
Altered HIV-1 mRNA Splicing Due to Drug-Resistance-Associated Mutations in Exon 2/2b

 ,
,
Abstract
:1. Introduction
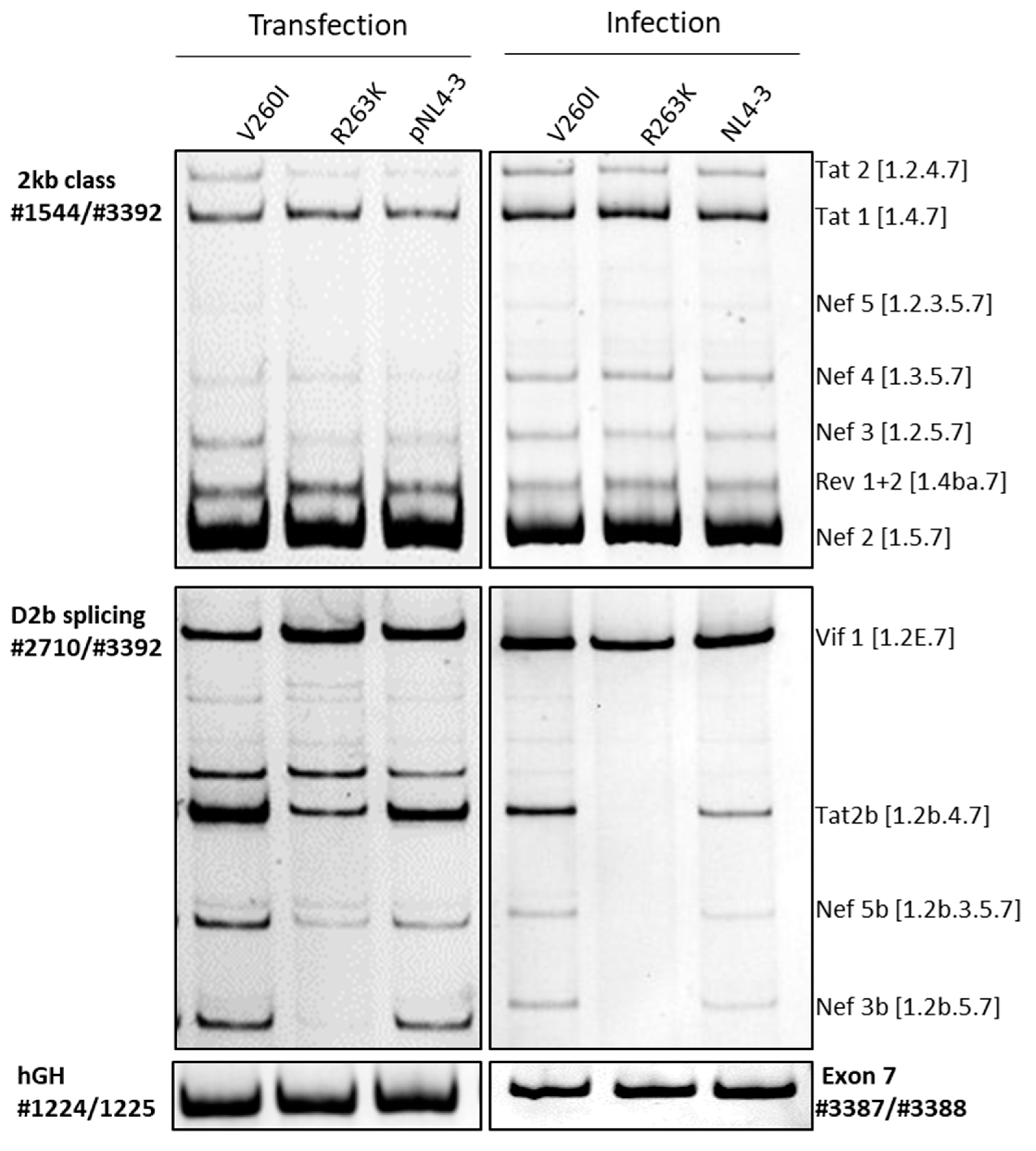
2. Results
3. Discussion
4. Materials and Methods
4.1. Proviral Plasmids
4.2. Three-Exon Minigenes
4.3. Expression Plasmids
4.4. Cell Culture, Infection, and Transfection
4.5. RNA Isolation and RT-PCR
4.6. Bioinformatic Tools
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buchholz, F.; Hauber, J. Antiviral therapy of persistent viral infection using genome editing. Curr. Opin. Virol. 2016, 20, 85–91. [Google Scholar] [CrossRef]
- Archin, N.M.; Sung, J.M.; Garrido, C.; Soriano-Sarabia, N.; Margolis, D.M. Eradicating HIV-1 infection: Seeking to clear a persistent pathogen. Nat. Rev. Genet. 2014, 12, 750–764. [Google Scholar] [CrossRef] [Green Version]
- Shang, H.-T.; Ding, J.-W.; Yu, S.-Y.; Wu, T.; Zhang, Q.-L.; Liang, F.-J. Progress and challenges in the use of latent HIV-1 reactivating agents. Acta Pharmacol. Sin. 2015, 36, 908–916. [Google Scholar] [CrossRef] [Green Version]
- Bradley, H.; Mattson, C.L.; Beer, L.; Huang, P.; Shouse, R.L. Increased antiretroviral therapy prescription and HIV viral suppression among persons receiving clinical care for HIV infection. AIDS 2016, 30, 2117–2124. [Google Scholar] [CrossRef]
- Maskew, M.; Bor, J.; MacLeod, W.; Carmona, S.; Sherman, G.G.; Fox, M.P. Adolescent Hiv Treatment in South Africa’s National Hiv Programme: A Retrospective Cohort Study. Lancet HIV 2019, 6, e760–e768. [Google Scholar] [CrossRef]
- Montaner, J.S.; Lima, V.D.; Harrigan, P.R.; Lourenço, L.; Yip, B.; Nosyk, B.; Wood, E.; Kerr, T.; Shannon, K.; Moore, D.; et al. Expansion of HAART Coverage Is Associated with Sustained Decreases in HIV/AIDS Morbidity, Mortality and HIV Transmission: The “HIV Treatment as Prevention” Experience in a Canadian Setting. PLoS ONE 2014, 9, e87872. [Google Scholar] [CrossRef]
- Keita, A.; Sereme, Y.; Pillet, S.; Coulibaly, S.; Diallo, F.; Pozzetto, B.; Thiero, T.A.; Bourlet, T. Impact of HIV-1 primary drug resistance on the efficacy of a first-line antiretroviral regimen in the blood of newly diagnosed individuals in Bamako, Mali. J. Antimicrob. Chemother. 2019, 74, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Nachega, J.B.; Marconi, V.C.; van Zyl, G.U.; Gardner, E.M.; Preiser, W.; Hong, S.Y.; Mills, E.J.; Gross, R. HIV Treatment Adherence, Drug Resistance, Virologic Failure: Evolving Concepts. Infect. Disord. Drug Targets 2011, 11, 167–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sertznig, H.; Hillebrand, F.; Erkelenz, S.; Schaal, H.; Widera, M. Behind the scenes of HIV-1 replication: Alternative splicing as the dependency factor on the quiet. Virology 2018, 516, 176–188. [Google Scholar] [CrossRef]
