
Resonance-Assisted Hydrogen Bond—Revisiting the Original Concept in the Context of Its Criticism in the Literature


Abstract
:1. Introduction
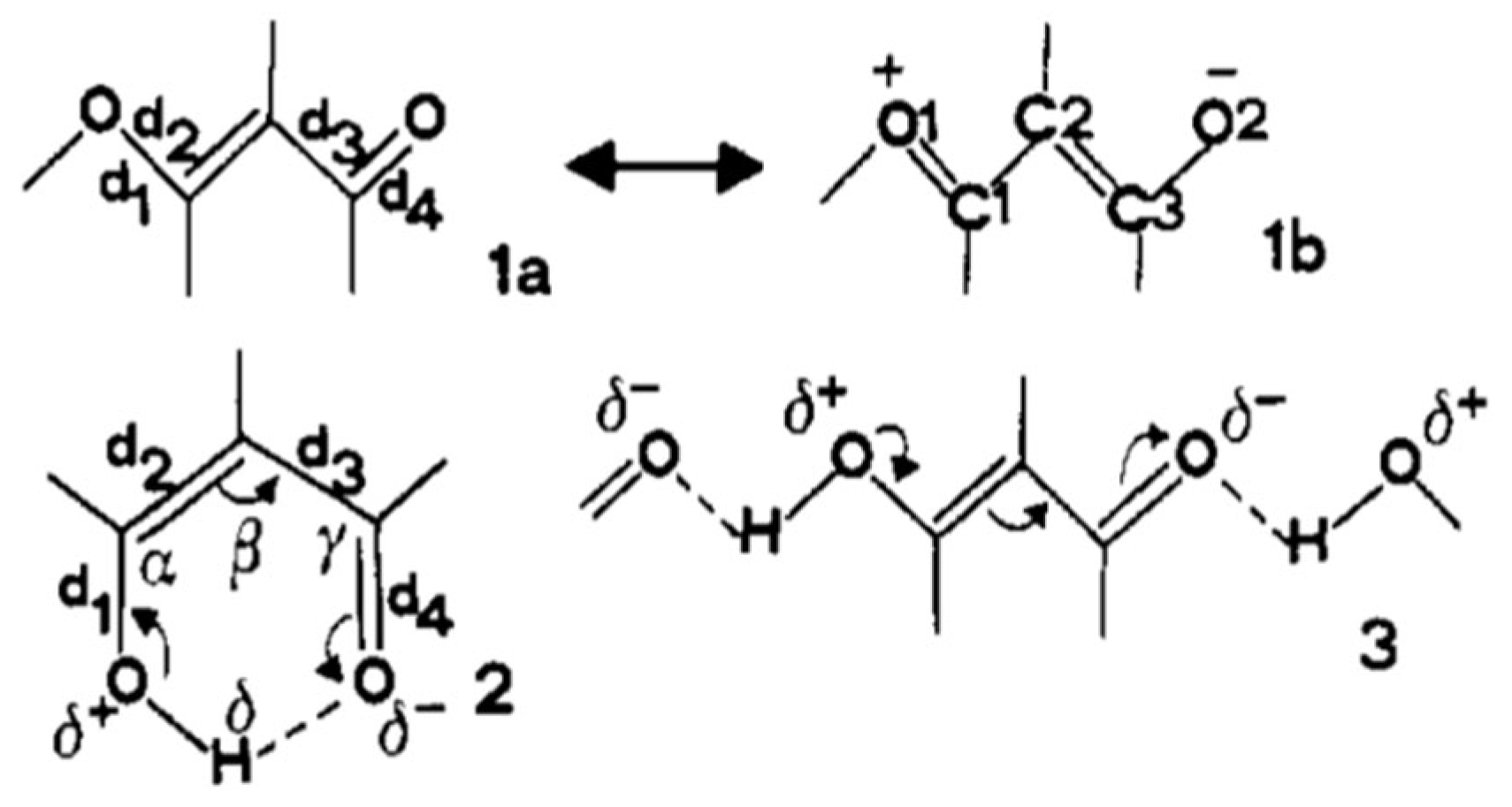
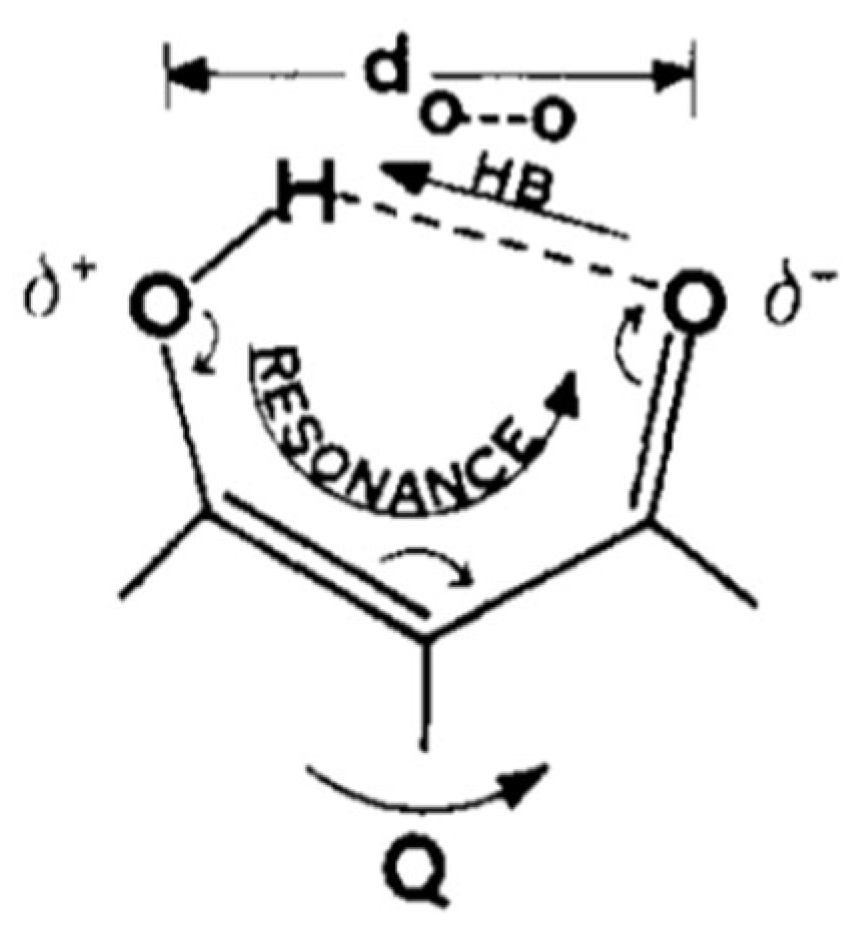
1.1. The Original Concept of RAHB
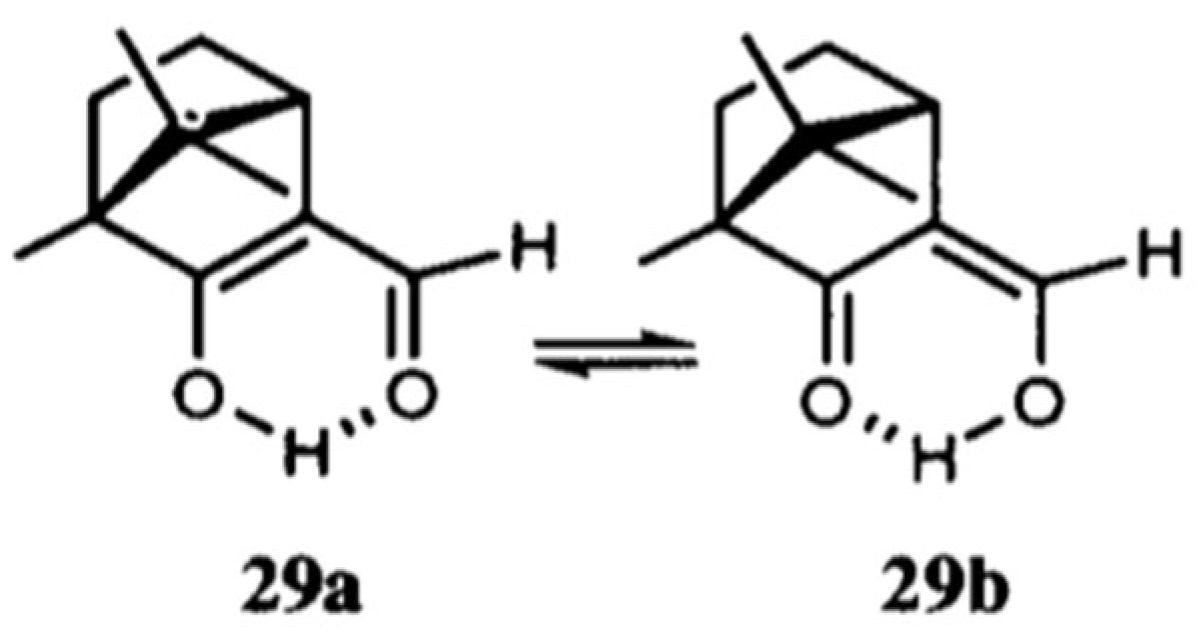
1.2. The Criticism of the RAHB Concept
2. Computational Methods
3. Results and Discussion
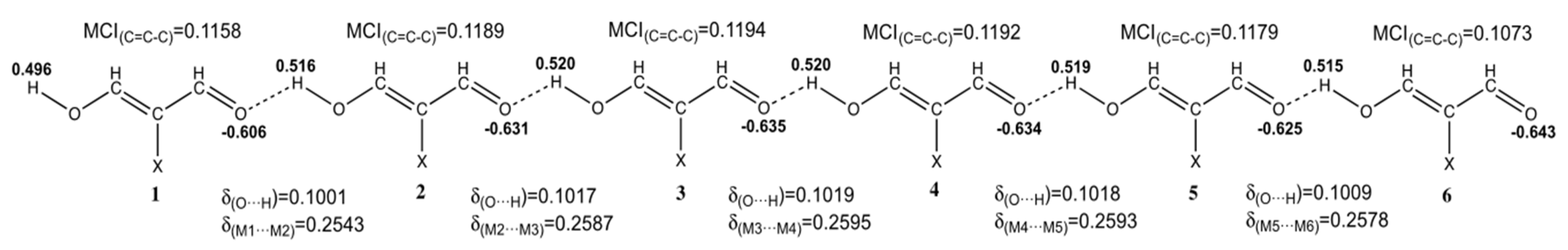
3.1. Intermolecular RAHB in View of Many-Body Interaction Theory
3.2. The Resonance in Intermolecular RAHB
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scheiner, S. Molecular Interactions: From van der Waals to Strongly Bound Complexes; Wiley Tutorial Series in Theoretical Chemistry; Wiley & Sons: New York, NY, USA, 1997. [Google Scholar]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: New York, NY, USA, 1999. [Google Scholar] [CrossRef]
- Gerlt, J.A.; Kreevoy, M.M.; Cleland, W.; Frey, P.A. Understanding enzymic catalysis: The importance of short, strong hydrogen bonds. Chem. Biol. 1997, 4, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Perrin, C.L.; Nielson, J.B. “Strong” hydrogen bonds in chemistry and biology. Annu. Rev. Phys. Chem. 1997, 48, 511–544. [Google Scholar] [CrossRef] [PubMed]
- Sobczyk, L.; Grabowski, S.J.; Krygowski, T.M. Interrelation between H-Bond and Pi-Electron Delocalization. Chem. Rev. 2005, 105, 3513–3560. [Google Scholar] [CrossRef] [PubMed]
- Krygowski, T.M.; Szatyłowicz, H. Interrelation between the substituent effects, π—Electron delocalization and H–bonding. Trends Org. Chem. 2006, 11, 37–53. [Google Scholar]
- Krygowski, T.M.; Szatylowicz, H.; Stasyuk, O.A.; Dominikowska, J.; Palusiak, M. Aromaticity from the viewpoint of molecular geometry: Application to planar systems. Chem. Rev. 2014, 114, 6383–6422. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed. Engl. 2003, 42, 1210–1250. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Beatty, A.M.; Lorimer, K.R. Charge-Assisted Hydrogen Bonds and Halogen-Halogen Interactions in Organic Salts: Benzylammonium Benzoates and Pentafluorobenzoates. Struct. Chem. 1999, 10, 229–242. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F. Intermolecular Interactions in Nonorganic Crystal Engineering. Acc. Chem. Res. 2000, 33, 601–608. [Google Scholar] [CrossRef]
- Braga, D.; Maini, L.; Grepioni, F.; De Cian, A.; Félix, O.; Fischer, J.; Hosseini, M.W. Charge-assisted N–H(+)···O(−) and O–H···O(−) hydrogen bonds control the supramolecular aggregation of ferrocenedicarboxylic acid and bis-amidines. New J. Chem. 2000, 24, 547–553. [Google Scholar] [CrossRef]
- Grepioni, F.; Cojazzi, G.; Draper, S.M.; Scully, N.; Braga, D. Crystal forms of hexafluorophosphate organometallic salts and importance of charge assisted C-H…F hydrogen bonds. Organometallics 1998, 17, 296–307. [Google Scholar] [CrossRef]
