
Assessing the Role of Calmodulin’s Linker Flexibility in Target Binding

Abstract
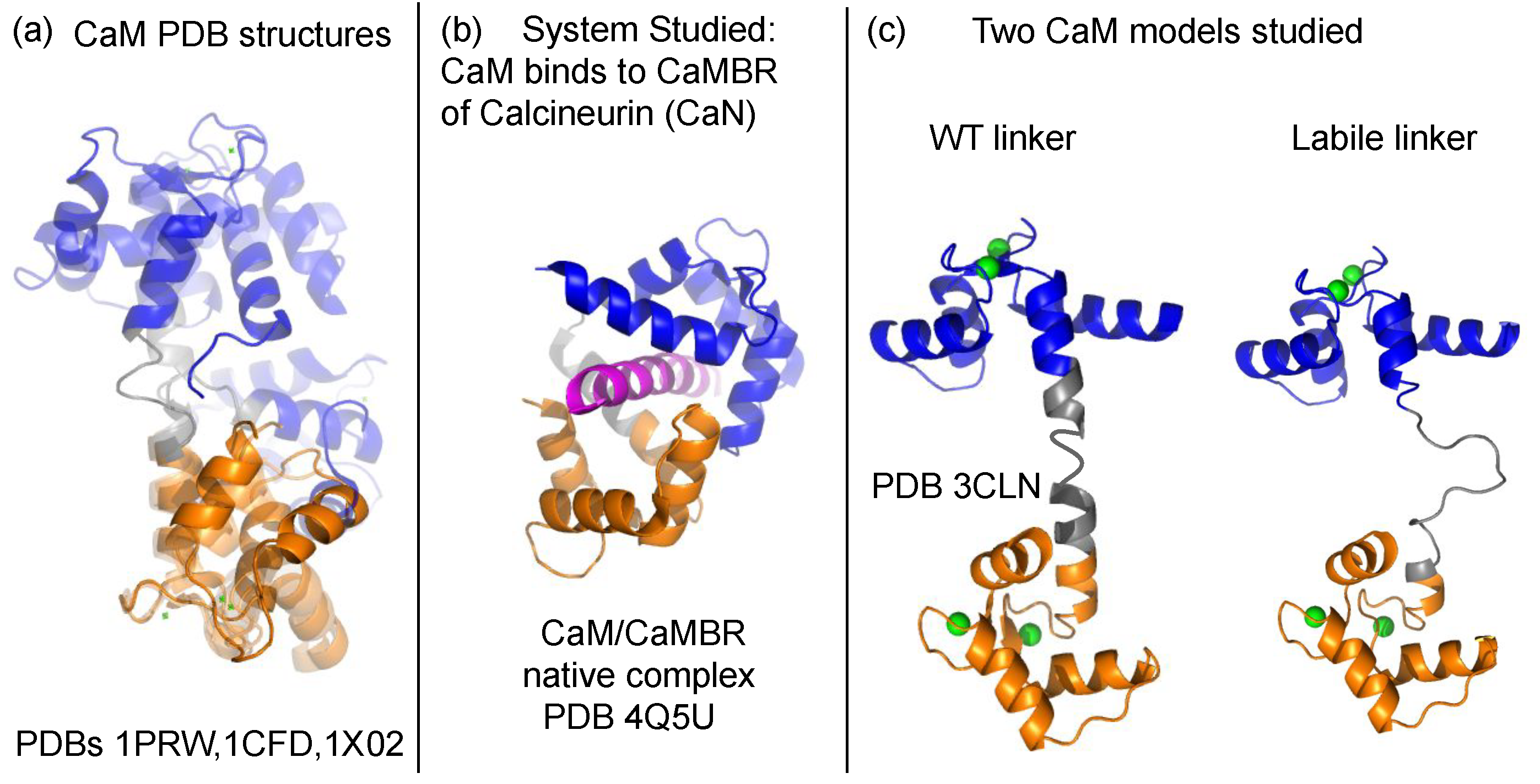
:1. Introduction
2. Materials and Methods
2.1. Martini CG Simulations
2.2. Analyses
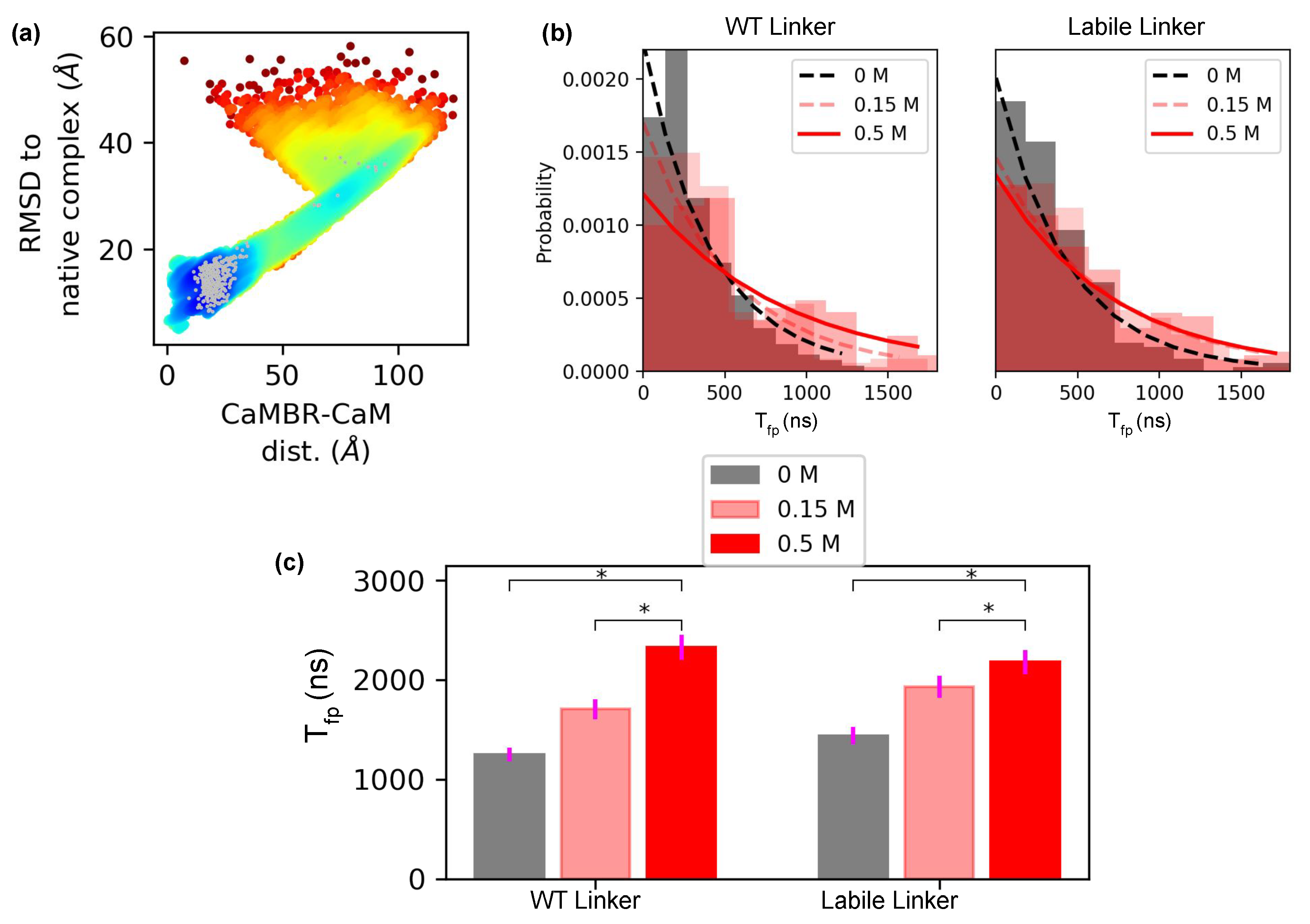
2.3. Association Rate Calculations Based on First Passage Time to Bound State
3. Results and Discussion
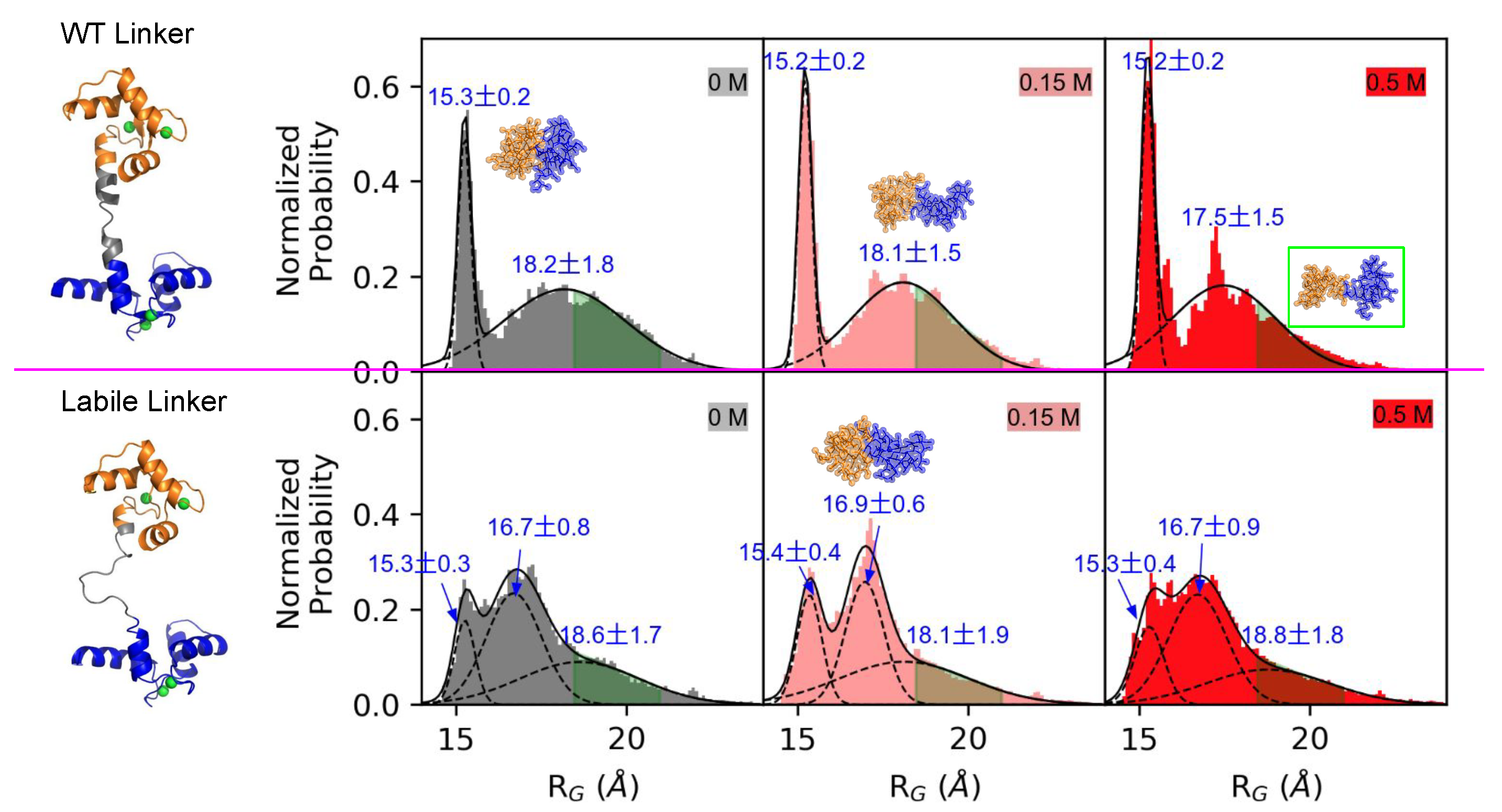
3.1. Linker Flexibility Impacts CaM/CaMBR Assembly
3.2. Linker Flexibility Determines the CaM Conformation Ensemble
3.3. Higher Linker Flexibility Attenuates the Sensitivity of the Association Rate to Ionic Strength
4. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CaM | Calmodulin |
| CaN | Calcineurin |
| CaMBR | CaM Binding Region |
| CG | Coarse-Grained |
| MD | Molecular Dynamics |
| COM | Center of Mass |
| RMSD | Root Mean Square Deviation |
References
- Kursula, P. The many structural faces of calmodulin: A multitasking molecular jackknife. Amino Acids 2014, 46, 2295–2304. [Google Scholar] [CrossRef]
- Munk, M.; Alcalde, J.; Lorentzen, L.; Villalobo, A.; Berchtold, M.W.; Panina, S. The impact of calmodulin on the cell cycle analyzed in a novel human cellular genetic system. Cell Calcium 2020, 88, 102207. [Google Scholar] [CrossRef] [PubMed]
- Shifman, J.M.; Mayo, S.L. Exploring the origins of binding specificity through the computational redesign of calmodulin. Proc. Natl. Acad. Sci. USA 2003, 100, 13274–13279. [Google Scholar] [CrossRef] [Green Version]
- Bayley, P.M.; Findlay, W.A.; Martin, S.R. Target recognition by calmodulin: Dissecting the kinetics and affinity of interaction using short peptide sequences. Protein Sci. 1996, 5, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Gsponer, J.; Christodoulou, J.; Cavalli, A.; Bui, J.M.; Richter, B.; Dobson, C.M.; Vendruscolo, M. A Coupled Equilibrium Shift Mechanism in Calmodulin-Mediated Signal Transduction. Structure 2008, 16, 736–746. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, S.E.; Yu, L.; Fowler, C.A.; Shea, M.A. Recognition of β-calcineurin by the domains of calmodulin: Thermodynamic and structural evidence for distinct roles. Proteins Struct. Funct. Bioinform. 2011, 79, 765–786. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Abrams, C.; Wang, L.; Gizzi, A.; He, L.; Lin, R.; Chen, Y.; Loll, P.J.; Pascal, J.M.; Zhang, J.F. Structural basis for calmodulin as a dynamic calcium sensor. Structure 2012, 20, 911–923. [Google Scholar] [CrossRef] [Green Version]