- Jayappa, K.D.; Ao, Z.; Yao, X. The HIV-1 passage from cytoplasm to nucleus: The process involving a complex exchange between the components of HIV-1 and cellular machinery to access nucleus and successful integration. Int. J. Biochem. Mol. Boil. 2012, 3, 70–85. [Google Scholar]
- Liu, R.-D.; Wu, J.; Shao, R.; Xue, Y.-H. Mechanism and factors that control HIV-1 transcription and latency activation. J. Zhejiang Univ. Sci. B 2014, 15, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karn, J.; Stoltzfus, C.M. Transcriptional and Posttranscriptional Regulation of HIV-1 Gene Expression. Cold Spring Harb. Perspect. Med. 2011, 2, a006916. [Google Scholar] [CrossRef]
- Purcell, D.F.; Martin, M.A. Alternative Splicing of Human Immunodeficiency Virus Type 1 Mrna Modulates Viral Protein Expression, Replication, and Infectivity. J. Virol. 1993, 67, 6365–6378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, M.A.; Soll, S.J.; Emery, A.; Blanco-Melo, D.; Swanstrom, R.; Bieniasz, P.D. Global synonymous mutagenesis identifies cis-acting RNA elements that regulate HIV-1 splicing and replication. PLoS Pathog. 2018, 14, e1006824. [Google Scholar] [CrossRef] [Green Version]
- Ocwieja, K.; Sherrill-Mix, S.; Mukherjee, R.; Custers-Allen, R.; David, P.; Brown, M.; Wang, S.; Link, D.R.; Olson, J.; Travers, K.; et al. Dynamic regulation of HIV-1 mRNA populations analyzed by single-molecule enrichment and long-read sequencing. Nucleic Acids Res. 2012, 40, 10345–10355. [Google Scholar] [CrossRef] [PubMed]
- Quang, N.N.; Goudey, S.; Ségéral, E.; Mohammad, A.; Lemoine, S.; Blugeon, C.; Versapuech, M.; Paillart, J.-C.; Berlioz-Torrent, C.; Emiliani, S.; et al. Dynamic nanopore long-read sequencing analysis of HIV-1 splicing events during the early steps of infection. Retrovirology 2020, 17, 25. [Google Scholar] [CrossRef]
- Emery, A.; Zhou, S.; Pollom, E.; Swanstrom, R. Characterizing HIV-1 Splicing by Using Next-Generation Sequencing. J. Virol. 2017, 91, e02515-16. [Google Scholar] [CrossRef] [Green Version]
- Freund, M.; Asang, C.; Kammler, S.; Konermann, C.; Krummheuer, J.; Hipp, M.; Meyer, I.; Gierling, W.; Theiss, S.; Preuss, T.; et al. A novel approach to describe a U1 snRNA binding site. Nucleic Acids Res. 2003, 31, 6963–6975. [Google Scholar] [CrossRef] [Green Version]
