
Transcriptome Analysis of Salt Stress in Hibiscus hamabo Sieb. et Zucc Based on Pacbio Full-Length Transcriptome Sequencing

 and
and
Abstract
:1. Introduction
2. Results
2.1. SMRT Sequencing, Data Processing, and Annotation
2.2. RNA-Seq Analysis during Salt Stress
2.3. DEGs Cluster Analysis
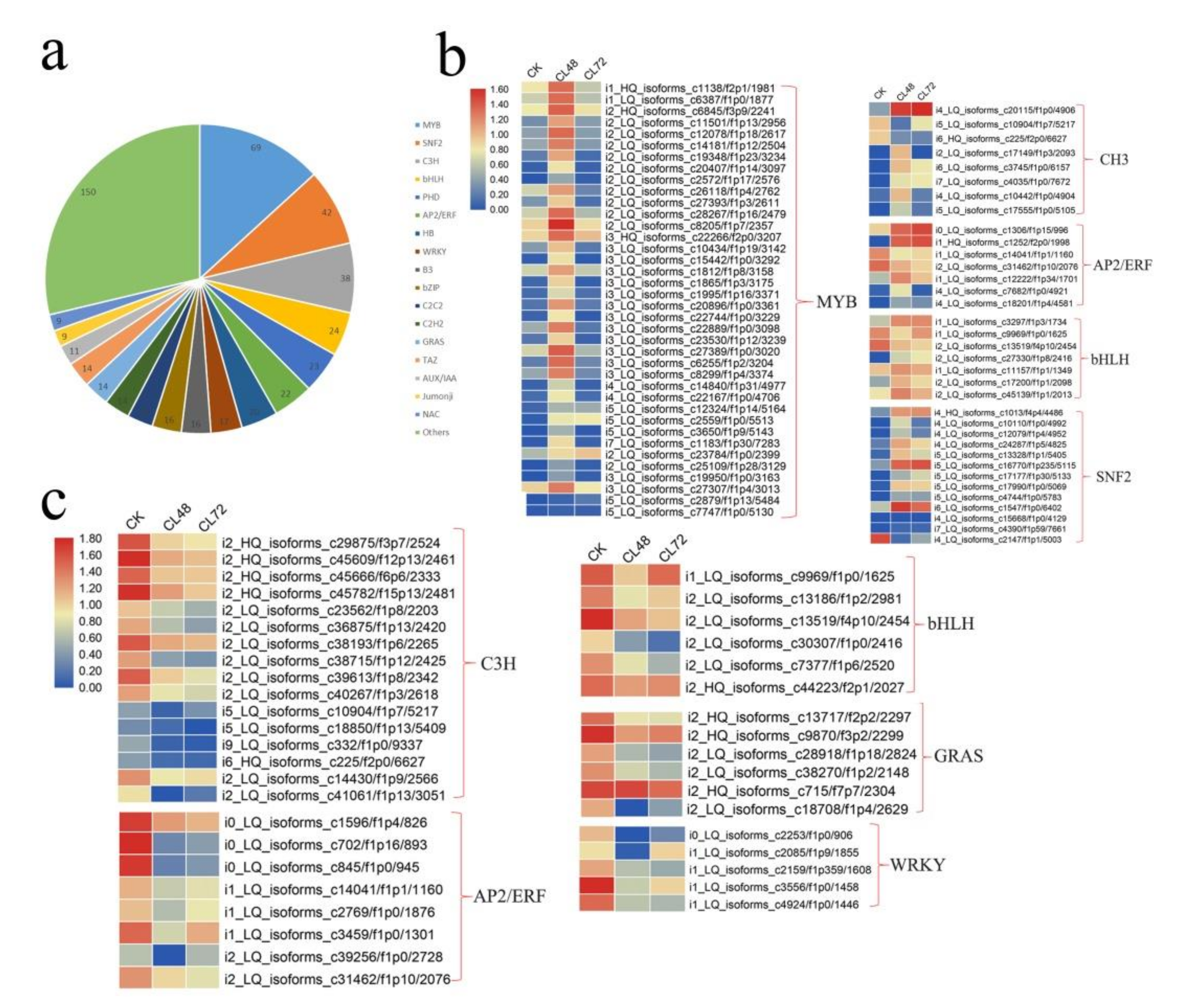
2.4. Genes Encoding Transcription Factors in Response to Salt Stress
2.5. Genes Related to Plant Hormones and Ion Signal Transduction Play an Important Role in the Response of H. Hamabo to Salt Stress
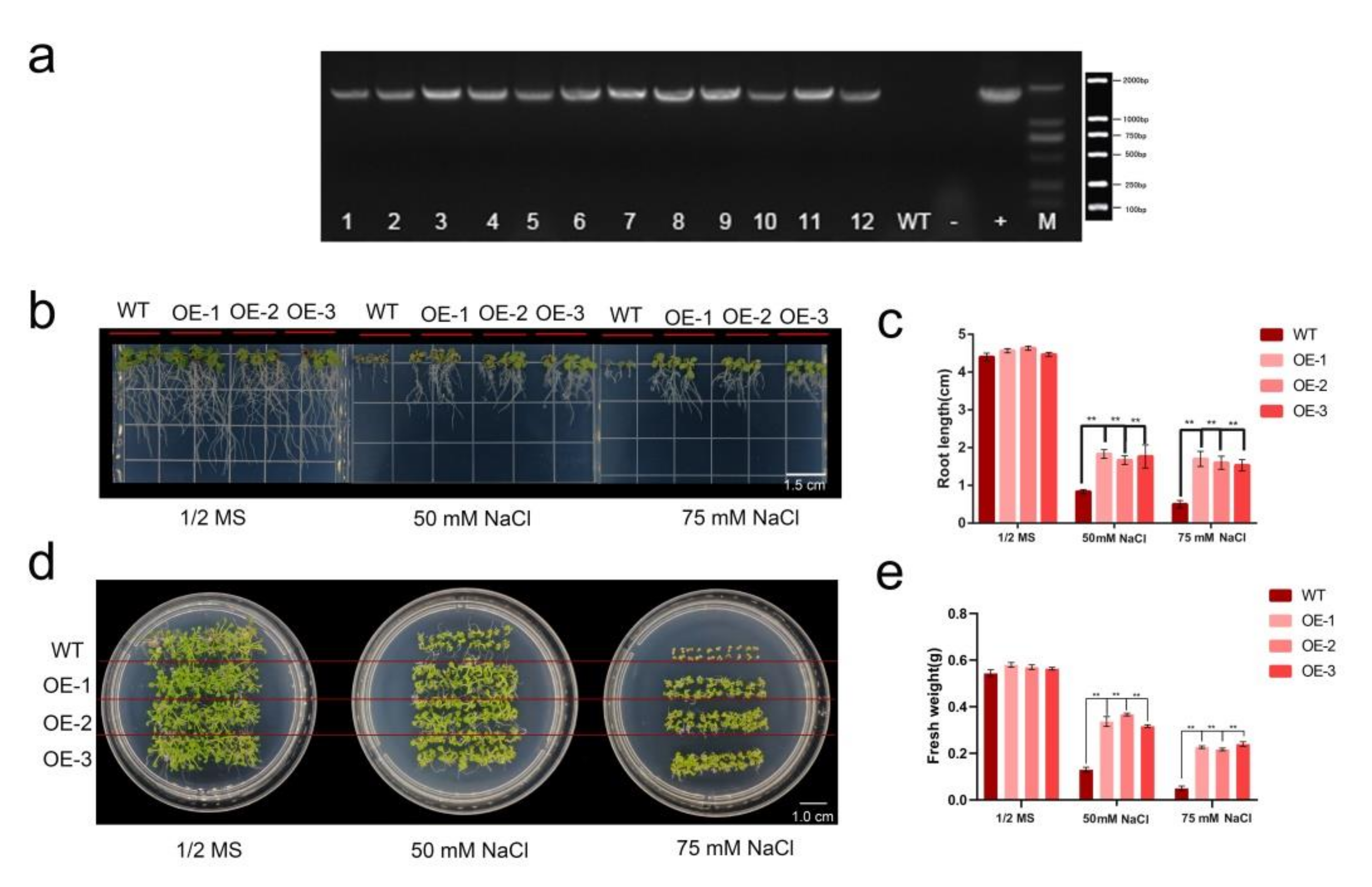
2.6. HhWRKY79 Overexpression Increases Transgenic A. Thaliana Salt Tolerance
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Salt Treatment
4.2. Iso-Seq Library RNA Preparation, Sequencing, and Analysis
4.3. RNA-Seq Library RNA Preparation, Sequencing, and Analysis
4.4. Differentially Expressed Gene Analysis
4.5. Generation of HhWRKY79 Transgenic A. Thaliana Plants
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liang, W.; Ma, X.; Wan, P.; Liu, L. Plant salt-tolerance mechanism: A review. Biochem. Biophys. Res. Commun. 2018, 495, 286–291. [Google Scholar] [CrossRef]
- van Zelm, E.; Zhang, Y.; Testerink, C. Salt tolerance mechanisms of plants. Annu. Rev. Plant. Biol. 2020, 71, 403–433. [Google Scholar] [CrossRef] [Green Version]
- Ni, L.; Wang, Z.; Guo, J.; Pei, X.; Liu, L.; Li, H.; Yuan, H.; Gu, C. Full-Length transcriptome sequencing and comparative transcriptome analysis to evaluate drought and salt stress in Iris lactea var. chinensis. Genes 2021, 12, 434. [Google Scholar] [CrossRef] [PubMed]
- Perrino, E.V.; Valerio, F.; Gannouchi, A.; Trani, A.; Mezzapesa, G. Ecological and plant community implication on essential oils composition in useful wild officinal species: A pilot case study in Apulia (Italy). Plants 2021, 10, 574. [Google Scholar] [CrossRef] [PubMed]
- Møller, I.S.; Tester, M. Salinity tolerance of Arabidopsis: A good model for cereals? Trends Plant Sci. 2007, 12, 534–540. [Google Scholar] [CrossRef]
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Organization of cis-acting regulatory elements in osmotic- and cold-stress-responsive promoters. Trends Plant Sci. 2005, 10, 88–94. [Google Scholar] [CrossRef]