- Van Petegem, F. Slaying a giant: Structures of calmodulin and protein kinase a bound to the cardiac ryanodine receptor. Cell Calcium 2019, 83, 102079. [Google Scholar] [CrossRef]
- De Diego, I.; Kuper, J.; Bakalova, N.; Kursula, P.; Wilmanns, M. Molecular basis of the death-associated protein kinase-calcium/calmodulin regulator complex. Sci. Signal. 2010, 3, ra6. [Google Scholar] [CrossRef]
- Marlow, M.S.; Dogan, J.; Frederick, K.K.; Valentine, K.G.; Wand, A.J. The role of conformational entropy in molecular recognition by calmodulin. Nat. Chem. Biol. 2010, 6, 352–358. [Google Scholar] [CrossRef] [Green Version]
- Shukla, D.; Peck, A.; Pande, V.S. Conformational heterogeneity of the calmodulin binding interface. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Chu, X.; Lu, H.P.; Wang, J. Molecular mechanism of multispecific recognition of Calmodulin through conformational changes. Proc. Natl. Acad. Sci. USA 2017, 114, E3927–E3934. [Google Scholar] [CrossRef] [Green Version]
- Fiorin, G.; Pastore, A.; Carloni, P.; Parrinello, M. Using metadynamics to understand the mechanism of calmodulin/target recognition at atomic detail. Biophys. J. 2006, 91, 2768–2777. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Jas, G.S.; Kuczera, K. Structure, dynamics and interaction with kinase targets: Computer simulations of calmodulin. Biochim. Biophys. Acta Proteins Proteom. 2004, 1697, 289–300. [Google Scholar] [CrossRef]
- Westerlund, A.M.; Delemotte, L. Effect of Ca2+ on the promiscuous target-protein binding of calmodulin. PLoS Comput. Biol. 2018, 14, e1006072. [Google Scholar] [CrossRef] [Green Version]
- Mahling, R.; Rahlf, C.R.; Hansen, S.C.; Hayden, M.R.; Shea, M.A. Ca2+-Saturated calmodulin binds tightly to the N-terminal domain of A-type fibroblast growth factor homologous factors. J. Biol. Chem. 2021, 100458. [Google Scholar] [CrossRef]
- Anthis, N.J.; Clore, G.M. The length of the calmodulin linker determines the extent of transient interdomain association and target affinity. J. Am. Chem. Soc. 2013, 135, 9648–9651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Peng, S.; Cossins, B.P.; Liao, X.; Chen, K.; Shao, Q.; Zhu, X.; Shi, J.; Zhu, W. Mapping central α-helix linker mediated conformational transition pathway of calmodulin via simple computational approach. J. Phys. Chem. B 2014, 118, 9677–9685. [Google Scholar] [CrossRef]
