakuraba, H. Characterization of two alpha-galactosidase mutants (Q279E and R301Q) found in an atypical variant of Fabry disease. Biochim. Biophys. Acta Mol. Basis Dis. 2000, 1501, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Yoshitama, T.; Nakao, S.; Takenaka, T.; Teraguchi, H.; Sasaki, T.; Kodama, C.; Tanaka, A.; Kisanuki, A.; Tei, C. Molecular genetic, biochemical, and clinical studies in three families with cardiac Fabry’s disease. Am. J. Cardiol. 2001, 87, 71–75. [Google Scholar] [CrossRef]
- Von Scheidt, W.; Eng, C.M.; Fitzmaurice, T.F.; Erdmann, E.; Hübner, G.; Olsen, E.G.; Christomanou, H.; Kandolf, R.; Bishop, D.F.; Desnick, R.J. An Atypical Variant of Fabry’s Disease with Manifestations Confined to the Myocardium. N. Engl. J. Med. 1991, 324, 395–399. [Google Scholar] [CrossRef]
- Sakuraba, H.; Oshima, A.; Fukuhara, Y.; Shimmoto, M.; Nagao, Y.; Bishop, D.F.; Desnick, R.J.; Suzuki, Y. Identification of point mutations in the alpha-galactosidase A gene in classical and atypical hemizygotes with Fabry disease. Am. J. Hum. Genet. 1990, 47, 784–789. [Google Scholar] [PubMed]
- Frustaci, A.; Chimenti, C.; Ricci, R.; Natale, L.; Russo, M.A.; Pieroni, M.; Eng, C.M.; Desnick, R.J. Improvement in Cardiac Function in the Cardiac Variant of Fabry’s Disease with Galactose-Infusion Therapy. N. Engl. J. Med. 2001, 345, 25–32. [Google Scholar] [CrossRef]
- Liang, K.-H.; Lu, Y.-H.; Niu, C.-W.; Chang, S.-K.; Chen, Y.-R.; Cheng, C.-Y.; Hsu, T.-R.; Yang, C.-F.; Nakamura, K.; Niu, D.-M. The Fabry disease-causing mutation, GLA IVS4+919G>A, originated in Mainland China more than 800 years ago. J. Hum. Genet. 2020, 65, 619–625. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Huang, C.-H.; Yu, H.-C.; Chong, K.-W.; Hsu, J.-H.; Lee, P.-C.; Cheng, K.-H.; Chiang, C.-C.; Ho, H.-J.; Lin, S.-P.; et al. Enzyme assay and clinical assessment in subjects with a Chinese hotspot late-onset Fabry mutation (IVS4 + 919G→A). J. Inherit. Metab. Dis. 2010, 33, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Oder, D.; Liu, D.; Hu, K.; Üçeyler, N.; Salinger, T.; Müntze, J.; Lorenz, K.; Kandolf, R.; Gröne, H.-J.; Sommer, C.; et al. α-Galactosidase A Genotype N215S Induces a Specific Cardiac Variant of Fabry Disease. Circ. Cardiovasc. Genet. 2017, 10, e001691. [Google Scholar] [CrossRef] [Green Version]
- Alharbi, F.J.; Baig, S.; Auray-Blais, C.; Boutin, M.; Ward, D.G.; Wheeldon, N.; Steed, R.; Dawson, C.; Hughes, D.; Geberhiwot, T. Globotriaosylsphingosine (Lyso-Gb3) as a biomarker for cardiac variant (N215S) Fabry disease. J. Inherit. Metab. Dis. 2018, 41, 239–247. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Liu, H.-C.; Huang, Y.-H.; Liao, H.-C.; Hsu, T.-R.; Shen, C.-I.; Li, S.-T.; Li, C.-F.; Lee, L.-H.; Lee, P.-C.; et al. Effects of enzyme replacement therapy for cardiac-type Fabry patients with a Chinese hotspot late-onset Fabry mutation (IVS4+919G>A). BMJ Open 2013, 3, e003146. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-C.; Lin, H.-Y.; Yang, C.-F.; Liao, H.-C.; Hsu, T.-R.; Lo, C.-W.; Chang, F.-P.; Huang, C.-K.; Lu, Y.-H.; Lin, S.-P.; et al. Globotriaosylsphingosine (lyso-Gb3) might not be a reliable marker for monitoring the long-term therapeutic outcomes of enzyme replacement therapy for late-onset Fabry patients with the Chinese hotspot mutation (IVS4+919G>A). Orphanet J. Rare Dis. 2014, 9, 111. [Google Scholar] [CrossRef] [Green Version]