- Yeo, G.; Burge, C.B. Maximum Entropy Modeling of Short Sequence Motifs with Applications to Rna Splicing Signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Stoltzfus, C.M.; Madsen, J.M. Role of Viral Splicing Elements and Cellular Rna Binding Proteins in Regulation of Hiv-1 Alternative Rna Splicing. Curr. HIV Res. 2006, 4, 43–55. [Google Scholar] [CrossRef]
- Kammler, S.; Otte, M.; Hauber, I.; Kjems, J.; Hauber, J.; Schaal, H. The strength of the HIV-1 3′ splice sites affects Rev function. Retrovirology 2006, 3, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asang, C.; Hauber, I.; Schaal, H. Insights into the selective activation of alternatively used splice acceptors by the human immunodeficiency virus type-1 bidirectional splicing enhancer. Nucleic Acids Res. 2008, 36, 1450–1463. [Google Scholar] [CrossRef]
- Ostermann, P.N.; Ritchie, A.; Ptok, J.; Schaal, H. Let it go: HIV-1 cis-acting repressive sequences. J. Virol. 2021, 95, e00342-00321. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.W.; Balachandran, A.; Ostrowski, M.A.; Cochrane, A. Digoxin Suppresses HIV-1 Replication by Altering Viral RNA Processing. PLoS Pathog. 2013, 9, e1003241. [Google Scholar] [CrossRef]
- Arias, A.; López, P.; Sánchez, R.; Yamamura, Y.; Rivera-Amill, V. Sanger and Next Generation Sequencing Approaches to Evaluate HIV-1 Virus in Blood Compartments. Int. J. Environ. Res. Public Health 2018, 15, 1697. [Google Scholar] [CrossRef] [Green Version]
- Brillen, A.L.; Walotka, L.; Hillebrand, F.; Müller, L.; Widera, M.; Theiss, S.; Schaal, H. Analysis of Competing Hiv-1 Splice Donor Sites Uncovers a Tight Cluster of Splicing Regulatory Elements within Exon 2/2b. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widera, M.; Erkelenz, S.; Hillebrand, F.; Krikoni, A.; Widera, D.; Kaisers, W.; Deenen, R.; Gombert, M.; Dellen, R.; Pfeiffer, T.; et al. An Intronic G Run within HIV-1 Intron 2 Is Critical for Splicing Regulation of vif mRNA. J. Virol. 2013, 87, 2707–2720. [Google Scholar] [CrossRef] [Green Version]
- Erkelenz, S.; Theiss, S.; Otte, M.; Widera, M.; Peter, J.O.; Schaal, H. Genomic HEXploring allows landscaping of novel potential splicing regulatory elements. Nucleic Acids Res. 2014, 42, 10681–10697. [Google Scholar] [CrossRef] [Green Version]