- Braga, D.; Draper, S.M.; Champeil, E.; Grepioni, F. Inorganic–organometallic crystal synthesis. The role of charge-assisted C–H…O and C–H…Cl hydrogen bonds in crystalline [(η5-C5H5)2Co][H2PO4]·3H2O and [(η6-C6H5Me)2Cr][Cl]. J. Organomet. Chem. 1999, 573, 73–77. [Google Scholar] [CrossRef]
- Gilli, G.; Gilli, P. Towards an unified hydrogen-bond theory. J. Mol. Struct. 2000, 552, 1–15. [Google Scholar] [CrossRef]
- Palusiak, M.; Janowska, I.; Zakrzewski, J.; Grabowski, S.J. Charge assisted N-H…I and C-H…I hydrogen bonding in (1R,2S)-1-(ferrocenylmethyl)-2-(methoxymethyl)pyrrolidinium iodide. Acta Crystallogr. C 2005, 61, m55–m57. [Google Scholar] [CrossRef]
- Bankiewicz, B.; Palusiak, M. The shape of the halogen atom—Anisotropy of electron distribution and its dependence on basis set and method used. Comput. Theor. Chem. 2011, 966, 113–119. [Google Scholar] [CrossRef]
- Bankiewicz, B.; Matczak, P.; Palusiak, M. Electron density characteristics in bond critical point (QTAIM) vs. interaction energy components (SAPT)—The case of charge-assisted hydrogen bonding. J. Phys. Chem. A 2012, 116, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Xantheas, S.S. Ab initio studies of cyclic water clusters (H2O)n, n = 1–6. II. Analysis of many-body interactions. J. Chem. Phys. 1994, 100, 7523–7534. [Google Scholar] [CrossRef]
- Gilli, G.; Bellucci, F.; Ferretti, V.; Bertolasi, V. Evidence for resonance-assisted hydrogen bonding from crystal-structure correlations on the enol form of the β-diketone fragment. J. Am. Chem. Soc. 1989, 111, 1023–1028. [Google Scholar] [CrossRef]
- Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G. Evidence for resonance-assisted hydrogen bonding. 2. Intercorrelation between crystal structure and spectroscopic parameters in eight intramolecularly hydrogen bonded 1,3-diaryl-1,3-propanedione enols. J. Am. Chem. Soc. 1991, 113, 4917–4925. [Google Scholar] [CrossRef]
- Bertolasi, V.; Ferretti, V.; Gilli, P.; Gilli, G.; Issa, Y.M.; Sherif, O.E. Intramolecular N–H⋯ O hydrogen bonding assisted by resonance. Part 2. Intercorrelation between structural and spectroscopic parameters for five 1,3-diketone arylhydrazones derived from dibenzoylmethane. J. Chem. Soc. Perkin Trans. 2 1993, 2223–2228. [Google Scholar] [CrossRef]
- Gilli, G.; Bertolasi, V.; Ferretti, V.; Gilli, P. Resonance-assisted hydrogen bonding. III. Formation of intermolecular hydrogen-bonded chains in crystals of β-diketone enols and its relevance to molecular association. Acta Crystallogr. B 1993, 49, 564–576. [Google Scholar] [CrossRef]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Evidence for resonance-assisted hydrogen bonding. 