- Chien, Y.; Chien, C.-S.; Chiang, H.-C.; Huang, W.-L.; Chou, S.-J.; Chang, W.-C.; Chang, Y.-L.; Leu, H.-B.; Chen, K.-H.; Wang, K.-L.; et al. Interleukin-18 deteriorates Fabry cardiomyopathy and contributes to the development of left ventricular hypertrophy in Fabry patients with GLA IVS4+919 G>A mutation. Oncotarget 2016, 7, 87161–87179. [Google Scholar] [CrossRef]
- Auray-Blais, C.; Lavoie, P.; Boutin, M.; Ntwari, A.; Hsu, T.-R.; Huang, C.-K.; Niu, D.-M. Biomarkers associated with clinical manifestations in Fabry disease patients with a late-onset cardiac variant mutation. Clin. Chim. Acta 2017, 466, 185–193. [Google Scholar] [CrossRef]
- Hsu, T.-R.; Sung, S.-H.; Chang, F.-P.; Yang, C.-F.; Liu, H.-C.; Lin, H.-Y.; Huang, C.-K.; Gao, H.-J.; Huang, Y.-H.; Liao, H.-C.; et al. Endomyocardial biopsies in patients with left ventricular hypertrophy and a common Chinese later-onset fabry mutation (IVS4 + 919G > A). Orphanet J. Rare Dis. 2014, 9, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, T.-R.; Hung, S.-C.; Chang, F.-P.; Yu, W.-C.; Sung, S.-H.; Hsu, C.-L.; Dzhagalov, I.; Yang, C.-F.; Chu, T.-H.; Lee, H.-J.; et al. Later Onset Fabry Disease, Cardiac Damage Progress in Silence: Experience with a Highly Prevalent Mutation. J. Am. Coll. Cardiol. 2016, 68, 2554–2563. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-T.; Sung, S.-H.; Liao, J.-N.; Hsu, T.-R.; Niu, D.-M.; Yu, W.-C. Cardiac manifestations in patients with classical or cardiac subtype of Fabry disease. J. Chin. Med. Assoc. 2020, 83, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Hsu, T.-R.; Hung, S.-C.; Yu, W.-C.; Chu, T.-H.; Yang, C.-F.; Bizjajeva, S.; Tiu, C.-M.; Niu, D.-M. A comparison of central nervous system involvement in patients with classical Fabry disease or the later-onset subtype with the IVS4+919G>A mutation. BMC Neurol. 2017, 17, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-J.; Hung, S.-C.; Hsu, T.-R.; Ko, S.-C.; Chui-Mei, T.; Huang, C.-C.; Niu, D.-M.; Lin, C.-P. Brain MR Imaging Findings of Cardiac-Type Fabry Disease with an IVS4+919G>A Mutation. Am. J. Neuroradiol. 2016, 37, 1044–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, O.; Gago, M.F.; Miltenberger-Miltenyi, G.; Sousa, N.; Cunha, D. Fabry Disease Therapy: State-of-the-Art and Current Challenges. Int. J. Mol. Sci. 2020, 22, 206. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- A Hughes, D.; Elliott, P.M.; Shah, J.; Zuckerman, J.; Coghlan, G.; Brookes, J.; Mehta, A.B. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson Fabry disease: A randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart 2008, 94, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.A.; Romero, M.Á.B.; Hollak, C.E.; Giugliani, R.; Deegan, P.B. Response of women with Fabry disease to enzyme replacement therapy: Comparison with men, using data from FOS—The Fabry Outcome Survey. Mol. Genet. Metab. 2011, 103, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, C.; Perrin, A.; Beck, M. Effectiveness of agalsidase alfa enzyme replacement in Fabry disease: Cardiac outcomes after 10 years’ treatment. Orphanet J. Rare Dis. 2015, 10, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baehner, F.; Kampmann, C.; Whybra, C.; Miebach, E.; Wiethoff, C.M.; Beck, M. Enzyme replacement therapy in heterozygous females with Fabry disease: Results of a phase IIIB study. J. Inherit. Metab. Dis. 2003, 26, 617–627. [Google Scholar] [CrossRef]