- VanBerkum, M.F.; George, S.E.; Means, A.R. Calmodulin activation of target enzymes. Consequences of deletions in the central helix. J. Biol. Chem. 1990, 265, 3750–3756. [Google Scholar] [CrossRef]
- Tidow, H.; Nissen, P. Structural diversity of calmodulin binding to its target sites. FEBS J. 2013, 280, 5551–5565. [Google Scholar] [CrossRef]
- Yamniuk, A.P.; Vogel, H.J. Calmodulin’s flexibility allows for promiscuity in its interactions with target proteins and peptides. Appl. Biochem. Biotechnol. Part B Mol. Biotechnol. 2004, 27, 33–57. [Google Scholar] [CrossRef]
- Smith, D.M.; Straatsma, T.P.; Squier, T.C. Retention of conformational entropy upon calmodulin binding to target peptides is driven by transient salt bridges. Biophys. J. 2012, 103, 1576–1584. [Google Scholar] [CrossRef] [Green Version]
- Katyal, P.; Yang, Y.; Fu, Y.J.; Iandosca, J.; Vinogradova, O.; Lin, Y. Binding and backbone dynamics of protein under topological constraint: Calmodulin as a model system. Chem. Commun. 2018, 54, 8917–8920. [Google Scholar] [CrossRef] [PubMed]
- Quintana, A.R.; Wang, D.; Forbes, J.E.; Waxham, M.N. Kinetics of calmodulin binding to calcineurin. Biochem. Biophys. Res. Commun. 2005, 334, 674–680. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, P.; Hoffman, L.; Tripathi, S.; Homouz, D.; Liu, Y.; Waxham, M.N.; Cheung, M.S. Protein recognition and selection through conformational and mutually induced fit. Proc. Natl. Acad. Sci. USA 2013, 110, 20545–20550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunlap, T.B.; Kirk, J.M.; Pena, E.A.; Yoder, M.S.; Creamer, T.P. Thermodynamics of binding by calmodulin correlates with target peptide α-helical propensity. Proteins Struct. Funct. Bioinform. 2013, 81, 607–612. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Gao, M.; Xiong, J.; Su, Z.; Huang, Y. Features of molecular recognition of intrinsically disordered proteins via coupled folding and binding. Protein Sci. 2019, 28, 1952–1965. [Google Scholar] [CrossRef]