4. Covalent nature of the strong homonuclear hydrogen bond. Study of the O-H--O system by crystal structure correlation methods. J. Am. Chem. Soc. 1994, 116, 909–915. [Google Scholar] [CrossRef]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Intramolecular N-H…O hydrogen bonding assisted by resonance. III. Structural studies of 1-ketone-2-arylhydrazone derivatives. Acta Crystallogr. B 1994, 50, 617–625. [Google Scholar] [CrossRef]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Intermolecular N–H⋯O hydrogen bonds assisted by resonance. Heteroconjugated systems as hydrogen-bond-strengthening functional groups. Acta Crystallogr. B 1995, 51, 1004–1015. [Google Scholar] [CrossRef] [Green Version]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Resonance-Assisted O-H … O Hydrogen Bonding: Its Role in the Crystalline Self-Recognition of β-Diketone Enols and its Structural and IR Characterization. Chem.—Eur. J. 1996, 2, 925–934. [Google Scholar] [CrossRef]
- Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G. Intramolecular O–H··· O hydrogen bonds assisted by resonance. Correlation between crystallographic data and 1 H NMR chemical shifts. J. Chem. Soc. Perkin Trans. 2 1997, 945–952. [Google Scholar] [CrossRef]
- Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G. Intermolecular NH⋯ O Hydrogen Bonding Assisted by Resonance. II. Self-Assembly of Hydrogen-Bonded Secondary Enaminones in Supramolecular Catemers. Acta Crystallogr. B 1998, 54, 50–65. [Google Scholar] [CrossRef]
- Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G.; Vaughan, K.; Jollimore, J.V. Interplay of hydrogen bonding and other molecular interactions in determining the crystal packing of a series of anti-beta-ketoarylhydrazones. Acta Crystallogr. B 1999, 55, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- Bertolasi, V.; Gilli, P.; Ferretti, V.; Gilli, G.; Vaughan, K. Interplay between steric and electronic factors in determining the strength of intramolecular resonance-assisted NH··· O hydrogen bond in a series of β-ketoarylhydrazones. New J. Chem. 1999, 23, 1261–1267. [Google Scholar] [CrossRef]
- Gilli, P.; Bertolasi, V.; Ferretti, V.; Gilli, G. Evidence for Intramolecular N−H···O Resonance-Assisted Hydrogen Bonding in β-Enaminones and Related Heterodienes. A Combined Crystal-Structural, IR and NMR Spectroscopic, and Quantum-Mechanical Investigation. J. Am. Chem. Soc. 2000, 122, 10405–10417. [Google Scholar] [CrossRef]
- Gilli, P.; Bertolasi, V.; Pretto, L.; Lyčka, A.; Gilli, G. The Nature of Solid-State N−H···O/O−H···N Tautomeric Competition in Resonant Systems. Intramolecular Proton Transfer in Low-Barrier Hydrogen Bonds Formed by the ···O=C−C=N−NH··· ⇄ ···HO−C=C−N=N··· Ketohydrazone−Azoenol System. A Variable-Temperature X-ray Crystallographic and DFT Computational Study. J. Am. Chem. Soc. 2002, 124, 13554–13567. [Google Scholar] [CrossRef] [PubMed]