- Whybra, C.; Miebach, E.; Mengel, E.; Gal, A.; Baron, K.; Beck, M.; Kampmann, C. A 4-year study of the efficacy and tolerability of enzyme replacement therapy with agalsidase alfa in 36 women with Fabry disease. Genet. Med. 2009, 11, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, M.; Hughes, D.; Kampmann, C.; Larroque, S.; Mehta, A.; Pintos-Morell, G.; Ramaswami, U.; West, M.; Wijatyk, A.; Giugliani, R.; et al. Long-term effectiveness of agalsidase alfa enzyme replacement in Fabry disease: A Fabry Outcome Survey analysis. Mol. Genet. Metab. Rep. 2015, 3, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, B.H.; Cho, J.H.; Kang, E.; Choi, J.-H.; Kim, G.-H.; Yoo, H.-W. Long-term enzyme replacement therapy for Fabry disease: Efficacy and unmet needs in cardiac and renal outcomes. J. Hum. Genet. 2016, 61, 923–929. [Google Scholar] [CrossRef]
- Germain, D.P.; Weidemann, F.; Abiose, A.; Patel, M.R.; Cizmarik, M.; Cole, J.A.; Beitner-Johnson, D.; Benistan, K.; Cabrera, G.; Charrow, J.; et al. Analysis of left ventricular mass in untreated men and in men treated with agalsidase-β: Data from the Fabry Registry. Genet. Med. 2013, 15, 958–965. [Google Scholar] [CrossRef] [Green Version]
- Motwani, M.; Banypersad, S.; Woolfson, P.; Waldek, S. Enzyme replacement therapy improves cardiac features and severity of Fabry disease. Mol. Genet. Metab. 2012, 107, 197–202. [Google Scholar] [CrossRef]
- Thurberg, B.L.; Fallon, J.T.; Mitchell, R.; Aretz, T.; Gordon, R.E.; O’Callaghan, M.W. Cardiac Microvascular Pathology in Fabry Disease: Evaluation of endomyocardial biopsies before and after enzyme replacement therapy. Circulation 2009, 119, 2561–2567. [Google Scholar] [CrossRef] [Green Version]
- Banikazemi, M.; Bultas, J.; Waldek, S.; Wilcox, W.R.; Whitley, C.B.; McDonald, M.; Finkel, R.; Packman, S.; Bichet, D.G.; Warnock, D.G.; et al. Agalsidase-Beta Therapy for Advanced Fabry Disease. Ann. Intern. Med. 2007, 146, 77–86. [Google Scholar] [CrossRef]
- Ortiz, A.; Abiose, A.; Bichet, D.G.; Cabrera, G.; Charrow, J.; Germain, D.P.; Hopkin, R.J.; Jovanovic, A.; Linhart, A.; Maruti, S.S.; et al. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase β: Data from the Fabry Registry. J. Med. Genet. 2016, 53, 495–502. [Google Scholar] [CrossRef]
- Hsu, T.-R.; Chang, F.-P.; Chu, T.-H.; Sung, S.-H.; Bizjajeva, S.; Yu, W.-C.; Niu, D.-M. Correlations between Endomyocardial Biopsies and Cardiac Manifestations in Taiwanese Patients with the Chinese Hotspot IVS4+919G>A Mutation: Data from the Fabry Outcome Survey. Int. J. Mol. Sci. 2017, 18, 119. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P.; Charrow, J.; Desnick, R.J.; Guffon, N.; Kempf, J.; Lachmann, R.H.; Lemay, R.; Linthorst, G.E.; Packman, S.; Scott, C.R.; et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J. Med. Genet. 2015, 52, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Feriozzi, S.; Linhart, A.; Ramaswami, U.; Kalampoki, V.; Gurevich, A.; Hughes, D.; Fabry Outcome Survey Study Group. Effects of Baseline Left Ventricular Hypertrophy and Decreased Renal Function on Cardiovascular and Renal Outcomes in Patients with Fabry Disease Treated with Agalsidase Alfa: A Fabry Outcome Survey Study. Clin. Ther. 2020, 42, 2321–2330.e0. [Google Scholar] [CrossRef]