- Erkelenz, S.; Mueller, W.F.; Evans, M.S.; Busch, A.; Schöneweis, K.; Hertel, K.J.; Schaal, H. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA 2013, 19, 96–102. [Google Scholar] [CrossRef] [Green Version]
- Ceccherini-Silberstein, F.; Malet, I.; D’Arrigo, R.; Antinori, A.; Marcelin, A.-G.; Perno, C.-F. Characterization and structural analysis of HIV-1 integrase conservation. Aids Rev. 2009, 11, 17–29. [Google Scholar]
- Lübke, N.; Jensen, B.; Hüttig, F.; Feldt, T.; Walker, A.; Thielen, A.; Däumer, M.; Obermeier, M.; Kaiser, R.; Knops, E.; et al. Failure of Dolutegravir First-Line ART with Selection of Virus Carrying R263K and G118R. N. Engl. J. Med. 2019, 381, 887–889. [Google Scholar] [CrossRef]
- Mesplède, T.; Leng, J.; Pham, H.T.; Liang, J.; Quan, Y.; Han, Y.; Wainberg, M.A. The R263K Dolutegravir Resistance-Associated Substitution Progressively Decreases HIV-1 Integration. mBio 2017, 8, e00157-17. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Flavell, S.; Ferns, B.; Frampton, D.; Edwards, S.G.; Miller, R.F.; Grant, P.; Nastouli, E.; Gupta, R. Development of the R263K Mutation to Dolutegravir in an HIV-1 Subtype D Virus Harboring 3 Class-Drug Resistance. Open Forum Infect. Dis. 2019, 6, 329. [Google Scholar] [CrossRef]
- Koma, T.; Doi, N.; Takemoto, M.; Watanabe, K.; Yamamoto, H.; Nakashima, S.; Adachi, A.; Nomaguchi, M. The Expression Level of HIV-1 Vif Is Optimized by Nucleotide Changes in the Genomic SA1D2prox Region during the Viral Adaptation Process. Viruses 2021, 13, 2079. [Google Scholar] [CrossRef]
- Mandal, D.; Exline, C.M.; Feng, Z.; Stoltzfus, C.M. Regulation of vif mRNA Splicing by Human Immunodeficiency Virus Type 1 Requires 5′ Splice Site D2 and an Exonic Splicing Enhancer To Counteract Cellular Restriction Factor APOBEC3G. J. Virol. 2009, 83, 6067–6078. [Google Scholar] [CrossRef] [Green Version]
- Lusso, P.; Cocchi, F.; Balotta, C.; Markham, P.D.; Louie, A.; Farci, P.; Pal, R.; Gallo, R.C.; Reitz, M.S., Jr. Growth of Macrophage-Tropic and Primary Human Immunodeficiency Virus Type 1 (Hiv-1) Isolates in a Unique Cd4+ T-Cell Clone (Pm1): Failure to Downregulate Cd4 and to Interfere with Cell-Line-Tropic Hiv-1. J. Virol. 1995, 69, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Stopak, K.; de Noronha, C.; Yonemoto, W.; Greene, W.C. HIV-1 Vif Blocks the Antiviral Activity of APOBEC3G by Impairing Both Its Translation and Intracellular Stability. Mol. Cell 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Exline, C.M.; Feng, Z.; Stoltzfus, C.M. Negative and Positive mRNA Splicing Elements Act Competitively to Regulate Human Immunodeficiency Virus Type 1 Vif Gene Expression. J. Virol. 