- Bertolasi, V.; Pretto, L.; Gilli, G.; Gilli, P. π-Bond cooperativity and anticooperativity effects in resonance-assisted hydrogen bonds (RAHBs). Acta Crystallogr. B 2006, 62, 850–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolasi, V.; Pretto, L.; Ferretti, V.; Gilli, P.; Gilli, G. Interplay between steric and electronic factors in determining the strength of intramolecular N-H…O resonance-assisted hydrogen bonds in β-enaminones. Acta Crystallogr. B 2006, 62, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Gilli, G.; Bertolasi, V.; Gilli, P. Solid-State N–H···O/O–H···N Tautomerism in Resonance-Assisted 1-(Arylazo)-2-Naphthols and Its Through-Space π* ← π Perturbation in TCNQ Cocrystals. A Variable-Temperature X-ray Crystal Study. Cryst. Growth Des. 2013, 13, 3308–3320. [Google Scholar] [CrossRef]
- Gilli, P.; Gilli, G. The Nature of the Hydrogen Bond: Outline of a Comprehensive Hydrogen Bond Theory; Oxford University Press: New York, NY, USA, 2009; Volume 23. [Google Scholar] [CrossRef] [Green Version]
- Ramos, M.; Alkorta, I.; Elguero, J. The Mills-Nixon effect on enol-enol tautomerism in beta-dicarbonyl compounds and on annular tautomerism in NH-pyrazoles: A semi-empirical study. Tetrahedron 1997, 53, 1403–1410. [Google Scholar] [CrossRef]
- Mills, W.H.; Nixon, I.G. Stereochemical influences on aromatic substitution. Substitution derivatives of 5-hydroxyhydrindene. J. Chem. Soc. 1930, 2510–2524. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Mó, O.; Yáñez, M.; Del Bene, J.E. Do coupling constants and chemical shifts provide evidence for the existence of resonance-assisted hydrogen bonds? Mol. Phys. 2004, 102, 2563–2574. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Romero-Montalvo, E.; Costales, A.; Pendas, A.M.; Rocha-Rinza, T. The nature of resonance-assisted hydrogen bonds: A quantum chemical topology perspective. Phys. Chem. Chem. Phys. 2016, 18, 26383–26390. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Sanz, P.; Mó, O.; Yáñez, M.; Elguero, J. Non-resonance-assisted hydrogen bonding in hydroxymethylene and aminomethylene cyclobutanones and cyclobutenones and their nitrogen counterparts. ChemPhysChem 2007, 8, 1950–1958. [Google Scholar] [CrossRef]
- Sanz, P.; Mó, O.; Yáñez, M.; Elguero, J. Resonance-assisted hydrogen bonds: A critical examination. Structure and stability of the enols of β-diketones and β-enaminones. J. Phys. Chem. A 2007, 111, 3585–3591. [Google Scholar] [CrossRef]
- Sanz, P.; Mó, O.; Yáñez, M.; Elguero, J. Bonding in tropolone, 2-aminotropone, and aminotroponimine: No evidence of resonance-assisted hydrogen-bond effects. Chem.—Eur. J. 2008, 14, 4225–4232. [Google Scholar] [CrossRef]