- Urano, K.; Kurihara, Y.; Seki, M.; Shinozaki, K. ’Omics’ analyses of regulatory networks in plant abiotic stress responses. Curr. Opin. Plant Biol. 2010, 13, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Xiao, Q.; Pan, J.Y.; Wu, J. Natural products from semi-mangrove flora: Source, chemistry and bioactivities. Nat. Prod. Rep. 2009, 26, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H. Distribution and ecology of the semi-mangrove, Hibiscus hamabo community in western Kyushu, Japan. Veg. Sci. 2017, 17, 81–88. [Google Scholar] [CrossRef]
- Wu, J.; Xiao, Q.; Xu, J.; Li, M.-Y.; Pan, J.-Y.; Yang, M.-H. Natural products from true mangrove flora: Source, chemistry and bioactivities. Nat. Pro. Rep. 2008, 25, 955–981. [Google Scholar] [CrossRef]
- Wang, Z.; Ni, L.; Guo, J.; Liu, L.; Li, H.; Yin, Y.; Gu, C. Phylogenetic and transcription analysis of Hibiscus hamabo sieb. et Zucc. WRKY transcription factors. DNA Cell Bio. 2020, 39, 1141–1154. [Google Scholar] [CrossRef] [PubMed]
- Qizhi, W.; Yu, C.; Hao, S.; Fuqin, G.; Youyi, Z.; Xu, F.; Ming, W. Research development on Hibiscus hamabo. Chin. Wild Plant Resour. 2012, 31, 8–10. [Google Scholar]
- Wang, Z.; Xu, X.; Ni, L.; Guo, J.; Gu, C. Efficient virus-induced gene silencing in Hibiscus hamabo Sieb. et Zucc. using Tobacco rattle virus. PeerJ 2019, 7, e7505. [Google Scholar] [CrossRef] [Green Version]
- Bo, P.-F.; Sun, X.-L.; Sun, T.-H.; Du, X.-H.; Yu, C.-Y. Antioxidative system and osmotic regulation in seedlings of annual Hibiscus hamabo Sieb. et Zucc. under salt stress. Acta Bot. Boreali-Occident. Sin. 2008, 28, 113–118. [Google Scholar]
- Wang, T.-T.; Yu, T.-F.; Fu, J.-D.; Su, H.-G.; Chen, J.; Zhou, Y.-B.; Chen, M.; Guo, J.; Ma, Y.-Z.; Wei, W.-L.; et al. Genome-wide analysis of the GRAS gene family and functional identification of GmGRAS37 in drought and salt tolerance. Front. Plant Sci. 2020, 11, 2024. [Google Scholar] [CrossRef]
- Ni, L.; Wang, Z.; Fu, Z.; Liu, D.; Yin, Y.; Li, H.; Gu, C. Genome-wide analysis of basic Helix-Loop-Helix family genes and expression analysis in response to drought and salt stresses in Hibiscus hamabo Sieb. et Zucc. Int. J. Mol. Sci. 2021, 22, 8748. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Li, G.; Xu, S.; Wu, W.; Chen, Q.; Shao, S.; Liu, M.; Wang, N.; Zhong, C.; He, Z.; et al. Genomic insights into molecular adaptation to intertidal environments in the mangrove Aegiceras corniculatum. New Phytol. 2021, 231, 2346–2358. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, P.; Mohanty, B.; Wijaya, E.; Lee, D.