
Deep Learning in Virtual Screening: Recent Applications and Developments


Abstract
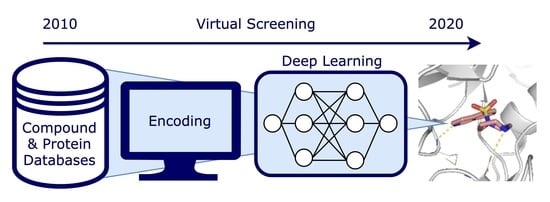
1. Introduction
1.1. Virtual Screening

1.2. Machine Learning and Deep Learning
1.3. Data Availability and Big Data
1.4. Deep Learning in Virtual Screening
2. Methods & Data
2.1. Encodings in Virtual Screening
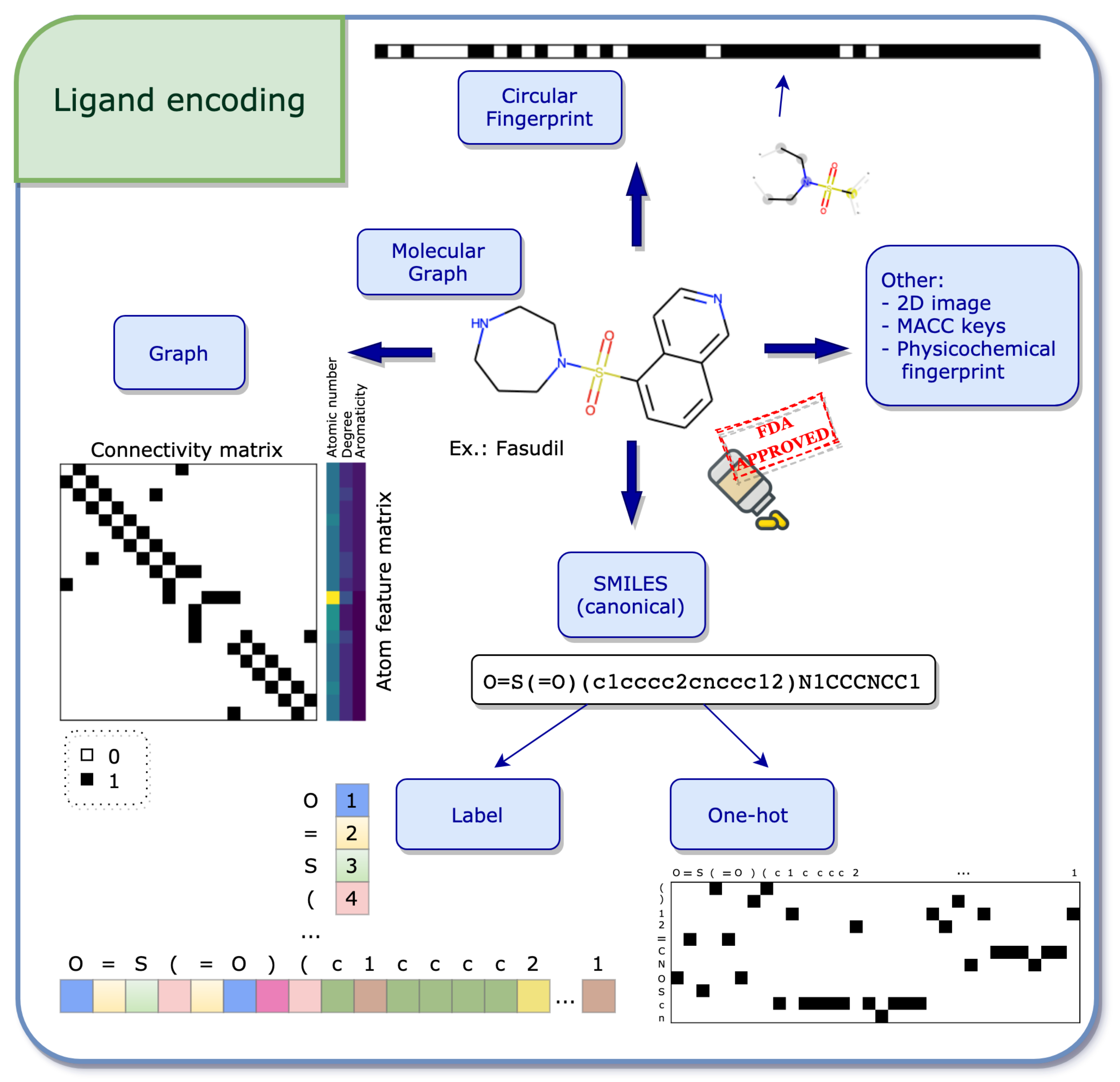
2.1.1. Ligand Encodings
- Graph
- SMILES
- Label and One-Hot Encoding
- Circular Fingerprint
- Other Encodings
2.1.2. Protein Encodings
- Protein Identifier
- Protein Sequence
- Z-Scales
- Domains and Motifs
- Structural Property Sequence
2.1.3. Complex Encodings
- Interaction Fingerprint
- 3D Grid
- Graph
- Other Encodings
2.2. Deep Learning Models in Virtual Screening
2.2.1. Supervised Deep Learning Models
2.2.1.1. Neural Networks
2.2.1.2. Convolutional Neural Networks
2.2.1.3. Recurrent Neural Networks
2.2.1.4. Graph Neural Networks
2.2.2. Model Evaluation Strategies and Metrics
2.3. Data Sets and Benchmarks in Virtual Screening
2.3.1. Structure-Based Data Sets
2.3.1.1. PDBbind
2.3.1.2. BindingDB
2.3.1.3. BindingMOAD
2.3.2. Bioactivity Data Sets
2.3.2.1. PubChem BioAssay
2.3.2.2. ChEMBL
2.3.2.3. Target-Family Specific Data Sets (Such as Kinases)
2.3.3. Benchmarking Data Sets
2.3.3.1. CASF
2.3.3.2. DUD(-E)
2.3.3.3. MUV
2.3.3.4. Benchmarking Set Collections
3. Recent Developments
3.1. Complex-Based Models
- Interaction Fingerprint-Based Studies
- 3D Grid-Based Studies
- Graph-Based Studies
- Other Studies
3.2. Pair-Based Models
- Ligand as SMILES
- Ligand as Fingerprint
- Ligand as Graph
4. Conclusions and Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| General | |
| HTS | High-throughput screening |
| VS | Virtual screening |
| SF | Scoring function |
| QSAR | Quantitative structure–activity relationship |
| PCM | Proteochemometric |
| FDA | Food and Drug Administration |
| Machine learning | |
| ML | Machine learning |
| DL | Deep learning |
| SVM | Support vector machine |
| RF | Random forest |
| NN | Neural network |
| ANN | Artificial neural network |
| MLP | Multilayer perceptron |
| CNN | Convolutional neural network |
| RNN | Recurrent neural network |
| GNN | Graph neural network |
| GCNN | Graph convolution neural network |
| GRU | Gated recurrent unit |
| GGNN | Gated graph neural network |
| GANN | Graph attention neural network |
| GAN | Generative adversarial network |
| Encoding | |
| SMILES | Simplified molecular input line entry system |
| ECFP | Extended-connectivity fingerprint |
| MCS | Maximum common substructure |
| IPF | Interaction fingerprint |
| ID | Identifier |
| Metrics | |
| MSE | Mean squared error |
| RMSE | Root mean squared error |
| RMSD | Root mean square deviation |
| ROC | Receiver operating characteristic |
| AUC | Area under the ROC curve |
| EF | Enrichment factor |
| Data | |
| PDB | Protein Data Bank |
| KIBA | Kinase inhibitor bioactivity |
| MUV | Maximum unbiased validation |
| TDC | Therapeutics Data Commons |
| DUD | Directory of useful decoys |
| CASF | Comparative assessment of scoring functions |
Appendix A. Figures

Appendix B. Evaluation Strategies and Metrics
Appendix B.1. Classification
Appendix B.2. Regression
Appendix B.3. Classification & Regression
References
- Berdigaliyev, N.; Aljofan, M. An overview of drug discovery and development. Future Med. Chem. 2020, 12, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Butkiewicz, M.; Wang, Y.; Bryant, S.; Lowe, E., Jr.; Weaver, D.; Meiler, J. High-Throughput Screening Assay Datasets from the PubChem Database. Chem. Inform. (Wilmington Del.) 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.; Stahl, M.T.; Murcko, M.A. Virtual screening—An overview. Drug Discov. Today 1998, 3, 160–178. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15–Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- MolPORT. Available online: https://www.molport.com (accessed on 2 March 2021).
- Enamine REAL. Available online: https://enamine.net/library-synthesis/real-compounds (accessed on 2 March 2021).
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing Pitfalls in Virtual Screening: A Critical Review. J. Chem. Inf. Model. 2012, 52, 867–881. [Google Scholar] [CrossRef]
- Kumar, A.; Zhang, K.Y. Hierarchical virtual screening approaches in small molecule drug discovery. Methods 2015, 71, 26–37. [Google Scholar] [CrossRef]