- Grosch, A.A.; van der Lubbe, S.C.C.; Fonseca Guerra, C. Nature of Intramolecular Resonance Assisted Hydrogen Bonding in Malonaldehyde and Its Saturated Analogue. J. Phys. Chem. A 2018, 122, 1813–1820. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Zhang, H.; Wu, W.; Mo, Y. A Critical Check for the Role of Resonance in Intramolecular Hydrogen Bonding (IMHB). Chem.—Eur. J. 2017, 23, 16885–16891. [Google Scholar] [CrossRef] [PubMed]
- Vatanparast, M.; Nekoei, A.R. RAHB concept and σ-skeleton in some oximes of 3-hydroxy fulvene; DFT, AIM, ELF and NBO studies. Struct. Chem. 2015, 26, 1039–1048. [Google Scholar] [CrossRef]
- Shapenova, D.S.; Shiryaev, A.A.; Bolte, M.; Kukułka, M.; Szczepanik, D.W.; Hooper, J.; Babashkina, M.G.; Mahmoudi, G.; Mitoraj, M.P.; Safin, D.A. Resonance Assisted Hydrogen Bonding Phenomenon Unveiled through Both Experiments and Theory: A New Family of Ethyl N-Salicylideneglycinate Dyes. Chem.—Eur. J. 2020, 26, 12987–12995. [Google Scholar] [CrossRef] [PubMed]
- Afonin, A.V.; Vashchenko, A.V. Quantitative decomposition of resonance-assisted hydrogen bond energy in β-diketones into resonance and hydrogen bonding (π-and σ-) components using molecular tailoring and function-based approaches. J. Comput. Chem. 2020, 41, 1285–1298. [Google Scholar] [CrossRef]
- Keykhaei, A.; Nowroozi, A. On the performance of molecular tailoring approach for estimation of the intramolecular hydrogen bond energies of RAHB systems: A comparative study. Struct. Chem. 2020, 31, 423–433. [Google Scholar] [CrossRef]
- Pareras, G.; Szczepanik, D.W.; Duran, M.; Solà, M.; Simon, S. Tuning the Strength of the Resonance-Assisted Hydrogen Bond in Acenes and Phenacenes with Two o-Hydroxyaldehyde Groups—The Importance of Topology. J. Org. Chem. 2019, 84, 15538–15548. [Google Scholar] [CrossRef]
- Rafat, R.; Nowroozi, A. Competition Between the Intramolecular Hydrogen Bond and the π-Electron Delocalization in Some RAHB Systems: A Theoretical Study. J. Struct. Chem. 2019, 60, 755–762. [Google Scholar] [CrossRef]
- Lin, X.; Jiang, X.; Wu, W.; Mo, Y. Induction, Resonance, and Secondary Electrostatic Interaction on Hydrogen Bonding in the Association of Amides and Imides. J. Org. Chem. 2018, 83, 13446–13453. [Google Scholar] [CrossRef]
- Nguyen, Y.H.; Lampkin, B.J.; Venkatesh, A.; Ellern, A.; Rossini, A.J.; VanVeller, B. Open-Resonance-Assisted Hydrogen Bonds and Competing Quasiaromaticity. J. Org. Chem. 2018, 83, 9850–9857. [Google Scholar] [CrossRef]
- Durlak, P.; Latajka, Z. Car–Parrinello and Path Integral Molecular Dynamics Study of the Proton Transfer in the Intramolecular Hydrogen Bonds in the Ketohydrazone–Azoenol System. J. Phys. Chem. B 2018, 122, 7862–7873. [Google Scholar] [CrossRef] [PubMed]