- Arends, M.; Biegstraaten, M.; Wanner, C.; Sirrs, S.; Mehta, A.; Elliott, P.M.; Oder, D.; Watkinson, O.T.; Bichet, D.G.; Khan, A.; et al. Agalsidase alfa versus agalsidase beta for the treatment of Fabry disease: An international cohort study. J. Med. Genet. 2018, 55, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bénichou, B.; Goyal, S.; Sung, C.; Norfleet, A.M.; O’Brien, F. A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol. Genet. Metab. 2009, 96, 4–12. [Google Scholar] [CrossRef]
- Lenders, M.; Stypmann, J.; Duning, T.; Schmitz, B.; Brand, S.-M.; Brand, E. Serum-Mediated Inhibition of Enzyme Replacement Therapy in Fabry Disease. J. Am. Soc. Nephrol. 2015, 27, 256–264. [Google Scholar] [CrossRef] [PubMed]
- van der Veen, S.; van Kuilenburg, A.; Hollak, C.; Kaijen, P.; Voorberg, J.; Langeveld, M. Antibodies against recombinant alpha-galactosidase A in Fabry disease: Subclass analysis and impact on response to treatment. Mol. Genet. Metab. 2019, 126, 162–168. [Google Scholar] [CrossRef]
- European Medicines Agency. 2016. Available online: https://www.ema.europa.eu/en/documents/product-information/galafold-epar-product-information_en.pdf (accessed on 15 February 2021).
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555. [Google Scholar] [CrossRef]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Feldt-Rasmussen, U.; Hughes, D.; Sunder-Plassmann, G.; Shankar, S.; Nedd, K.; Olivotto, I.; Ortiz, D.; Ohashi, T.; Hamazaki, T.; Skuban, N.; et al. Long-term efficacy and safety of migalastat treatment in Fabry disease: 30-month results from the open-label extension of the randomized, phase 3 ATTRACT study. Mol. Genet. Metab. 2020, 131, 219–228. [Google Scholar] [CrossRef]
- Müntze, J.; Salinger, T.; Gensler, D.; Wanner, C.; Nordbeck, P. Treatment of hypertrophic cardiomyopathy caused by cardiospecific variants of Fabry disease with chaperone therapy. Eur. Heart J. 2018, 39, 1861–1862. [Google Scholar] [CrossRef] [Green Version]
- Müntze, J.; Gensler, D.; Maniuc, O.; Liu, D.; Cairns, T.; Oder, D.; Hu, K.; Lorenz, K.; Frantz, S.; Wanner, C.; et al. Oral Chaperone Therapy Migalastat for Treating Fabry Disease: Enzymatic Response and Serum Biomarker Changes After 1 Year. Clin. Pharmacol. Ther. 2019, 105, 1224–1233. [Google Scholar] [CrossRef] [Green Version]
- Riccio, E.; AFFIINITY Group; Zanfardino, M.; Ferreri, L.; Santoro, C.; Cocozza, S.; Capuano, I.; Imbriaco, M.; Feriozzi, S.; Pisani, A. Switch from enzyme replacement therapy to oral chaperone migalastat for treating fabry disease: Real-life data. Eur. J. Hum. Genet. 2020, 28, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Nordbeck, P.; Kurschat, C.; Karabul, N.; Kaufeld, J.; Hennermann, J.B.; Patten, M.; Cybulla, M.; Müntze, J.; Üçeyler, N.; et al. Treatment of Fabry’s Disease with Migalastat: Outcome From a Prospective Observational Multicenter Study (FAMOUS). Clin. Pharmacol. Ther. 2020, 108, 326–337. [Google Scholar] [CrossRef]