- Brooijmans, N.; Kuntz, I.D. Molecular Recognition and Docking Algorithms. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 335–373. [Google Scholar] [CrossRef]
- Sulimov, V.B.; Kutov, D.C.; Sulimov, A.V. Advances in Docking. Curr. Med. Chem. 2020, 26, 7555–7580. [Google Scholar] [CrossRef]
- Fischer, A.; Smieško, M.; Sellner, M.; Lill, M.A. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef]
- Klebe, G. Virtual ligand screening: Strategies, perspectives and limitations. Drug Discov. Today 2006, 11, 580–594. [Google Scholar] [CrossRef]
- Kolodzik, A.; Schneider, N.; Rarey, M. Structure-Based Virtual Screening. In Applied Chemoinformatics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2018; Chapter 6.8; pp. 313–331. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- Li, J.; Fu, A.; Zhang, L. An Overview of Scoring Functions Used for Protein–Ligand Interactions in Molecular Docking. Interdiscip. Sci. Comput. Life Sci. 2019, 11, 320–328. [Google Scholar] [CrossRef]
- Shen, C.; Ding, J.; Wang, Z.; Cao, D.; Ding, X.; Hou, T. From machine learning to deep learning: Advances in scoring functions for protein–ligand docking. WIREs Comput. Mol. Sci. 2019, 10. [Google Scholar] [CrossRef]
- Ain, Q.U.; Aleksandrova, A.; Roessler, F.D.; Ballester, P.J. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 405–424. [Google Scholar] [CrossRef]
- Sunseri, J.; Koes, D.R. Pharmit: Interactive exploration of chemical space. Nucleic Acids Res. 2016, 44, W442–W448. [Google Scholar] [CrossRef]
- Schaller, D.; Šribar, D.; Noonan, T.; Deng, L.; Nguyen, T.N.; Pach, S.; Machalz, D.; Bermudez, M.; Wolber, G. Next generation 3D pharmacophore modeling. WIREs Comput. Mol. Sci. 2020, 10, e1468. [Google Scholar] [CrossRef]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef]
- Sydow, D.; Burggraaff, L.; Szengel, A.; van Vlijmen, H.W.T.; IJzerman, A.P.; van Westen, G.J.P.; Volkamer, A. Advances and Challenges in Computational Target Prediction. J. Chem. Inf. Model. 2019, 59, 1728–1742. [Google Scholar] [CrossRef]
- Lapinsh, M.; Prusis, P.; Gutcaits, A.; Lundstedt, T.; Wikberg, J.E. Development of proteo-chemometrics: A novel technology for the analysis of drug-receptor interactions. Biochim. Biophys. Acta (BBA) Gen. Subj. 2001, 1525, 180–190. [Google Scholar] [CrossRef]
- Van Westen, G.J.P.; Wegner, J.K.; IJzerman, A.P.; van Vlijmen, H.W.T.; Bender, A. Proteochemometric modeling as a tool to design selective compounds and for extrapolating to novel targets. Med. Chem. Commun. 2011, 2, 16–30. [Google Scholar] [CrossRef]
- Geppert, H.; Humrich, J.; Stumpfe, D.; Gärtner, T.; Bajorath, J. Ligand Prediction from Protein Sequence and Small Molecule Information Using Support Vector Machines and Fingerprint Descriptors. J. Chem. Inf. Model. 2009, 49, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Ramsundar, B.; Feinberg, E.N.; Gomes, J.; Geniesse, C.; Pappu, A.S.; Leswing, K.; Pande, V. MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 2018, 9, 513–530. [Google Scholar] [CrossRef]
- Oladipupo, T. Types of Machine Learning Algorithms; IntechOpen: London, UK, 2010. [Google Scholar] [CrossRef]
- Rosenblatt, F. Principles of Neurodynamics: Perceptrons and the Theory of Brain Mechanisms; Technical Report; Cornell Aeronautical Lab Inc.: Buffalo, NY, USA, 1961. [Google Scholar]
- Breiman, L.; Friedman, J.; Stone, C.J.; Olshen, R.A. Classification and Regression Trees; CRC Press: Boca Raton, FL, USA, 1984. [Google Scholar]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Bisong, E. Google Colaboratory. In Building Machine Learning and Deep Learning Models on Google Cloud Platform: A Comprehensive Guide for Beginners; Apress: Berkeley, CA, USA, 2019; pp. 59–64. [Google Scholar] [CrossRef]
- Goodfellow, I.; Bengio, Y.; Courville, A. Deep Learning; MIT Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Kuznetsova, A.; Rom, H.; Alldrin, N.; Uijlings, J.; Krasin, I.; Pont-Tuset, J.; Kamali, S.; Popov, S.; Malloci, M.; Kolesnikov, A.; et al. The Open Images Dataset V4. Int. J. Comput. Vis. 2020, 128, 1956–1981. [Google Scholar] [CrossRef]
- LeCun, Y.; Cortes, C. MNIST Handwritten Digit Database. 2010. Available online: http://yann.lecun.com/exdb/mnist/ (accessed on 2 March 2021).