- Pareras, G.; Palusiak, M.; Duran, M.; Solà, M.; Simon, S. Tuning the Strength of the Resonance-Assisted Hydrogen Bond in o-Hydroxybenzaldehyde by Substitution in the Aromatic Ring. J. Phys. Chem. A 2018, 122, 2279–2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Deng, G.; Zheng, Y.-Z.; Xu, J.; Ashraf, H.; Yu, Z.-W. Evidences for Cooperative Resonance-Assisted Hydrogen Bonds in Protein Secondary Structure Analogs. Sci. Rep. 2016, 6, 36932. [Google Scholar] [CrossRef]
- Nakhaei, E.; Nowroozi, A. On the performance of resonance assisted hydrogen bond theory in malonaldehyde derivatives. Comput. Theor. Chem. 2016, 1096, 27–32. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Bankiewicz, B.; Czarnocki, Z.; Palusiak, M. Quasi-aromaticity—What does it mean? Tetrahedron 2015, 71, 4895–4908. [Google Scholar] [CrossRef]
- Rusinska-Roszak, D. Intramolecular O–H···O=C Hydrogen Bond Energy via the Molecular Tailoring Approach to RAHB Structures. J. Phys. Chem. A 2015, 119, 3674–3687. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M. Blue-shifting intramolecular C-H…O(S) contacts in sterically strained systems. J. Mol. Struct. Theochem. 2007, 820, 118–127. [Google Scholar] [CrossRef]
- Jabłoński, M.; Kaczmarek, A.; Sadlej, A.J. Estimates of the Energy of Intramolecular Hydrogen Bonds. J. Phys. Chem. A 2006, 110, 10890–10898. [Google Scholar] [CrossRef]
- Grabowski, S.J. Intramolecular Hydrogen Bond Energy and Its Decomposition—O–H∙∙∙O Interactions. Crystals 2021, 11, 5. [Google Scholar] [CrossRef]
- Trujillo, C.; Sánchez-Sanz, G.; Alkorta, I.; Elguero, J.; Mó, O.; Yáñez, M. Resonance assisted hydrogen bonds in open-chain and cyclic structures of malonaldehyde enol: A theoretical study. J. Mol. Struct. 2013, 1048, 138–151. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. V. Core-valence basis sets for boron through neon. J. Chem. Phys. 1995, 103, 4572–4585. [Google Scholar] [CrossRef] [Green Version]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Domagała, M.; Lutyńska, A.; Palusiak, M. Extremely Strong Halogen Bond. The Case of a Double-Charge-Assisted Halogen Bridge. J. Phys. Chem. A 2018, 122, 5484–5492. [Google Scholar] [CrossRef] [PubMed]
- Domagała, M.; Matczak, P.; Palusiak, M. Halogen bond, hydrogen bond and N⋯C interaction—On interrelation among these three noncovalent interactions. Comput. Theor. Chem. 2012, 998, 26–33. [Google Scholar] [CrossRef]
- Allen, F.H.; Bruno, I.J. Bond lengths in organic and metal-organic compounds revisited: X—H bond lengths from neutron diffraction data. Acta Crystallogr. B 2010, 66, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Petersson, G.A.; Al-Laham, M.A. A complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atoms. J. Chem. Phys. 1991, 94, 6081–6090. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Keith, T.A. AimAll—(Version 17.11.14); TK Gristmill Software: Overland Park, KS, USA, 2017; Available online: http://aim.tkgristmill.com (accessed on 15 December 2021).