- Dogan, J.; Gianni, S.; Jemth, P. The binding mechanisms of intrinsically disordered proteins. Phys. Chem. Chem. Phys. 2014, 16, 6323–6331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollica, L.; Bessa, L.M.; Hanoulle, X.; Jensen, M.R.; Blackledge, M.; Schneider, R. Binding mechanisms of intrinsically disordered proteins: Theory, simulation, and experiment. Front. Mol. Biosci. 2016, 3, 52. [Google Scholar] [CrossRef] [Green Version]
- Das, P.; Matysiak, S.; Mittal, J. Looking at the Disordered Proteins through the Computational Microscope. ACS Cent. Sci. 2018, 4, 534–542. [Google Scholar] [CrossRef]
- Collins, A.P.; Anderson, P.C. Complete Coupled Binding-Folding Pathway of the Intrinsically Disordered Transcription Factor Protein Brinker Revealed by Molecular Dynamics Simulations and Markov State Modeling. Biochemistry 2018, 57, 4404–4420. [Google Scholar] [CrossRef]
- Saglam, A.S.; Chong, L.T. Protein-protein binding pathways and calculations of rate constants using fully-continuous, explicit-solvent simulations. Chem. Sci. 2019, 10, 2360–2372. [Google Scholar] [CrossRef] [Green Version]
- Souza, P.C.; Thallmair, S.; Conflitti, P.; Ramírez-Palacios, C.; Alessandri, R.; Raniolo, S.; Limongelli, V.; Marrink, S.J. Protein–ligand binding with the coarse-grained Martini model. Nat. Commun. 2020, 11, 3714. [Google Scholar] [CrossRef]
- Levy, Y.; Wolynes, P.G.; Onuchic, J.N. Protein topology determines binding mechanism. Proc. Natl. Acad. Sci. USA 2004, 101, 511–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.R.; Chen, J.; Wu, Y. Predicting Protein-protein Association Rates using Coarse-grained Simulation and Machine Learning. Sci. Rep. 2017, 7, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Chu, X.; Wang, J. Position-, disorder-, and salt-dependent diffusion in binding-coupled-folding of intrinsically disordered proteins. Phys. Chem. Chem. Phys. 2019, 21, 5634–5645. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Cyert, M.S. Identifying New Substrates and Functions for an Old Enzyme: Calcineurin. Cold Spring Harb. Perspect. Biol. 2020, 12, a035436. [Google Scholar] [CrossRef] [PubMed]
- Rusnak, F.; Mertz, P. Calcineurin: Form and function. Physiol. Rev. 2000, 80, 1483–1521. [Google Scholar] [CrossRef]
- Cook, E.C.; Creamer, T.P. Influence of electrostatic forces on the association kinetics and conformational ensemble of an intrinsically disordered protein. Proteins Struct. Funct. Bioinform. 2020. [Google Scholar] [CrossRef]