-Y.; Lim, T.-M.; Lin, Q.; Xu, J.; Loh, C.-S.; Kumar, P.P. Transcriptomics analysis of salt stress tolerance in the roots of the mangrove Avicennia officinalis. Sci. Rep. 2017, 7, 10031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, W.; Wang, T.; Wei, W.; Lou, S.; Lan, F.; Zhu, S.; Li, Q.; Ji, G.; Lin, C.; Wu, X. The full-length transcriptome of Spartina alterniflora reveals the complexity of high salt tolerance in monocotyledonous halophyte. Plant Cell Physiol. 2020, 61, 882–896. [Google Scholar] [CrossRef]
- Wang, Z.; Ni, L.; Hua, J.; Liu, L.; Yin, Y.; Li, H.; Gu, C. Transcriptome analysis reveals regulatory framework for salt and drought tolerance in Hibiscus hamabo siebold & zuccarini. Forests 2021, 12, 454. [Google Scholar] [CrossRef]
- Sun, M.; Huang, D.; Zhang, A.; Khan, I.; Yan, H.; Wang, X.; Zhang, X.; Zhang, J.; Huang, L. Transcriptome analysis of heat stress and drought stress in pearl millet based on Pacbio full-length transcriptome sequencing. BMC Plant Biol. 2020, 20, 323. [Google Scholar] [CrossRef]
- Zhu, J.-K. Abiotic stress signaling and responses in plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Ding, Y.; Yang, Y.; Song, C.; Gong, Z. Protein kinases in plant responses to drought, salt, and cold stress. J. Integr. Plant Biol. 2021, 63, 53–78. [Google Scholar] [CrossRef]
- Luo, D.; Zhou, Q.; Wu, Y.; Chai, X.; Liu, W.; Wang, Y.; Yang, Q.; Wang, Z.; Liu, Z. Full-length transcript sequencing and comparative transcriptomic analysis to evaluate the contribution of osmotic and ionic stress components towards salinity tolerance in the roots of cultivated alfalfa (Medicago sativa L.). BMC Plant Biol. 2019, 19, 32. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Zhou, Q.; Ma, L.; Xie, W.; Wang, Y.; Hu, X.; Liu, Z. Novel polymorphic expressed-sequence tag–simple-sequence repeat markers in Campeiostachys nutans for genetic diversity analyses. Crop Sci. 2015, 55, 2712–2718. [Google Scholar] [CrossRef] [Green Version]
- Dong, R.; Dong, D.; Luo, D.; Zhou, Q.; Chai, X.; Zhang, J.; Xie, W.; Liu, W.; Dong, Y.; Wang, Y. Transcriptome analyses reveal candidate pod shattering-associated genes involved in the pod ventral sutures of common vetch (Vicia sativa L.). Front. Plant Sci. 2017, 8, 649. [Google Scholar] [CrossRef]