- kaggle. Available online: https://www.kaggle.com/ (accessed on 2 March 2021).
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2018, 47, D930–D940. [Google Scholar] [CrossRef]
- ChEMBL. Available online: https://www.ebi.ac.uk/chembl/ (accessed on 2 March 2021).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2020, 49, D437–D451. [Google Scholar] [CrossRef]
- RCSB PDB. Available online: http://www.rcsb.org/stats/growth/growth-released-structures (accessed on 2 March 2021).
- Berman, H.M.; Vallat, B.; Lawson, C.L. The data universe of structural biology. IUCrJ 2020, 7, 630–638. [Google Scholar] [CrossRef]
- Helliwell, J.R. New developments in crystallography: Exploring its technology, methods and scope in the molecular biosciences. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Ajay; Walters, P.W.; Murcko, M.A. Can We Learn to Distinguish between “Drug-like” and “Nondrug-like” Molecules? J. Med. Chem. 1998, 41, 3314–3324. [Google Scholar] [CrossRef]
- Burden, F.R.; Winkler, D.A. Robust QSAR Models Using Bayesian Regularized Neural Networks. J. Med. Chem. 1999, 42, 3183–3187. [Google Scholar] [CrossRef] [PubMed]
- Burden, F.R.; Ford, M.G.; Whitley, D.C.; Winkler, D.A. Use of Automatic Relevance Determination in QSAR Studies Using Bayesian Neural Networks. J. Chem. Inf. Comput. Sci. 2000, 40, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Baskin, I.I.; Winkler, D.; Tetko, I.V. A renaissance of neural networks in drug discovery. Expert Opin. Drug Discov. 2016, 11, 785–795. [Google Scholar] [CrossRef]
- Carpenter, K.A.; Cohen, D.S.; Jarrell, J.T.; Huang, X. Deep learning and virtual drug screening. Future Med. Chem. 2018, 10, 2557–2567. [Google Scholar] [CrossRef]
- Ellingson, S.R.; Davis, B.; Allen, J. Machine learning and ligand binding predictions: A review of data, methods, and obstacles. Biochim. Biophys. Acta (BBA) Gen. Subj. 2020, 1864, 129545. [Google Scholar] [CrossRef]
- D’Souza, S.; Prema, K.; Balaji, S. Machine learning models for drug–target interactions: Current knowledge and future directions. Drug Discov. Today 2020, 25, 748–756. [Google Scholar] [CrossRef]
- Li, H.; Sze, K.H.; Lu, G.; Ballester, P.J. Machine-learning scoring functions for structure-based drug lead optimization. WIREs Comput. Mol. Sci. 2020, 10. [Google Scholar] [CrossRef]
- Li, H.; Sze, K.H.; Lu, G.; Ballester, P.J. Machine-learning scoring functions for structure-based virtual screening. WIREs Comput. Mol. Sci. 2020, 11. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Atas, H.; Martin, M.J.; Cetin-Atalay, R.; Atalay, V.; Doğan, T. Recent applications of deep learning and machine intelligence on in silico drug discovery: Methods, tools and databases. Briefings Bioinform. 2018, 20, 1878–1912. [Google Scholar] [CrossRef]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef]
- Xu, Y.; Verma, D.; Sheridan, R.P.; Liaw, A.; Ma, J.; Marshall, N.M.; McIntosh, J.; Sherer, E.C.; Svetnik, V.; Johnston, J.M. Deep Dive into Machine Learning Models for Protein Engineering. J. Chem. Inf. Model. 2020, 60, 2773–2790. [Google Scholar] [CrossRef]
- Bond, J.E.; Kokosis, G.; Ren, L.; Selim, M.A.; Bergeron, A.; Levinson, H. Wound Contraction Is Attenuated by Fasudil Inhibition of Rho-Associated Kinase. Plast. Reconstr. Surg. 2011, 128, 438e–450e. [Google Scholar] [CrossRef]
- Carles, F.; Bourg, S.; Meyer, C.; Bonnet, P. PKIDB: A Curated, Annotated and Updated Database of Protein Kinase Inhibitors in Clinical Trials. Molecules 2018, 23, 908. [Google Scholar] [CrossRef]
- Torng, W.; Altman, R.B. Graph Convolutional Neural Networks for Predicting Drug-Target Interactions. J. Chem. Inf. Model. 2019, 59, 4131–4149. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Bjerrum, E.J. SMILES Enumeration as Data Augmentation for Neural Network Modeling of Molecules. arXiv 2017, arXiv:1703.07076. [Google Scholar]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep drug–target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef]
- Kimber, T.B.; Engelke, S.; Tetko, I.V.; Bruno, E.; Godin, G. Synergy Effect between Convolutional Neural Networks and the Multiplicity of SMILES for Improvement of Molecular Prediction. arXiv 2018, arXiv:1812.04439. [Google Scholar]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- RDKit: Open-source cheminformatics. Available online: http://www.rdkit.org (accessed on 2 March 2021).