- Kar, P.; Mirams, G.R.; Christian, H.C.; Parekh, A.B. Control of NFAT Isoform Activation and NFAT-Dependent Gene Expression through Two Coincident and Spatially Segregated Intracellular Ca2+ Signals. Mol. Cell 2016, 64, 746–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, B.J.; Stewart, B.D.; Vaughan, D.D.; Bachstetter, A.D.; Kekenes-Huskey, P.M. Simulation of P2X-mediated calcium signalling in microglia. J. Physiol. 2019, 597, 799–818. [Google Scholar] [CrossRef] [Green Version]
- Ueda, Y.; Taketomi, H.; Gō, N. Studies on protein folding, unfolding, and fluctuations by computer simulation. II. A. Three-dimensional lattice model of lysozyme. Biopolymers 1978, 17, 1531–1548. [Google Scholar] [CrossRef]
- Marrink, S.J.; Tieleman, D.P. Perspective on the martini model. Chem. Soc. Rev. 2013, 42, 6801–6822. [Google Scholar] [CrossRef] [Green Version]
- Takada, S.; Kanada, R.; Tan, C.; Terakawa, T.; Li, W.; Kenzaki, H. Modeling Structural Dynamics of Biomolecular Complexes by Coarse-Grained Molecular Simulations. Acc. Chem. Res. 2015, 48, 3026–3035. [Google Scholar] [CrossRef]
- Sterpone, F.; Derreumaux, P.; Melchionna, S. Protein simulations in fluids: Coupling the OPEP coarse-grained force field with hydrodynamics. J. Chem. Theory Comput. 2015, 11, 1843–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, Y.S.; Bugg, C.E.; Cook, W.J. Structure of calmodulin refined at 2.2 Å resolution. J. Mol. Biol. 1988, 204, 191–204. [Google Scholar] [CrossRef]
- Dunlap, T.B.; Guo, H.F.; Cook, E.C.; Holbrook, E.; Rumi-Masante, J.; Lester, T.E.; Colbert, C.L.; Vander Kooi, C.W.; Creamer, T.P. Stoichiometry of the Calcineurin Regulatory Domain-Calmodulin Complex. Biochemistry 2014, 53, 5779–5790. [Google Scholar] [CrossRef]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S.J. The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef] [PubMed]
- De Jong, D.H.; Singh, G.; Bennett, W.F.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved parameters for the martini coarse-grained protein force field. J. Chem. Theory Comput. 2013, 9, 687–697. [Google Scholar] [CrossRef]
- Marrink, S.J.; De Vries, A.H.; Mark, A.E. Coarse Grained Model for Semiquantitative Lipid Simulations. J. Phys. Chem. B 2004, 108, 750–760. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Touw, W.G.; Baakman, C.; Black, J.; Te Beek, T.A.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Periole, X.; Cavalli, M.; Marrink, S.J.; Ceruso, M.A. Combining an elastic network with a coarse-grained molecular force field: Structure, dynamics, and intermolecular recognition. J. Chem. Theory Comput. 2009, 5, 2531–2543. [Google Scholar] [CrossRef] [Green Version]