- Kramer, J.; Bijnens, B.; Störk, S.; Ritter, C.O.; Liu, D.; Ertl, G.; Wanner, C.; Weidemann, F. Left Ventricular Geometry and Blood Pressure as Predictors of Adverse Progression of Fabry Cardiomyopathy. PLoS ONE 2015, 10, e0140627. [Google Scholar] [CrossRef] [PubMed]
- Tahir, H.; Jackson, L.L.; Warnock, D.G. Antiproteinuric Therapy and Fabry Nephropathy: Sustained Reduction of Proteinuria in Patients Receiving Enzyme Replacement Therapy with Agalsidase-beta. J. Am. Soc. Nephrol. 2007, 18, 2609–2617. [Google Scholar] [CrossRef] [Green Version]
- Warnock, D.G.; Thomas, C.P.; Vujkovac, B.; Campbell, R.C.; Charrow, J.; A Laney, D.; Jackson, L.L.; Wilcox, W.R.; Wanner, C. Antiproteinuric therapy and Fabry nephropathy: Factors associated with preserved kidney function during agalsidase-beta therapy. J. Med. Genet. 2015, 52, 860–866. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef] [PubMed]
- Knuuti, J.; Wijns, W.; Achenbach, S.; Agewall, S.; Barbato, E.; Bax, J.J.; Capodanno, D.; Cuisset, T.; Deaton, C.; Dickstein, K.; et al. 2019 ESC Guidelines for the diagnosis and management of chronic coronary syndromes. Eur. Heart J. 2020, 41, 407–477. [Google Scholar] [CrossRef]
- Fukuda, Y.; Onishi, T.; Suzuki, A.; Tanaka, H.; Fukuzawa, K.; Yoshida, A.; Kawai, H.; Hirata, K.-I. Follow-up of Cardiac Fabry Disease Treated by Cardiac Resynchronization Therapy. CASE 2017, 1, 134–137. [Google Scholar] [CrossRef] [Green Version]
- Meghji, Z.; Nguyen, A.; Miranda, W.R.; Geske, J.B.; Schaff, H.V.; Peck, D.S.; Newman, D.B. Surgical septal myectomy for relief of dynamic obstruction in Anderson-Fabry Disease. Int. J. Cardiol. 2019, 292, 91–94. [Google Scholar] [CrossRef]
- Magage, S.; Linhart, A.; Bultas, J.; Vojacek, J.; Mates, M.; Palecek, T.; Popelová, J.; Tintera, J.; Aschermann, M.; Goldman, M.E.; et al. Fabry Disease: Percutaneous Transluminal Septal Myocardial Ablation Markedly Improved Symptomatic Left Ventricular Hypertrophy and Outflow Tract Obstruction in a Classically Affected Male. Echocardiography 2005, 22, 333–339. [Google Scholar] [CrossRef]
- Liu, D.; Hu, K.; Schmidt, M.; Müntze, J.; Maniuc, O.; Gensler, D.; Oder, D.; Salinger, T.; Weidemann, F.; Ertl, G.; et al. Value of the CHA2DS2-VASc score and Fabry-specific score for predicting new-onset or recurrent stroke/TIA in Fabry disease patients without atrial fibrillation. Clin. Res. Cardiol. 2018, 107, 1111–1121. [Google Scholar] [CrossRef]
- Fine, N.M.; Wang, Y.; Khan, A. Acute Decompensated Heart Failure After Initiation of Amiodarone in a Patient With Anderson-Fabry Disease. Can. J. Cardiol. 2019, 35, 104.e5–104.e7. [Google Scholar] [CrossRef]
- Halliwell, W.H. Cationic Amphiphilic Drug-Induced Phospholipidosis. Toxicol. Pathol. 1997, 25, 53–60. [Google Scholar] [CrossRef]
- Qian, P.; Ross, D.; Tchan, M.; Sadick, N.; Tchan, M. A patient with recurrent disabling atrial fibrillation and Fabry cardiomyopathy successfully treated with single ring pulmonary vein isolation. Int. J. Cardiol. 2015, 182, 375–376. [Google Scholar] [CrossRef]
- Hagège, A.; Réant, P.; Habib, G.; Damy, T.; Barone-Rochette, G.; Soulat, G.; Donal, E.; Germain, D.P. Fabry disease in cardiology practice: Literature review and expert point of view. Arch. Cardiovasc. Dis. 2019, 112, 278–287. [Google Scholar] [CrossRef]
- Cantor, W.J.; Daly, P.; Iwanochko, M.; Clarke, J.T.; Cusimano, R.J.; Butany, J. Cardiac transplantation for Fabry’s disease. Can J. Cardiol. 1998, 14, 81–84. [Google Scholar] [PubMed]