- Hassan, M.; Brown, R.D.; Varma-O’Brien, S.; Rogers, D. Cheminformatics analysis and learning in a data pipelining environment. Mol. Divers. 2006, 10, 283–299. [Google Scholar] [CrossRef] [PubMed]
- Kundu, I.; Paul, G.; Banerjee, R. A machine learning approach towards the prediction of protein-ligand binding affinity based on fundamental molecular properties. RSC Adv. 2018, 8, 12127–12137. [Google Scholar] [CrossRef]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Winter, R.; Montanari, F.; Noé, F.; Clevert, D.A. Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chem. Sci. 2019, 10, 1692–1701. [Google Scholar] [CrossRef]
- Rifaioglu, A.S.; Nalbat, E.; Atalay, V.; Martin, M.J.; Cetin-Atalay, R.; Doğan, T. DEEPScreen: High performance drug–target interaction prediction with convolutional neural networks using 2-D structural compound representations. Chem. Sci. 2020, 11, 2531–2557. [Google Scholar] [CrossRef]
- Murray, R.K.; Bender, D.A.; Botham, K.M.; Kennelly, P.J.; Rodwell, V.W.; Weil, P.A. Harper’s Illustrated Biochemistry, Twenty-Eighth Edition; McGraw-Hill Medical McGraw-Hill Distributor: New York, NY, USA, 2009. [Google Scholar]
- Sorgenfrei, F.A.; Fulle, S.; Merget, B. Kinome-wide profiling prediction of small molecules. ChemMedChem 2018, 13, 495–499. [Google Scholar] [CrossRef]
- Hellberg, S.; Sjoestroem, M.; Skagerber, B.; Wold, S. Peptide quantitative structure-activity relationships, multivariate approach. J. Med. Chem. 1987, 30, 1126–1135. [Google Scholar] [CrossRef]
- Sigrist, C.J.A.; de Castro, E.; Cerutti, L.; Cuche, B.A.; Hulo, N.; Bridge, A.; Bougueleret, L.; Xenarios, I. New and continuing developments at PROSITE. Nucleic Acids Res. 2012, 41, D344–D347. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2013, 42, D222–D230. [Google Scholar] [CrossRef]
- Karimi, M.; Wu, D.; Wang, Z.; Shen, Y. DeepAffinity: Interpretable deep learning of compound–protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 2019, 35, 3329–3338. [Google Scholar] [CrossRef]
- Magnan, C.N.; Baldi, P. SSpro/ACCpro 5: Almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics 2014, 30, 2592–2597. [Google Scholar] [CrossRef] [PubMed]
- De Freitas, R.F.; Schapira, M. A systematic analysis of atomic protein–ligand interactions in the PDB. MedChemComm 2017, 8, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Chuaqui, C.; Singh, J. Structural Interaction Fingerprint (SIFt): A Novel Method for Analyzing Three-Dimensional Protein-Ligand Binding Interactions. J. Med. Chem. 2004, 47, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Radifar, M.; Yuniarti, N.; Istyastono, E.P. PyPLIF: Python-based Protein-Ligand Interaction Fingerprinting. Bioinformation 2013, 9, 325–328. [Google Scholar] [CrossRef]
- DaSilva, F.; Desaphy, J.; Rognan, D. IChem: A Versatile Toolkit for Detecting, Comparing, and Predicting Protein-Ligand Interactions. ChemMedChem 2017, 13, 507–510. [Google Scholar] [CrossRef]
- Jasper, J.B.; Humbeck, L.; Brinkjost, T.; Koch, O. A novel interaction fingerprint derived from per atom score contributions: Exhaustive evaluation of interaction fingerprint performance in docking based virtual screening. J. Cheminform. 2018, 10. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Chupakhin, V.; Marcou, G.; Gaspar, H.; Varnek, A. Simple Ligand–Receptor Interaction Descriptor (SILIRID) for alignment-free binding site comparison. Comput. Struct. Biotechnol. J. 2014, 10, 33–37. [Google Scholar] [CrossRef]
- Pérez-Nueno, V.I.; Rabal, O.; Borrell, J.I.; Teixidó, J. APIF: A New Interaction Fingerprint Based on Atom Pairs and Its Application to Virtual Screening. J. Chem. Inf. Model. 2009, 49, 1245–1260. [Google Scholar] [CrossRef]
- Sato, T.; Honma, T.; Yokoyama, S. Combining Machine Learning and Pharmacophore-Based Interaction Fingerprint for in Silico Screening. J. Chem. Inf. Model. 2009, 50, 170–185. [Google Scholar] [CrossRef]
- Desaphy, J.; Raimbaud, E.; Ducrot, P.; Rognan, D. Encoding Protein–Ligand Interaction Patterns in Fingerprints and Graphs. J. Chem. Inf. Model. 2013, 53, 623–637. [Google Scholar] [CrossRef]
- Da, C.; Kireev, D. Structural Protein–Ligand Interaction Fingerprints (SPLIF) for Structure-Based Virtual Screening: Method and Benchmark Study. J. Chem. Inf. Model. 2014, 54, 2555–2561. [Google Scholar] [CrossRef]
- Wójcikowski, M.; Kukiełka, M.; Stepniewska-Dziubinska, M.M.; Siedlecki, P. Development of a protein–ligand extended connectivity (PLEC) fingerprint and its application for binding affinity predictions. Bioinformatics 2018, 35, 1334–1341. [Google Scholar] [CrossRef]
- Wallach, I.; Dzamba, M.; Heifets, A. AtomNet: A deep convolutional neural network for bioactivity prediction in structure-based drug discovery. arXiv 2015, arXiv:1510.02855. [Google Scholar]
- Stepniewska-Dziubinska, M.M.; Zielenkiewicz, P.; Siedlecki, P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics 2018, 34, 3666–3674. [Google Scholar] [CrossRef]