- Yesylevskyy, S.O.; Schäfer, L.V.; Sengupta, D.; Marrink, S.J. Polarizable water model for the coarse-grained MARTINI force field. PLoS Comput. Biol. 2010, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Páll, S.; Zhmurov, A.; Bauer, P.; Abraham, M.; Lundborg, M.; Gray, A.; Hess, B.; Lindahl, E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 2020, 153, 134110. [Google Scholar] [CrossRef]
- Wassenaar, T.A.; Pluhackova, K.; Böckmann, R.A.; Marrink, S.J.; Tieleman, D.P. Going backward: A flexible geometric approach to reverse transformation from coarse grained to atomistic models. J. Chem. Theory Comput. 2014, 10, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.X. Rate theories for biologists. Q. Rev. Biophys. 2010, 43, 219–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buch, I.; Giorgino, T.; De Fabritiis, G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc. Natl. Acad. Sci. USA 2011, 108, 10184–10189. [Google Scholar] [CrossRef] [Green Version]
- Efron, B.; Tibshirani, R.J. An Introduction to the Bootstrap; Chapman & Hall/CRC: Boca Raton, FL, USA, 1994. [Google Scholar]
- Schreiber, G.; Haran, G.; Zhou, H.X. Fundamental aspects of protein—Protein association kinetics. Chem. Rev. 2009, 109, 839–860. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Cook, E.C.; Creamer, T.P.; Kekenes-Huskey, P.M. Electrostatic control of calcineurin’s intrinsically-disordered regulatory domain binding to calmodulin. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2651–2659. [Google Scholar] [CrossRef] [PubMed]
- Kurut, A.; Persson, B.A.; Åkesson, T.; Forsman, J.; Lund, M. Anisotropic interactions in protein mixtures: Self assembly and phase behavior in aqueous solution. J. Phys. Chem. Lett. 2012, 3, 731–734. [Google Scholar] [CrossRef] [Green Version]
- Lund, M. Anisotropic protein-protein interactions due to ion binding. Colloids Surf. B Biointerfaces 2016, 137, 17–21. [Google Scholar] [CrossRef]