2008, 82, 3921–3931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomaguchi, M.; Doi, N.; Sakai, Y.; Ode, H.; Iwatani, Y.; Ueno, T.; Matsumoto, Y.; Miyazaki, Y.; Masuda, T.; Adachi, A. Natural Single-Nucleotide Variations in the HIV-1 Genomic SA1prox Region Can Alter Viral Replication Ability by Regulating Vif Expression Levels. J. Virol. 2016, 90, 4563–4578. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.J.; Zhao, X.Z.; Passos, D.O.; Lyumkis, D.; Burke, T.R.; Hughes, S.H. Integrase Strand Transfer Inhibitors Are Effective Anti-Hiv Drugs. Viruses 2021, 13, 205. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.-Y.; Grant, P.M.; Tzou, P.L.; Barrow, G.; Harrigan, P.R.; Ioannidis, J.P.A.; Shafer, R.W. A systematic review of the genetic mechanisms of dolutegravir resistance. J. Antimicrob. Chemother. 2019, 74, 3135–3149. [Google Scholar] [CrossRef]
- Dlamini, Z.; Hull, R. Can the HIV-1 splicing machinery be targeted for drug discovery? HIV AIDS 2017, 9, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Hillebrand, F.; Ostermann, P.N.; Müller, L.; Degrandi, D.; Erkelenz, S.; Widera, M.; Pfeffer, K.; Schaal, H. Gymnotic Delivery of LNA Mixmers Targeting Viral SREs Induces HIV-1 mRNA Degradation. Int. J. Mol. Sci. 2019, 20, 1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpinski, J.; Hauber, I.; Chemnitz, J.; Schäfer, C.; Paszkowski-Rogacz, M.; Chakraborty, D.; Beschorner, N.; Hofmann-Sieber, H.; Lange, U.C.; Grundhoff, A.; et al. Directed evolution of a recombinase that excises the provirus of most HIV-1 primary isolates with high specificity. Nat. Biotechnol. 2016, 34, 401–409. [Google Scholar] [CrossRef]
- Jessen, H.; Allen, T.; Streeck, H. How a Single Patient Influenced HIV—15-Year Follow-up. N. Engl. J. Med. 2014, 370, 682–683. [Google Scholar] [CrossRef] [Green Version]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müßig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-Term Control of Hiv by Ccr5 Delta32/Delta32 Stem-Cell Transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.K.; Peppa, D.; Hill, A.L.; Galvez, C.; Salgado, M.; Pace, M.; McCoy, L.E.; Griffith, S.A.; Thornhill, J.; Alrubayyi, A.; et al. Evidence for Hiv-1 Cure after Ccr5delta32/Delta32 Allogeneic Haemopoietic Stem-Cell Transplantation 30 Months Post Analytical Treatment Interruption: A Case Report. Lancet HIV 2020, 7, e340–e347. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| RT-PCR: |