- Sunseri, J.; King, J.E.; Francoeur, P.G.; Koes, D.R. Convolutional neural network scoring and minimization in the D3R 2017 community challenge. J. Comput.-Aided Mol. Des. 2018, 33, 19–34. [Google Scholar] [CrossRef]
- Jiménez, J.; Škalič, M.; Martínez-Rosell, G.; Fabritiis, G.D. KDEEP: Protein–Ligand Absolute Binding Affinity Prediction via 3D-Convolutional Neural Networks. J. Chem. Inf. Model. 2018, 58, 287–296. [Google Scholar] [CrossRef]
- Li, Y.; Rezaei, M.A.; Li, C.; Li, X. DeepAtom: A Framework for Protein-Ligand Binding Affinity Prediction. In Proceedings of the 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, USA, 18–21 November 2019. [Google Scholar] [CrossRef]
- Skalic, M.; Martínez-Rosell, G.; Jiménez, J.; Fabritiis, G.D. PlayMolecule BindScope: Large scale CNN-based virtual screening on the web. Bioinformatics 2018, 35, 1237–1238. [Google Scholar] [CrossRef]
- Erdas-Cicek, O.; Atac, A.O.; Gurkan-Alp, A.S.; Buyukbingol, E.; Alpaslan, F.N. Three-Dimensional Analysis of Binding Sites for Predicting Binding Affinities in Drug Design. J. Chem. Inf. Model. 2019, 59, 4654–4662. [Google Scholar] [CrossRef]
- Lim, J.; Ryu, S.; Park, K.; Choe, Y.J.; Ham, J.; Kim, W.Y. Predicting Drug–Target Interaction Using a Novel Graph Neural Network with 3D Structure-Embedded Graph Representation. J. Chem. Inf. Model. 2019, 59, 3981–3988. [Google Scholar] [CrossRef]
- Feinberg, E.N.; Sur, D.; Wu, Z.; Husic, B.E.; Mai, H.; Li, Y.; Sun, S.; Yang, J.; Ramsundar, B.; Pande, V.S. PotentialNet for Molecular Property Prediction. ACS Cent. Sci. 2018, 4, 1520–1530. [Google Scholar] [CrossRef]
- Cang, Z.; Wei, G.W. TopologyNet: Topology based deep convolutional and multi-task neural networks for biomolecular property predictions. PLoS Comput. Biol. 2017, 13, e1005690. [Google Scholar] [CrossRef]
- Zhu, F.; Zhang, X.; Allen, J.E.; Jones, D.; Lightstone, F.C. Binding Affinity Prediction by Pairwise Function Based on Neural Network. J. Chem. Inf. Model. 2020, 60, 2766–2772. [Google Scholar] [CrossRef]
- Pereira, J.C.; Caffarena, E.R.; dos Santos, C.N. Boosting Docking-Based Virtual Screening with Deep Learning. J. Chem. Inf. Model. 2016, 56, 2495–2506. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R.; Friedman, J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction; Springer: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Webel, H.E.; Kimber, T.B.; Radetzki, S.; Neuenschwander, M.; Nazaré, M.; Volkamer, A. Revealing cytotoxic substructures in molecules using deep learning. J. Comput.-Aided Mol. Des. 2020, 34, 731–746. [Google Scholar] [CrossRef]
- Simonyan, K.; Zisserman, A. Very Deep Convolutional Networks for Large-Scale Image Recognition. arXiv 2015, arXiv:1409.1556. [Google Scholar]
- Krizhevsky, A.; Sutskever, I.; Hinton, G.E. ImageNet classification with deep convolutional neural networks. Commun. ACM 2017, 60, 84–90. [Google Scholar] [CrossRef]
- Szegedy, C.; Liu, W.; Jia, Y.; Sermanet, P.; Reed, S.; Anguelov, D.; Erhan, D.; Vanhoucke, V.; Rabinovich, A. Going deeper with convolutions. In Proceedings of the 2015 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Boston, MA, USA, 7–12 June 2015. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, J. Introduction to Graph Neural Networks. Synth. Lect. Artif. Intell. Mach. Learn. 2020, 14, 1–127. [Google Scholar] [CrossRef]
- Li, Y.; Tarlow, D.; Brockschmidt, M.; Zemel, R. Gated Graph Sequence Neural Networks. arXiv 2017, arXiv:1511.05493. [Google Scholar]
- Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Liò, P.; Bengio, Y. Graph Attention Networks. arXiv 2018, arXiv:1710.10903. [Google Scholar]
- Zhou, J.; Cui, G.; Zhang, Z.; Yang, C.; Liu, Z.; Sun, M. Graph Neural Networks: A Review of Methods and Applications. arXiv 2018, arXiv:1812.08434. [Google Scholar]
- Wieder, O.; Kohlbacher, S.; Kuenemann, M.; Garon, A.; Ducrot, P.; Seidel, T.; Langer, T. A compact review of molecular property prediction with graph neural networks. Drug Discovery Today Technol. 2020. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2020, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Su, M.; Han, L.; Liu, J.; Yang, Q.; Li, Y.; Wang, R. Forging the Basis for Developing Protein–Ligand Interaction Scoring Functions. Accounts Chem. Res. 2017, 50, 302–309. [Google Scholar] [CrossRef]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2015, 44, D1045–D1053. [Google Scholar] [CrossRef] [PubMed]
- BindingDB. Available online: https://www.bindingdb.org/bind/index.jsp (accessed on 2 March 2021).
- Ahmed, A.; Smith, R.D.; Clark, J.J.; Dunbar, J.B.; Carlson, H.A. Recent improvements to Binding MOAD: A resource for protein–ligand binding affinities and structures. Nucleic Acids Res. 2014, 43, D465–D469. [Google Scholar] [CrossRef]
- Smith, R.D.; Clark, J.J.; Ahmed, A.; Orban, Z.J.; Dunbar, J.B.; Carlson, H.A. Updates to Binding MOAD (Mother of All Databases): Polypharmacology Tools and Their Utility in Drug Repurposing. J. Mol. Biol. 2019, 431, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/ (accessed on 2 March 2021).