- Walton, S.D.; Chakravarthy, H.; Shettigar, V.; O’Neil, A.J.; Siddiqui, J.K.; Jones, B.R.; Tikunova, S.B.; Davis, J.P. Divergent soybean calmodulins respond similarly to calcium transients: Insight into differential target regulation. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallon, J.L.; Quiocho, F.A. A closed compact structure of native Ca2+-calmodulin. Structure 2003, 11, 1303–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delfino, F.; Porozov, Y.; Stepanov, E.; Tamazian, G.; Tozzini, V. Structural Transition States Explored With Minimalist Coarse Grained Models: Applications to Calmodulin. Front. Mol. Biosci. 2019, 6, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokh, D.B.; Amaral, M.; Bomke, J.; Grädler, U.; Musil, D.; Buchstaller, H.P.; Dreyer, M.K.; Frech, M.; Lowinski, M.; Vallee, F.; et al. Estimation of Drug-Target Residence Times by τ-Random Acceleration Molecular Dynamics Simulations. J. Chem. Theory Comput. 2018, 14, 3859–3869. [Google Scholar] [CrossRef]
- Pan, A.C.; Xu, H.; Palpant, T.; Shaw, D.E. Quantitative Characterization of the Binding and Unbinding of Millimolar Drug Fragments with Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 3372–3377. [Google Scholar] [CrossRef]
- Bruce, N.J.; Ganotra, G.K.; Kokh, D.B.; Sadiq, S.K.; Wade, R.C. New approaches for computing ligand–receptor binding kinetics. Curr. Opin. Struct. Biol. 2018, 49, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Winquist, J.; Geschwindner, S.; Xue, Y.; Gustavsson, L.; Musil, D.; Deinum, J.; Danielson, U.H. Identification of structural-kinetic and structural-thermodynamic relationships for thrombin inhibitors. Biochemistry 2013, 52, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Sugase, K.; Dyson, H.J.; Wright, P.E. Conformational propensities of intrinsically disordered proteins influence the mechanism of binding and folding. Proc. Natl. Acad. Sci. USA 2015, 112, 9614–9619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, W.T.; Clarke, J.; Shammas, S.L.; Wang, J. Role of non-native electrostatic interactions in the coupled folding and binding of PUMA with Mcl-1. PLoS Comput. Biol. 2017, 13, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Gerlach, G.J.; Carrock, R.; Stix, R.; Stollar, E.J.; Aurelia Ball, K. A disordered encounter complex is central to the yeast Abp1p SH3 domain binding pathway. PLoS Comput. Biol. 2020, 16, e1007815. [Google Scholar] [CrossRef]