- Matito, E. ESI-3D: Electron Sharing Indices Program for 3D Molecular Space Partitioning; Institute of Computational Chemistry and Catalysis, University of Girona: Catalonia, Spain, 2006; Available online: http://iqcc.udg.edu/~eduard/ESI/ (accessed on 15 December 2021).
- Matito, E.; Duran, M.; Solà, M. The aromatic fluctuation index (FLU): A new aromaticity index based on electron delocalization. J. Chem. Phys. 2004, 122, 014109. [Google Scholar] [CrossRef] [Green Version]
- Herman, K.M.; Heindel, J.P.; Xantheas, S.S. The many-body expansion for aqueous systems revisited: III. Hofmeister ion–water interactions. Phys. Chem. Chem. Phys. 2021, 23, 11196–11210. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.P.; Yu, Q.; Bowman, J.M.; Xantheas, S.S. Benchmark Electronic Structure Calculations for H3O+(H2O)n, n = 0–5, Clusters and Tests of an Existing 1,2,3-Body Potential Energy Surface with a New 4-Body Correction. J. Chem. Theor. Comput. 2018, 14, 4553–4566. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Xantheas, S.S. Structures, Energetics, and Spectroscopic Fingerprints of Water Clusters n = 2–24. In Handbook of Computational Chemistry; Leszczynski, J., Kaczmarek-Kedziera, A., Puzyn, T.G., Papadopoulos, M., Reis, H.K., Shukla, M., Eds.; Springer: Cham, Switzerland, 2017; pp. 1139–1173. [Google Scholar] [CrossRef]
- Iwata, S.; Bandyopadhyay, P.; Xantheas, S.S. Cooperative Roles of Charge Transfer and Dispersion Terms in Hydrogen-Bonded Networks of (H2O)n, n = 6, 11, and 16. J. Phys. Chem. A 2013, 117, 6641–6651. [Google Scholar] [CrossRef]
- Yoo, S.; Xantheas, S.S. Structures, Energetics, and Spectroscopic Fingerprints of Water Clusters n = 2–24. In Handbook of Computational Chemistry; Leszczynski, J., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 761–792. [Google Scholar] [CrossRef]
- Dominikowska, J.; Palusiak, M. Halogen-halogen interactions in view of many-body approach. Chem. Phys. Lett. 2013, 583, 8–13. [Google Scholar] [CrossRef]
- Fradera, X.; Poater, J.; Simon, S.; Duran, M.; Solà, M. Electron-pairing analysis from localization and delocalization indices in the framework of the atoms-in-molecules theory. Theor. Chem. Acc. 2002, 108, 214–224. [Google Scholar] [CrossRef]
- Giambiasi, M.; de Giambiasi, M.S.; dos Santos Silva, C.D.; de Figueiredo, A.P. Multicenter bond indices as a measure of aromaticity. Phys. Chem. Chem. Phys. 2000, 2, 3381–3392. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, 72, 171–179. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | 2 | 3 | 4 | 5 | 6 | n→∞ | |
|---|---|---|---|---|---|---|---|
| −12.89 | −14.29 | −15.01 | −15.45 | −15.73 | −16.48 | ||
| I | −13.24 | −13.41 | −13.52 | −13.59 | −13.77 | ||
| −1.06 | −1.60 | −1.93 | −2.14 | −2.87 | |||
| −13.38 | −14.83 | −15.59 | −16.05 | −16.36 | −17.14 | ||
| II | −13.73 | −13.90 | −14.01 | −14.08 | −14.26 | ||
| −1.10 | −0.68 | −2.04 | −2.28 | −3.08 | |||
| −12.92 | −14.33 | −15.12 | −15.64 | −15.95 | −16.94 | ||
| III | −13.26 | −13.47 | −13.61 | −13.71 | −14.04 | ||
| −1.07 | −1.65 | −2.03 | −2.24 | −3.47 | |||