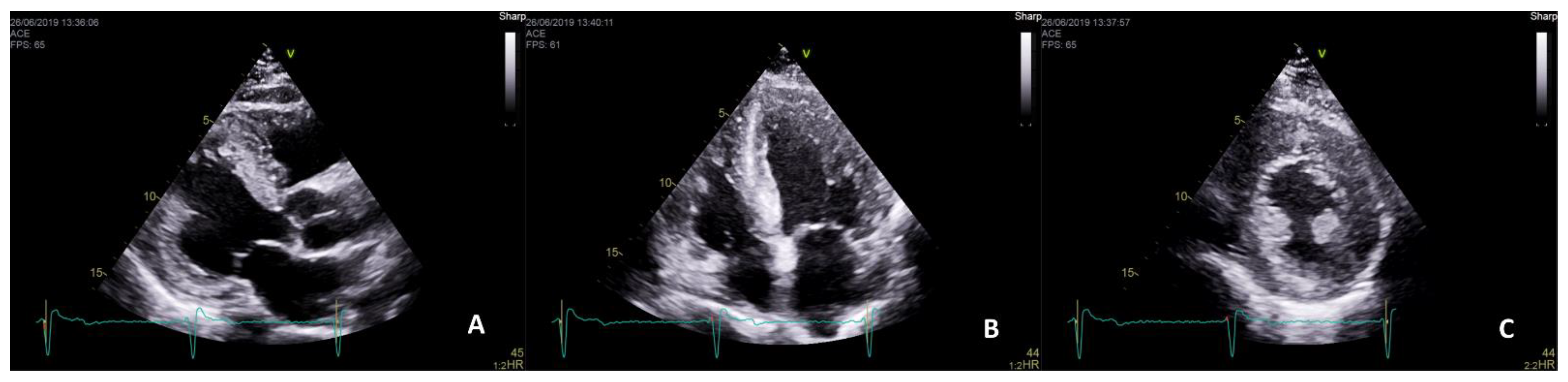
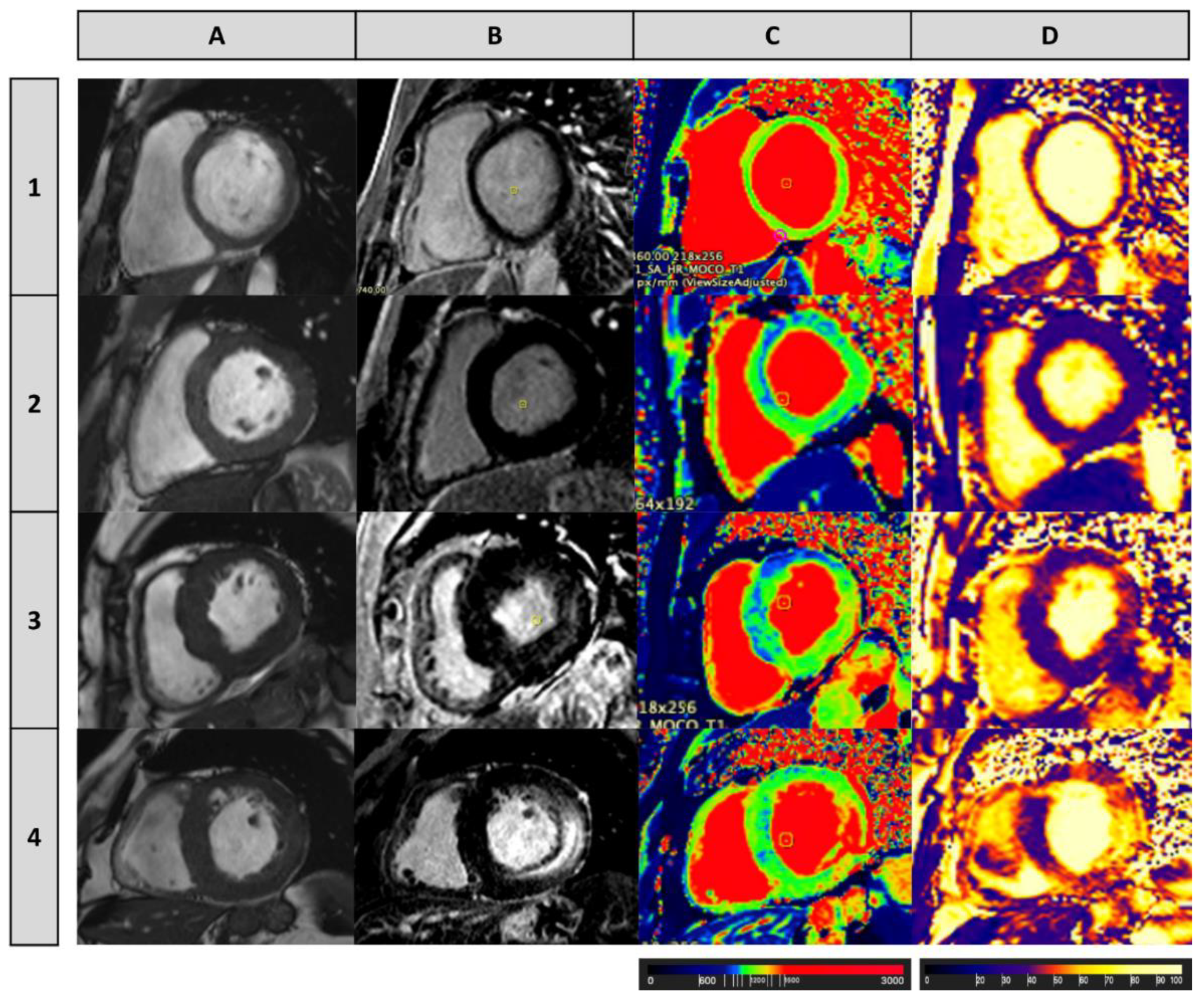
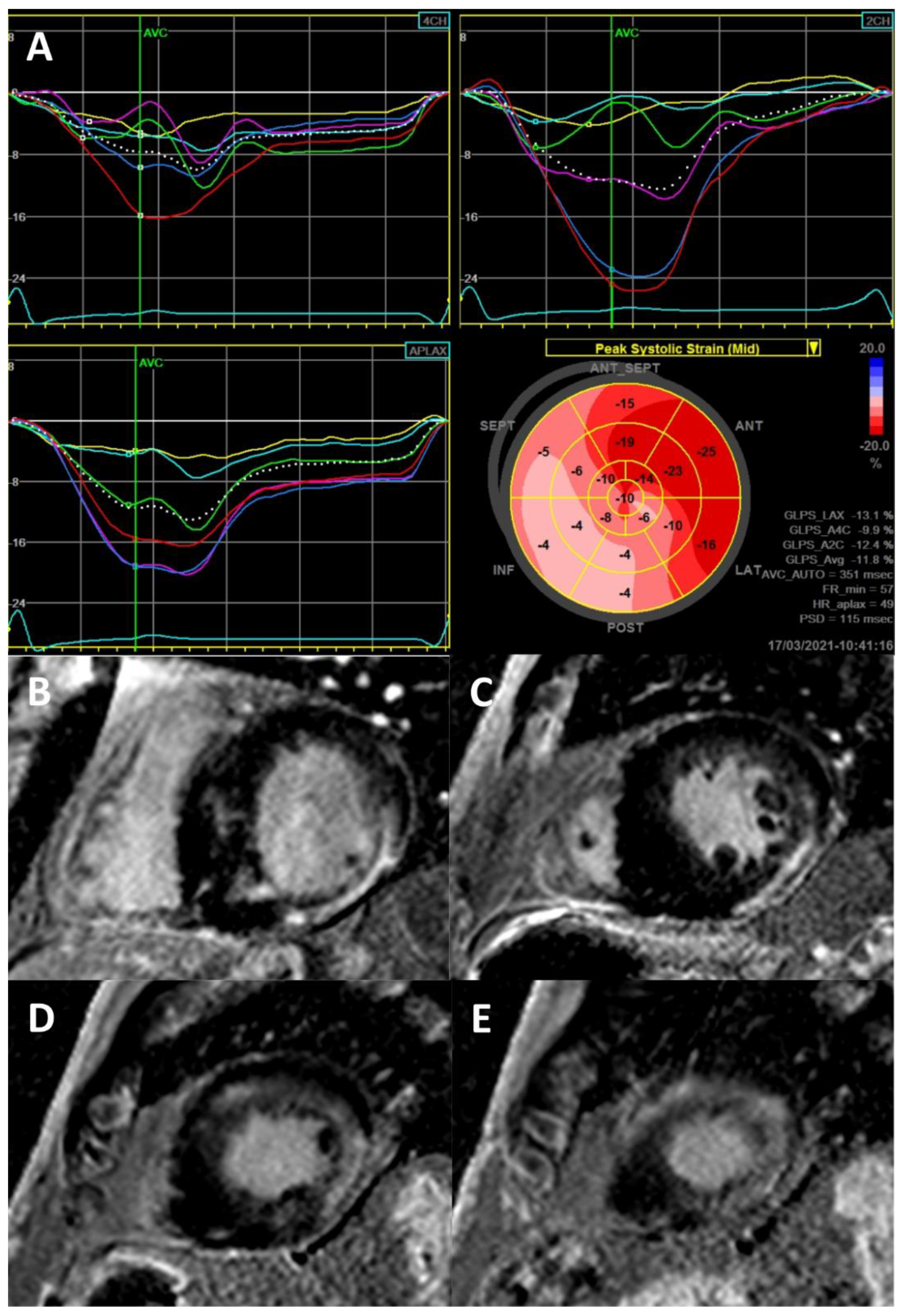

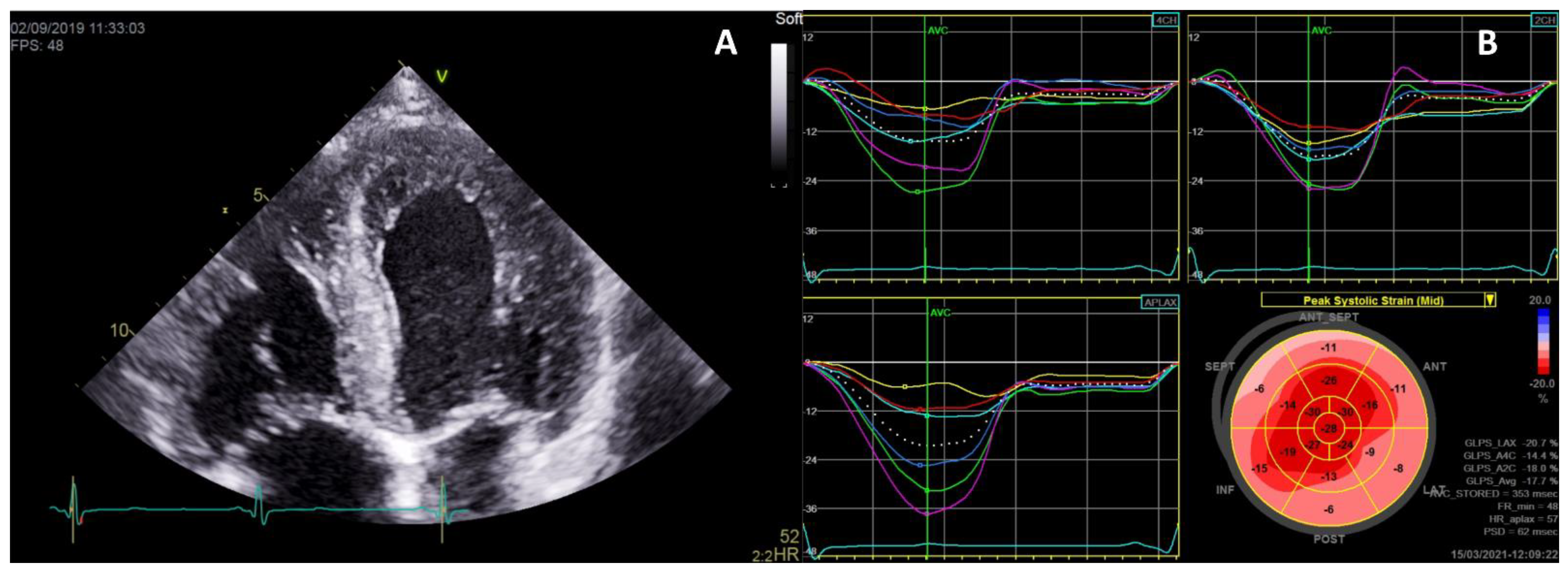

| Cardiac Manifestations | Frequencies | References |
|---|---|---|
| Cardiac signs or symptoms |
|
|
| Cardiac symptoms as presenting symptoms |
|
|
| Cardiac Manifestations | Frequencies | References |
|---|---|---|
| LVH |
|
|
|
| |
In late-onset phenotype with predominant cardiac involvement
|
| |
| LGE |
|
|
In late-onset phenotype with predominant cardiac involvement
|
| |
| LV diastolic dysfunction |
|
|
In late-onset phenotype with predominant cardiac involvement
|
| |
| LV systolic dysfunction (reduced EF) |
|
|
| Latent LVOT obstruction |
|
|
| RV hypertrophy |
| |
| Heart Failure (or Dyspnoea) |
|
|
|
| |
|
| |
In late-onset phenotype with predominant cardiac involvement
|
|
| Cardiac Endpoints | Predictors/Factors Associated to Outcome | References |
|---|---|---|
| Composite endpoint of VT, bradycardia requiring device implantation, severe heart failure or cardiac death | Predictors
|
|
| Composite endpoint of new onset atrial fibrillation, NYHA ≥ III symptoms, device insertion for bradycardia or cardiac death | Predictors
|
|
| Composite endpoint of myocardial infarction, heart failure, or cardiac-related death | Predictor
|
|
| Composite endpoint of death, myocardial infarction, cerebral vascular accident, exacerbation of heart failure, arrythmia, or implantation of permanent pacemaker or cardioverter-defibrillator | Associated factor
|
|
| Composite endpoint of sudden death, arrhythmia or pacing device insertion | Predictor
|
|
| Composite of cardiac death, malignant VT, atrial fibrillation or severe heart failure | Predictors
|
|
| Recommendations for the Diagnosis and Monitoring of Cardiac Manifestations in FD |
| ECG |
| A standard 12-lead ECG is recommended in all adult patients at first clinical evaluation, every 6–12 months and when there is development of new symptoms. |
| Echocardiogram |
| Echocardiogram is recommended in all patients at baseline, every 12–24 months and with the development of new symptoms. |
| Exercise echocardiography |
| Exercise echocardiography should be performed in all symptomatic patients to exclude latent obstruction and exercise-induced mitral regurgitation. |
| Cardiac MRI |
| Cardiac MRI should be considered in all adult patients at baseline to assess cardiac morphology and function and myocardial fibrosis; and may be considered, every 2–5 years in patients without cardiac abnormalities and every 2–3 years in patients with progressive disease, in order to assess progression of fibrosis and cardiac function. T1 mapping may also be considered to detect early cardiac involvement or to help in the differential diagnosis of LVH. |
| Holter monitoring |
| A 24 h-Holter monitoring should be considered in all adult patients at first clinical evaluation, every 6–12 months and when there is development of new symptoms. |
| ILR |
| A prolonged Holter monitoring or preferably an ILR should be considered in patients with recurrent episodes of unexplained syncope. An ILR may also be considered in patients with palpitations or recent stroke and negative Holter monitoring. |