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef]
- Kooistra, A.J.; Volkamer, A. Kinase-Centric Computational Drug Development. In Annual Reports in Medicinal Chemistry; Elsevier: Amsterdam, The Netherlands, 2017; pp. 197–236. [Google Scholar] [CrossRef]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef]
- Tang, J.; Szwajda, A.; Shakyawar, S.; Xu, T.; Hintsanen, P.; Wennerberg, K.; Aittokallio, T. Making Sense of Large-Scale Kinase Inhibitor Bioactivity Data Sets: A Comparative and Integrative Analysis. J. Chem. Inf. Model. 2014, 54, 735–743. [Google Scholar] [CrossRef]
- Sieg, J.; Flachsenberg, F.; Rarey, M. In need of bias control: Evaluating chemical data for machine learning in structure-based virtual screening. J. Chem. Inf. Model. 2019, 59, 947–961. [Google Scholar] [CrossRef]
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Comparative assessment of scoring functions: The CASF-2016 update. J. Chem. Inf. Model. 2018, 59, 895–913. [Google Scholar] [CrossRef]
- Rodgers, J.L.; Nicewander, W.A. Thirteen Ways to Look at the Correlation Coefficient. Am. Stat. 1988, 42, 59–66. [Google Scholar] [CrossRef]
- Spearman, C. The Proof and Measurement of Association between Two Things. Am. J. Psychol 1904, 15, 72–101. [Google Scholar] [CrossRef]
- Glasser, G.J.; Winter, R.F. Critical Values of the Coefficient of Rank Correlation for Testing the Hypothesis of Independence. Biometrika 1961, 48, 444. [Google Scholar] [CrossRef]
- Wells, R.D.; Bond, J.S.; Klinman, J.; Masters, B.S.S. (Eds.) RMSD, Root-Mean-Square Deviation. In Molecular Life Sciences: An Encyclopedic Reference; Springer: New York, NY, USA, 2018; p. 1078. [Google Scholar] [CrossRef]
- Truchon, J.F.; Bayly, C.I. Evaluating Virtual Screening Methods: Good and Bad Metrics for the “Early Recognition” Problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy ofdocking with a new scoring function, efficient optimization, andmultithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking Sets for Molecular Docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Rohrer, S.G.; Baumann, K. Maximum Unbiased Validation (MUV) Data Sets for Virtual Screening Based on PubChem Bioactivity Data. J. Chem. Inf. Model. 2009, 49, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Fu, T.; Gao, W.; Zhao, Y.; Roohani, Y.; Leskovec, J.; Coley, C.; Xiao, C.; Sun, J.; Zitnik, M. Therapeutics Data Commons: Machine Learning Datasets for Therapeutics. Available online: https://tdcommons.ai (accessed on 2 March 2021).
- Riniker, S.; Landrum, G.A. Open-source platform to benchmark fingerprints for ligand-based virtual screening. J. Cheminform. 2013, 5, 1758–2946. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, P.; Qiao, P. The Virtual Screening of the Drug Protein with a Few Crystal Structures Based on the Adaboost-SVM. Comput. Math. Methods Med. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wan, X.; Xing, J.; Tan, X.; Li, X.; Wang, Y.; Zhao, J.; Wu, X.; Liu, X.; Li, Z.; et al. Deep Neural Network Classifier for Virtual Screening Inhibitors of (S)-Adenosyl-L-Methionine (SAM)-Dependent Methyltransferase Family. Front. Chem. 2019, 7. [Google Scholar] [CrossRef]
- Imrie, F.; Bradley, A.R.; van der Schaar, M.; Deane, C.M. Protein Family-Specific Models Using Deep Neural Networks and Transfer Learning Improve Virtual Screening and Highlight the Need for More Data. J. Chem. Inf. Model. 2018, 58, 2319–2330. [Google Scholar] [CrossRef]
- Sato, A.; Tanimura, N.; Honma, T.; Konagaya, A. Significance of Data Selection in Deep Learning for Reliable Binding Mode Prediction of Ligands in the Active Site of CYP3A4. Chem. Pharm. Bull. 2019, 67, 1183–1190. [Google Scholar] [CrossRef]
- Nguyen, D.D.; Gao, K.; Wang, M.; Wei, G.W. MathDL: Mathematical deep learning for D3R Grand Challenge 4. J. Comput.-Aided Mol. Des. 2019, 34, 131–147. [Google Scholar] [CrossRef]
- Cang, Z.; Mu, L.; Wei, G.W. Representability of algebraic topology for biomolecules in machine learning based scoring and virtual screening. PLoS Comput. Biol. 2018, 14, e1005929. [Google Scholar] [CrossRef]
- Zheng, L.; Fan, J.; Mu, Y. OnionNet: A Multiple-Layer Intermolecular-Contact-Based Convolutional Neural Network for Protein–Ligand Binding Affinity Prediction. ACS Omega 2019, 4, 15956–15965. [Google Scholar] [CrossRef]
- Mordalski, S.; Kosciolek, T.; Kristiansen, K.; Sylte, I.; Bojarski, A.J. Protein binding site analysis by means of structural interaction fingerprint patterns. Bioorganic Med. Chem. Lett. 2011, 21, 6816–6819. [Google Scholar] [CrossRef]
- Desaphy, J.; Bret, G.; Rognan, D.; Kellenberger, E. sc-PDB: A 3D-database of ligandable binding sites—10 years on. Nucleic Acids Res. 2014, 43, D399–D404. [Google Scholar] [CrossRef]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring with smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef]
- Huang, G.; Liu, Z.; Van Der Maaten, L.; Weinberger, K.Q. Densely connected convolutional networks. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, Honolulu, HI, USA, 21–26 July 2017; pp. 4700–4708. [Google Scholar] [CrossRef]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein–Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef]