- Chu, W.T.; Shammas, S.L.; Wang, J. Charge Interactions Modulate the Encounter Complex Ensemble of Two Differently Charged Disordered Protein Partners of KIX. J. Chem. Theory Comput. 2020, 16, 3856–3868. [Google Scholar] [CrossRef]
- Kovacs, E.; Tóth, J.; Vé Rtessy, B.G.; Liliom, K. Dissociation of calmodulin-target peptide complexes by the lipid mediator sphingosylphosphorylcholine: Implications in calcium signaling. J. Biol. Chem. 2010, 285, 1799–1808. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Kim, J.C.; Lee, M.S.; Do Heo, W.; Seo, H.Y.; Yoon, H.W.; Hong, J.C.; Lee, S.Y.; Bahk, J.D.; Hwang, I.; et al. Identification of a novel divergent calmodulin isoform from soybean which has differential ability to activate calmodulin-dependent enzymes. J. Biol. Chem. 1995, 270, 21806–21812. [Google Scholar] [CrossRef] [Green Version]
- Jensen, H.H.; Brohus, M.; Nyegaard, M.; Overgaard, M.T. Human calmodulin mutations. Front. Mol. Neurosci. 2018, 11, 396. [Google Scholar] [CrossRef] [Green Version]
- Benaim, G.; Villalobo, A. Phosphorylation of calmodulin: Functional implications. Eur. J. Biochem. 2002, 269, 3619–3631. [Google Scholar] [CrossRef]
- Towns, J.; Cockerill, T.; Dahan, M.; Foster, I.; Gaither, K.; Grimshaw, A.; Hazlewood, V.; Lathrop, S.; Lifka, D.; Peterson, G.D.; et al. XSEDE: Accelerating Scientific Discovery. Comput. Sci. Eng. 2014, 16, 62–74. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT Linker | Labile Linker | |||||
|---|---|---|---|---|---|---|
| 0 M | 0.15 M | 0.5 M | 0 M | 0.15 M | 0.5 M | |
| T (ns) | 1247.78 ± 0.05 | 1708.28 ± 0.06 | 2326.98 ± 0.05 | 1441.32 ± 0.06 | 1930.28 ± 0.06 | 2178.93 ± 0.06 |
| (10 M s, 300 K) a | 16.29 ± 0.92 | 11.90 ± 0.70 | 8.73 ± 0.46 | 14.10 ± 0.85 | 10.53 ± 0.60 | 9.33 ± 0.53 |
| Expt. (10 M s, 310 K) b | 2.2 ± 0.44 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, B.; Kekenes-Huskey, P.M. Assessing the Role of Calmodulin’s Linker Flexibility in Target Binding. Int. J. Mol. Sci. 2021, 22, 4990. https://doi.org/10.3390/ijms22094990
Sun B, Kekenes-Huskey PM. Assessing the Role of Calmodulin’s Linker Flexibility in Target Binding. International Journal of Molecular Sciences. 2021; 22(9):4990. https://doi.org/10.3390/ijms22094990
Chicago/Turabian StyleSun, Bin, and Peter M. Kekenes-Huskey. 2021. "Assessing the Role of Calmodulin’s Linker Flexibility in Target Binding" International Journal of Molecular Sciences 22, no. 9: 4990. https://doi.org/10.3390/ijms22094990