| Cardiopulmonary exercise testing |
| Cardiopulmonary exercise testing should be considered in patients with exercise intolerance. |
| Coronary angiography |
| Coronary angiography (or CT coronary angiography) is recommended in all patients with angina CCS class ≥ II. Invasive coronary angiography is recommended in adult survivors of cardiac arrest, in patients with sustained VT and in patients with severe stable angina (CCS class III) or unstable angina. |
| BNP/NT-proBNP |
| Measurement of plasma BNP/NT-proBNP is recommended in symptomatic patients with suspected heart failure. |
| High-sensitivity troponin |
| High-sensitivity troponin may be considered to assess disease severity. |
| Renal function |
| Regular assessment of renal function and albuminuria/proteinuria is recommended in all patients. |
| Endomyocardial biopsy |
| When a genetic variant of uncertain significance is found in the GLA gene, an endomyocardial biopsy with electron microscopy should be considered, particularly in females or in patients with high residual enzyme activity (>10%) and low lyso-GB3 levels, in order to exclude FD as the cause of LVH. |
| Recommendations for the Supportive Treatment of Cardiac Manifestations in FD |
| Angiotensin Converting Enzyme Inhibitors or Angiotensin II Receptor Blockers 1,2 |
| Angiotensin converting enzyme inhibitors (or angiotensin II receptor blockers, if not tolerated) should be used in patients with LV systolic dysfunction and heart failure [25,51,145]. |
| Beta-blockers 3 |
| Beta-blockers should be considered in patients with heart failure and LV systolic dysfunction; or in patients with angina [25,51,145,207,208]. Beta-blockers are recommended to relieve LVOT obstruction symptoms and to control the rate of atrial fibrillation/flutter [25,51,145]. |
| Mineralocorticoid receptor antagonists 1 |
| Mineralocorticoid receptor antagonists should be considered in patients with heart failure and LV systolic dysfunction [51,145]. |
| Loop diuretics |
| Loop diuretics should be considered to treat symptoms of congestion in patients with heart failure [134,207]. |
| Calcium channel blockers |
| Dihydropyridines2 should be considered for the treatment of angina [134,208]. Verapamil3 is recommended in patients with LVOT obstruction symptoms and should be considered in patients with angina [51,145,208]. Diltiazem3 should be considered in patients with LVOT obstruction symptoms or angina [51,145,208]. |
| Ivabradine 3 |
| Ivabradine should be considered for the treatment of heart failure or angina, according to ESC guidelines [145,207,208]. |
| Antiplatelet therapy |
| Antiplatelet therapy should be started in patients who suffered a stroke or myocardial infarction [134]. |
| Anticoagulation |
| Anticoagulation should be immediately started once atrial fibrillation or flutter is detected [145]. Direct oral anticoagulants (DOACs) should be considered as the first-line choice in patients without contra-indications [145]. |
| Anti-arrhythmic drugs |
| Amiodarone should be avoided in FD [213,214]. Dronedarone is contra-indicated in patients with heart failure (NYHA class III–IV) and renal failure (eGFR < 30mL/min) [145]. Sotalol, flecainide and propafenone are contra-indicated in patients with heart failure [145]. |