- Jubb, H.C.; Higueruelo, A.P.; Ochoa-Montaño, B.; Pitt, W.R.; Ascher, D.B.; Blundell, T.L. Arpeggio: A Web Server for Calculating and Visualising Interatomic Interactions in Protein Structures. J. Mol. Biol. 2017, 429, 365–371. [Google Scholar] [CrossRef]
- Ballester, P.J.; Mitchell, J.B.O. A machine learning approach to predicting protein–ligand binding affinity with applications to molecular docking. Bioinformatics 2010, 26, 1169–1175. [Google Scholar] [CrossRef]
- Parks, C.D.; Gaieb, Z.; Chiu, M.; Yang, H.; Shao, C.; Walters, W.P.; Jansen, J.M.; McGaughey, G.; Lewis, R.A.; Bembenek, S.D.; et al. D3R grand challenge 4: Blind prediction of protein–ligand poses, affinity rankings, and relative binding free energies. J. Comput.-Aided Mol. Des. 2020, 34, 99–119. [Google Scholar] [CrossRef]
- Li, H.; Leung, K.S.; Wong, M.H.; Ballester, P.J. Improving AutoDock Vina Using Random Forest: The Growing Accuracy of Binding Affinity Prediction by the Effective Exploitation of Larger Data Sets. Mol. Inform. 2015, 34, 115–126. [Google Scholar] [CrossRef]
- Zhang, H.; Liao, L.; Saravanan, K.M.; Yin, P.; Wei, Y. DeepBindRG: A deep learning based method for estimating effective protein–ligand affinity. PeerJ 2019, 7, e7362. [Google Scholar] [CrossRef]
- Öztürk, H.; Ozkirimli, E.; Özgür, A. WideDTA: Prediction of drug-target binding affinity. arXiv 2019, arXiv:1902.04166. [Google Scholar]
- Tian, K.; Shao, M.; Wang, Y.; Guan, J.; Zhou, S. Boosting compound-protein interaction prediction by deep learning. Methods 2016, 110, 64–72. [Google Scholar] [CrossRef]
- Lee, I.; Keum, J.; Nam, H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 2019, 15, e1007129. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Li, Z.; Zhang, S.; Wang, S.; Wang, X.; Yuan, Q.; Wei, Z. Drug–target affinity prediction using graph neural network and contact maps. RSC Adv. 2020, 10, 20701–20712. [Google Scholar] [CrossRef]
- Feng, Q.; Dueva, E.V.; Cherkasov, A.; Ester, M. PADME: A Deep Learning-based Framework for Drug-Target Interaction Prediction. arXiv 2018, arXiv:1807.09741. [Google Scholar]
- Van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug–target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef]
- He, T.; Heidemeyer, M.; Ban, F.; Cherkasov, A.; Ester, M. SimBoost: A read-across approach for predicting drug–target binding affinities using gradient boosting machines. J. Cheminform. 2017, 9. [Google Scholar] [CrossRef]
- Woźniak, M.; Wołos, A.; Modrzyk, U.; Górski, R.L.; Winkowski, J.; Bajczyk, M.; Szymkuć, S.; Grzybowski, B.A.; Eder, M. Linguistic measures of chemical diversity and th “keywords” of molecular collections. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Sigrist, C.J.A.; Cerutti, L.; de Castro, E.; Langendijk-Genevaux, P.S.; Bulliard, V.; Bairoch, A.; Hulo, N. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res. 2009, 38, D161–D166. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2006, 35, D198–D201. [Google Scholar] [CrossRef]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2013, 42, D1091–D1097. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016, 45, D353–D361. [Google Scholar] [CrossRef]
- Southan, C.; Sharman, J.L.; Benson, H.E.; Faccenda, E.; Pawson, A.J.; Alexander, S.; Buneman, O.P.; Davenport, A.P.; McGrath, J.C.; Peters, J.A.; et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: Towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res. 2015, 44, D1054–D1068. [Google Scholar] [CrossRef]
- Bagley, S.C.; Altman, R.B. Characterizing the microenvironment surrounding protein sites. Protein Sci. 1995, 4, 622–635. [Google Scholar] [CrossRef]
- Michel, M.; Menéndez Hurtado, D.; Elofsson, A. PconsC4: Fast, accurate and hassle-free contact predictions. Bioinformatics 2018, 35, 2677–2679. [Google Scholar] [CrossRef]
- Cao, D.S.; Xu, Q.S.; Liang, Y.Z. propy: A tool to generate various modes of Chou’s PseAAC. Bioinformatics 2013, 29, 960–962. [Google Scholar] [CrossRef]
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep Neural Nets as a Method for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef]
- Ballester, P.J.; Schreyer, A.; Blundell, T.L. Does a More Precise Chemical Description of Protein–Ligand Complexes Lead to More Accurate Prediction of Binding Affinity? J. Chem. Inf. Model. 2014, 54, 944–955. [Google Scholar] [CrossRef]
- Wallach, I.; Heifets, A. Most Ligand-Based Classification Benchmarks Reward Memorization Rather than Generalization. J. Chem. Inf. Model. 2018, 58, 916–932. [Google Scholar] [CrossRef]
- Göller, A.H.; Kuhnke, L.; Montanari, F.; Bonin, A.; Schneckener, S.; ter Laak, A.; Wichard, J.; Lobell, M.; Hillisch, A. Bayer’s in silico ADMET platform: A journey of machine learning over the past two decades. Drug Discov. Today 2020, 25, 1702–1709. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3. [Google Scholar] [CrossRef]
- Chen, L.; Cruz, A.; Ramsey, S.; Dickson, C.J.; Duca, J.S.; Hornak, V.; Koes, D.R.; Kurtzman, T. Hidden bias in the DUD-E dataset leads to misleading performance of deep learning in structure-based virtual screening. PLoS ONE 2019, 14, e0220113. [Google Scholar] [CrossRef]
- Jiménez-Luna, J.; Skalic, M.; Weskamp, N.; Schneider, G. Coloring Molecules with Explainable Artificial Intelligence for Preclinical Relevance Assessment. J. Chem. Inf. Model. 2021. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Cortés-Ciriano, I. Artificial intelligence in drug discovery: What is realistic, what are illusions? Part 1: Ways to make an impact, and why we are not there yet. Drug Discov. Today 2020. [Google Scholar] [CrossRef]