| #1554 CTTGAAAGCGAAAGTAAAGC |
| #2710 AAGGGGCAGTAGTAATACAA |
| #3392 CGTCCCAGATAAGTGCTAAGG |
| #1224 TCTTCCAGCCTCCCATCAGCGTTTGG |
| #1225 CAACAGAAATCCAACCTAGAGCTGCT |
| #2649 TCCACCACCGTCTTCTTTAG |
| #6253 CAACCAAACAACCTTAGGGGA |
| Cloning: |
| (a) Proviral plasmids |
| #3632 (d) TGGATGCTTCCAGGGCTC |
| #5553 (a) CTGGCAGAAAACAGGGAGATT |
| V260I as (b) CTTTGCTTTTCTTCTTGGTATTACTTTTATGTCACTATTATC |
| V260I s (c) GTGACATAAAAGTAATACCAAGAAGAAAAGCAAAGATCATCAG |
| R263K as (b) GATGATCTTTGCTTTTTTTCTTGGCACTACTTTTATG |
| R263K s (c) GTAGTGCCAAGAAAAAAAGCAAAGATCATCAGG |
| R263R s (c) GTAGTGCCAAGACGGAAAGCAAAGATCATCAGG |
| R263R as (b) GATGATCTTTGCTTTCCGTCTTGGCACTACTTTTATG |
| (b) Three-exon-minigene |
| #6040 GGGGCTAGCGCAGTAGTAATACAAGATAATAGTGACATAAAAGTAATACCAAGAAGAAAAGCAAAGATCAT |
| #6041 GGGGCTAGCGCAGTAGTAATACAAGATAATAGTGACATAAAAGTAGTGCCAAGAAAAAAAGCAAAGATCATCAGGGATTAT |
| #6042 GGGGCTAGCGCAGTAGTAATACAAGATAATAGTGACATAAAAGTAGTCCCAAGAAGAAAAGCAAAGATCAT |
| #6043 GGGGCTAGCGCAGTAGTAATACAAGATAATAGTGACATAAAAGTAGTGCCAAGACGGAAAGCAAAGATCATCAGGGATTAT |
| #5941 GGACAGTGGCTGACAGT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, L.; Moskorz, W.; Brillen, A.-L.; Hillebrand, F.; Ostermann, P.N.; Kiel, N.; Walotka, L.; Ptok, J.; Timm, J.; Lübke, N.; et al. Altered HIV-1 mRNA Splicing Due to Drug-Resistance-Associated Mutations in Exon 2/2b. Int. J. Mol. Sci. 2022, 23, 156. https://doi.org/10.3390/ijms23010156
Müller L, Moskorz W, Brillen A-L, Hillebrand F, Ostermann PN, Kiel N, Walotka L, Ptok J, Timm J, Lübke N, et al. Altered HIV-1 mRNA Splicing Due to Drug-Resistance-Associated Mutations in Exon 2/2b. International Journal of Molecular Sciences. 2022; 23(1):156. https://doi.org/10.3390/ijms23010156
Chicago/Turabian StyleMüller, Lisa, Wiebke Moskorz, Anna-Lena Brillen, Frank Hillebrand, Philipp Niklas Ostermann, Niklas Kiel, Lara Walotka, Johannes Ptok, Jörg Timm, Nadine Lübke, and et al. 2022. "Altered HIV-1 mRNA Splicing Due to Drug-Resistance-Associated Mutations in Exon 2/2b" International Journal of Molecular Sciences 23, no. 1: 156. https://doi.org/10.3390/ijms23010156
APA StyleMüller, L., Moskorz, W., Brillen, A.-L., Hillebrand, F., Ostermann, P. N., Kiel, N., Walotka, L., Ptok, J., Timm, J., Lübke, N., & Schaal, H. (2022). Altered HIV-1 mRNA Splicing Due to Drug-Resistance-Associated Mutations in Exon 2/2b. International Journal of Molecular Sciences, 23(1), 156. https://doi.org/10.3390/ijms23010156