- Zhou, Q.; Luo, D.; Ma, L.; Xie, W.; Wang, Y.; Wang, Y.; Liu, Z. Development and cross-species transferability of EST-SSR markers in Siberian wildrye (Elymus sibiricus L.) using Illumina sequencing. Sci. Rep. 2016, 6, 20549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, D.; Jalmi, S.K.; Bhagat, P.K.; Verma, N.; Sinha, A.K. A bHLH transcription factor, MYC2, imparts salt intolerance by regulating proline biosynthesis in Arabidopsis. FEBS J. 2020, 287, 2560–2576. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, Y.; Cai, H.; Guo, M.; Chai, M.; She, Z.; Ye, L.; Cheng, Y.; Wang, B.; Qin, Y. The bZIP transcription factor GmbZIP15 negatively regulates salt-and drought-stress responses in soybean. Int. J. Mol. Sci. 2020, 21, 7778. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, G.; Sun, C.; Sui, N. Research advances of MYB transcription factors in plant stress resistance and breeding. Plant Signal. Behav. 2019, 14, 1613131. [Google Scholar] [CrossRef]
- Ma, X.; Liang, W.; Gu, P.; Huang, Z. Salt tolerance function of the novel C2H2-type zinc finger protein TaZNF in wheat. Plant Physiol. Bioch. 2016, 106, 129–140. [Google Scholar] [CrossRef]
- Qu, Y.; Nong, Q.; Jian, S.; Lu, H.; Zhang, M.; Xia, K. An AP2/ERF Gene, HuERF1, from pitaya (Hylocereus undatus) positively regulates salt tolerance. Int. J. Mol. Sci. 2020, 21, 4586. [Google Scholar] [CrossRef]
- Lv, B.; Wu, Q.; Wang, A.; Li, Q.; Dong, Q.; Yang, J.; Zhao, H.; Wang, X.; Chen, H.; Li, C. A WRKY transcription factor, FtWRKY46, from Tartary buckwheat improves salt tolerance in transgenic Arabidopsis thaliana. Plant Physiol. Bioch. 2020, 147, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-K. Salt and drought stress signal transduction in plants. Ann. Rev. Plant Biol. 2002, 53, 247–273. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Liu, H.; Han, G.; Cai, R.; Pan, F.; Xiang, Y. A moso bamboo WRKY gene PeWRKY83 confers salinity tolerance in transgenic Arabidopsis plants. Sci. Rep. 2017, 7, 11721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Q.; Zheng, W.; Duan, D.; Huang, D.; Wang, Q.; Liu, C.; Li, C.; Gong, X.; Li, C.; Mao, K. MdWRKY30, a group IIa WRKY gene from apple, confers tolerance to salinity and osmotic stresses in transgenic apple callus and Arabidopsis seedlings. Plant Sci. 2020, 299, 110611. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Song, A.; Gao, C.; Wang, L.; Wang, Y.; Sun, J.; Jiang, J.; Chen, F.; Chen, S. Chrysanthemum WRKY gene CmWRKY17 negatively regulates salt stress tolerance in transgenic chrysanthemum and Arabidopsis plants. Plant Cell Rep. 2015, 34, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Wang, X.; Chen, M.; Chai, T.; Wang, H. A WRKY transcription factor, PcWRKY33, from Polygonum cuspidatum reduces salt tolerance in transgenic Arabidopsis thaliana. Plant Cell Rep. 2018, 37, 1033–1048. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Liu, L.; Song, A.; Liu, Z.; Zhang, Y.; Huang, S. Iris lactea var. chinensis (Fisch.) cysteine-rich gene llCDT1 enhances cadmium tolerance in yeast cells and Arabidopsis thaliana. Ecotoxicol. Environ. Safe 2018, 157, 67–72. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pacbio | |
|---|---|
| Subreads base (G) | 9.92 |
| Subreads number | 3,287,061 |
| Average subreads length | 3017 |
| CCS | 274,380 |
| 5′-primer | 244,651 |
| 3′-primer | 245,645 |
| Poly-A | 206,861 |
| Flnc | 177,383 |
| Average flnc read length | 3829 |
| Number of isoform | 121,091 |
| Number of unigene | 94,562 |
| Mean_length | 4206 |
| Min_length | 161 |
| Max_length | 14,547 |
| ExN50 (consensus) | 3766 |
| ExN90 (consensus) | 7144 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, L.; Wang, Z.; Liu, X.; Wu, S.; Hua, J.; Yin, Y.; Li, H.; Gu, C. Transcriptome Analysis of Salt Stress in Hibiscus hamabo Sieb. et Zucc Based on Pacbio Full-Length Transcriptome Sequencing. Int. J. Mol. Sci. 2022, 23, 138. https://doi.org/10.3390/ijms23010138
Ni L, Wang Z, Liu X, Wu S, Hua J, Yin Y, Li H, Gu C. Transcriptome Analysis of Salt Stress in Hibiscus hamabo Sieb. et Zucc Based on Pacbio Full-Length Transcriptome Sequencing. International Journal of Molecular Sciences. 2022; 23(1):138. https://doi.org/10.3390/ijms23010138
Chicago/Turabian StyleNi, Longjie, Zhiquan Wang, Xiangdong Liu, Shuting Wu, Jianfeng Hua, Yunlong Yin, Huogen Li, and Chunsun Gu. 2022. "Transcriptome Analysis of Salt Stress in Hibiscus hamabo Sieb. et Zucc Based on Pacbio Full-Length Transcriptome Sequencing" International Journal of Molecular Sciences 23, no. 1: 138. https://doi.org/10.3390/ijms23010138
APA StyleNi, L., Wang, Z., Liu, X., Wu, S., Hua, J., Yin, Y., Li, H., & Gu, C. (2022). Transcriptome Analysis of Salt Stress in Hibiscus hamabo Sieb. et Zucc Based on Pacbio Full-Length Transcriptome Sequencing. International Journal of Molecular Sciences, 23(1), 138. https://doi.org/10.3390/ijms23010138