- Bender, A.; Cortes-Ciriano, I. Artificial intelligence in drug discovery: What is realistic, what are illusions? Part 2: A discussion of chemical and biological data. Drug Discov. Today 2021. [Google Scholar] [CrossRef]
- Nguyen, H.; Case, D.A.; Rose, A.S. NGLview–interactive molecular graphics for Jupyter notebooks. Bioinformatics 2017, 34, 1241–1242. [Google Scholar] [CrossRef] [PubMed]
- Wójcikowski, M.; Zielenkiewicz, P.; Siedlecki, P. Open Drug Discovery Toolkit (ODDT): A new open-source player in the drug discovery field. J. Cheminform. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. The PyMOL Molecular Graphics System; Version 1.8; Schrödinger LLC: New York, NY, USA, 2015. [Google Scholar]
- Bradley, A.P. The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recognit. 1997, 30, 1145–1159. [Google Scholar] [CrossRef]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21. [Google Scholar] [CrossRef]
- Kvålseth, T.O. Cautionary Note about R2. Am. Stat. 1985, 39, 279–285. [Google Scholar] [CrossRef]
- Ash, A.; Shwartz, M. R2: A useful measure of model performance when predicting a dichotomous outcome. Stat. Med. 1999, 18, 375–384. [Google Scholar] [CrossRef]
- Benesty, J.; Chen, J.; Huang, Y.; Cohen, I. Pearson Correlation Coefficient; Springer: Berlin, Germany, 2009; pp. 1–4. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Size and Content 1 | Availability 2 |
|---|---|---|
| PDBbind v.2019 | structures + activities: general: 21,382; refined: 4852; core: 285 | http://www.pdbbind.org.cn |
| BindingDB | 2823 structures + activities 2,229,892 activities | https://www.bindingdb.org |
| BindingMOAD 2019 | 38,702 structures 15,964 structures + activities | https://bindingmoad.org |
| PubChem BioAssay 2020 | >280 million activities | https://pubchem.ncbi.nlm.nih.gov |
| ChEMBL v.28 | 17,276,334 activities | https://www.ebi.ac.uk/chembl |
| Name | Size | Data Source | Label | Availability |
|---|---|---|---|---|
| CASF-2016 | 57 targets 285 complexes | PDBbind | affinitiy | http://www.pdbbind.org.cn/casf.php |
| DUD-E | 102 targets 22,886 actives 50 decoys per active | PubChem, ZINC | active/decoy | http://dude.docking.org |
| MUV | 17 targets ∼90,000 compounds | PubChem, ZINC | active/decoy | MUV@TU Braunschweig 1 |
| Year | Name | Complex Encoding 1 | ML/DL Model | Framework |
|---|---|---|---|---|
| 2010 | Sato et al. [84] | IFP | SVM, RF, MLP | class. |
| 2016 | Wang et al. [135] | IFP | Adaboost-SVM | class. |
| 2019 | Li et al. [136] | IFP | MLP | class. |
| 2018 | gnina [90] | 3D grid | CNN | class. |
| 2018 | KDEEP [91] | 3D grid | CNN | reg. |
| 2018 | Pafnucy [89] | 3D grid | CNN | reg. |
| 2018 | DenseFS [137] | 3D grid | CNN | class. |
| 2019 | DeepAtom [92] | 3D grid | CNN | reg. |
| 2019 | Sato et al. [138] | 3D grid | CNN | class. |
| 2019 | Erdas-Cicek et al. [94] | 3D grid | CNN | reg. |
| 2019 | BindScope [93] | 3D grid | CNN | class. |
| 2018 | PotentialNet [96] | graph | GGNN | reg. |
| 2019 | Lim et al. [95] | graph | GANN | class. |
| 2017 | TopologyNet [97] | topol. | CNN | reg. |
| 2019 | Math-DL [139] | topol. | GAN, CNN | reg. |
| 2018 | Cang et al. [140] | topol. | CNN | reg. |
| 2016 | DeepVS [99] | atom contexts | CNN | class. |
| 2019 | OnionNet [141] | atom pairs | CNN | reg. |
| 2020 | Zhu et al. [98] | atom pairs | MLP | reg. |
| Year | Name | Ligand Encoding 1 | Protein Encoding 1 | ML/DL Model | Framework |
|---|---|---|---|---|---|
| 2018 | DeepDTA [60] | SMILES | full seq. | CNN | reg. |
| 2019 | WideDTA [152] | SMILES & MCS | full seq. & domains/motifs | CNN | reg. |
| 2019 | DeepAffinity [74] | SMILES | struct. property seq. | RNN+CNN | reg. |
| 2016 | DL-CPI [153] | substructure FP | domains | MLP | class. |
| 2018 | Kundu et al. [65] | div. feat. FP | div. feat. FP | RF & SVM & MLP | reg. |
| 2018 | Sorgenfrei et al. [70] | Morgan FP | z-scales | RF | class. |
| 2019 | DeepConv-DTI [154] | Morgan FP | full seq. | CNN | class. |
| 2019 | Torng and Altman [57] | graph | graph | GCNN | class. |
| 2020 | DGraphDTA [155] | graph | graph | GCNN | reg. |
| 2018 | PADME [156] | graph (or Morgan FP) | seq. comp. | GCNN (or MLP) | reg. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimber, T.B.; Chen, Y.; Volkamer, A. Deep Learning in Virtual Screening: Recent Applications and Developments. Int. J. Mol. Sci. 2021, 22, 4435. https://doi.org/10.3390/ijms22094435
Kimber TB, Chen Y, Volkamer A. Deep Learning in Virtual Screening: Recent Applications and Developments. International Journal of Molecular Sciences. 2021; 22(9):4435. https://doi.org/10.3390/ijms22094435
Chicago/Turabian StyleKimber, Talia B., Yonghui Chen, and Andrea Volkamer. 2021. "Deep Learning in Virtual Screening: Recent Applications and Developments" International Journal of Molecular Sciences 22, no. 9: 4435. https://doi.org/10.3390/ijms22094435
APA StyleKimber, T. B., Chen, Y., & Volkamer, A. (2021). Deep Learning in Virtual Screening: Recent Applications and Developments. International Journal of Molecular Sciences, 22(9), 4435. https://doi.org/10.3390/ijms22094435