| Management of cardiovascular risk factors |
| Control of cardiovascular risk factors, including arterial hypertension, diabetes and dyslipidaemia, is indicated [134]. |
| Pacemaker |
| Pacemaker may be required to treat symptomatic bradycardia or symptomatic/advanced cardiac blocks, according to ESC guidelines [134,145]. Dual chamber pacemakers should be implanted unless patients are in permanent atrial fibrillation [145]. |
| ICD |
| ICD implantation is recommended in patients who suffered sudden cardiac arrest due to VT/fibrillation or sustained VT causing syncope/haemodynamic compromise and have a life expectancy >1 year [209]. ICD implantation should be considered in patients with advanced hypertrophy and fibrosis, who require pacemaker implantation and have a life expectancy >1 year [145]. ICD implantation may be considered in patients with severe LVH and advanced fibrosis or non-sustained VT, who have a life expectancy >1 year [145]. ICD implantation is recommended in patients with heart failure (NYHA class II-III) and LV ejection fraction ≤35%, despite ≥3 months of optimal treatment, who have a life expectancy >1 year [207]. |
| CRT |
| CRT should be considered in patients with LV ejection fraction ≤35%, according to ESC guidelines [207]. CRT-P should be considered in symptomatic patients with a pacing indication, LV ejection fraction <50% and QRS duration >120ms [145]. |
| Septal reduction therapy (myectomy/alcohol ablation therapy) |
| Septal reduction therapy is recommended in patients with a resting or provoked LVOT gradient ≥50 mm Hg, who are in NYHA class III–IV, despite maximum tolerated medical therapy [51,210,211]. Septal reduction therapy should be considered in patients with a resting or provoked LVOT gradient ≥50 mm Hg, who suffer recurrent exertional syncope, despite maximum tolerated medical therapy [51,210,211]. |
| Heart transplantation |
| Heart transplantation should be considered in patients with advanced heart failure with severe LV dysfunction and NYHA class III–IV despite optimal medical therapy, or intractable ventricular arrhythmia, depending on the extension of the extracardiac involvement by the disease [134,217]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azevedo, O.; Cordeiro, F.; Gago, M.F.; Miltenberger-Miltenyi, G.; Ferreira, C.; Sousa, N.; Cunha, D. Fabry Disease and the Heart: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 4434. https://doi.org/10.3390/ijms22094434
Azevedo O, Cordeiro F, Gago MF, Miltenberger-Miltenyi G, Ferreira C, Sousa N, Cunha D. Fabry Disease and the Heart: A Comprehensive Review. International Journal of Molecular Sciences. 2021; 22(9):4434. https://doi.org/10.3390/ijms22094434
Chicago/Turabian StyleAzevedo, Olga, Filipa Cordeiro, Miguel Fernandes Gago, Gabriel Miltenberger-Miltenyi, Catarina Ferreira, Nuno Sousa, and Damião Cunha. 2021. "Fabry Disease and the Heart: A Comprehensive Review" International Journal of Molecular Sciences 22, no. 9: 4434. https://doi.org/10.3390/ijms22094434
APA StyleAzevedo, O., Cordeiro, F., Gago, M. F., Miltenberger-Miltenyi, G., Ferreira, C., Sousa, N., & Cunha, D. (2021). Fabry Disease and the Heart: A Comprehensive Review. International Journal of Molecular Sciences, 22(9), 4434. https://doi.org/10.3390/ijms22094434