| −12.96 | −14.99 | −15.48 | −16.21 | −16.65 | −17.89 | ||
| IV | −13.51 | −13.90 | −11.69 | −14.38 | −14.84 | ||
| −0.98 | −1.58 | −2.03 | −2.28 | −3.27 | |||
| −6.88 | −7.71 | −7.18 | −7.15 | −7.19 | −7.30 | ||
| V | −7.63 | −6.94 | −6.88 | −6.89 | −6.93 | ||
| −0.08 | −0.24 | −0.27 | −0.30 | −0.38 |
| #n | 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|---|
| I | 1 | 0.0925 | |||||
| 2 | 0.1033 | 0.0952 | |||||
| 3 | 0.1046 | 0.1060 | 0.0957 | ||||
| 4 | 0.1050 | 0.1072 | 0.1065 | 0.0959 | |||
| 5 | 0.1051 | 0.1075 | 0.1077 | 0.1067 | 0.0960 | ||
| 6 | 0.1052 | 0.1077 | 0.1081 | 0.1079 | 0.1069 | 0.0960 | |
| II | 1 | 0.1030 | |||||
| 2 | 0.1137 | 0.1063 | |||||
| 3 | 0.1152 | 0.1169 | 0.1070 | ||||
| 4 | 0.1156 | 0.1184 | 0.1176 | 0.1072 | |||
| 5 | 0.1158 | 0.1188 | 0.1190 | 0.1178 | 0.1073 | ||
| 6 | 0.1158 | 0.1189 | 0.1194 | 0.1192 | 0.1179 | 0.1073 | |
| III | 1 | 0.0947 | |||||
| 2 | 0.1025 | 0.0999 | |||||
| 3 | 0.1032 | 0.1077 | 0.1008 | ||||
| 4 | 0.1035 | 0.1082 | 0.1085 | 0.1012 | |||
| 5 | 0.1036 | 0.1085 | 0.1090 | 0.1088 | 0.1013 | ||
| 6 | 0.1036 | 0.1086 | 0.1093 | 0.1093 | 0.1089 | 0.1013 | |
| IV | 1 | 0.1036 | |||||
| 2 | 0.1168 | 0.1076 | |||||
| 3 | 0.1189 | 0.1222 | 0.1088 | ||||
| 4 | 0.1198 | 0.1247 | 0.1236 | 0.1091 | |||
| 5 | 0.1202 | 0.1254 | 0.1264 | 0.1246 | 0.1093 | ||
| 6 | 0.1208 | 0.1256 | 0.1269 | 0.1269 | 0.1243 | 0.1056 | |
| V | 1 | 0.0302 | |||||
| 2 | 0.0308 | 0.0301 | |||||
| 3 | 0.0308 | 0.0309 | 0.0302 | ||||
| 4 | 0.0307 | 0.0308 | 0.0310 | 0.0341 | |||
| 5 | 0.0307 | 0.0308 | 0.0310 | 0.0342 | 0.0301 | ||
| 6 | 0.0307 | 0.0308 | 0.0308 | 0.0341 | 0.0308 | 0.0301 |
| #n | HO | O···HO | O···HO | O···HO | O···HO | O···HO | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | 1 | 0.6245 | ||||||||||
| 2 | 0.6132 | 0.0939 | 0.4523 | |||||||||
| 3 | 0.6104 | 0.0945 | 0.4416 | 0.0949 | 0.4444 | |||||||
| 4 | 0.6094 | 0.0947 | 0.4389 | 0.0957 | 0.4336 | 0.0952 | 0.4423 | |||||
| 5 | 0.6090 | 0.0948 | 0.4380 | 0.0959 | 0.4310 | 0.0959 | 0.4315 | 0.0953 | 0.4416 | |||
| 6 | 0.6088 | 0.0948 | 0.4376 | 0.0959 | 0.4300 | 0.0961 | 0.4289 | 0.0960 | 0.4307 | 0.0954 | 0.4412 | |
| II | 1 | 0.6310 | ||||||||||
| 2 | 0.6197 | 0.0990 | 0.4603 | |||||||||
| 3 | 0.6170 | 0.0999 | 0.4497 | 0.1004 | 0.4523 | |||||||
| 4 | 0.6161 | 0.1001 | 0.4473 | 0.1014 | 0.4417 | 0.1008 | 0.4504 | |||||
| 5 | 0.6158 | 0.1001 | 0.4463 | 0.1016 | 0.4392 | 0.1017 | 0.4396 | 0.1009 | 0.4495 | |||
| 6 | 0.6156 | 0.1001 | 0.4460 | 0.1017 | 0.4384 | 0.1019 | 0.4372 | 0.1018 | 0.4389 | 0.1009 | 0.4492 | |
| III | 1 | 0.6349 | ||||||||||
| 2 | 0.6223 | 0.0925 | 0.4653 | |||||||||
| 3 | 0.6189 | 0.0937 | 0.4536 | 0.0934 | 0.4582 | |||||||
| 4 | 0.6176 | 0.0941 | 0.4506 | 0.0948 | 0.4463 | 0.0940 | 0.4558 | |||||
| 5 | 0.6170 | 0.0941 | 0.4493 | 0.0952 | 0.4434 | 0.0952 | 0.4440 | 0.0941 | 0.4549 | |||
| 6 | 0.6167 | 0.0943 | 0.4489 | 0.0953 | 0.4421 | 0.0957 | 0.4410 | 0.0954 | 0.4431 | 0.0942 | 0.4545 | |
| IV | 1 | 0.6161 | ||||||||||
| 2 | 0.6048 | 0.0957 | 0.4659 | |||||||||
| 3 | 0.6011 | 0.0968 | 0.4548 | 0.1005 | 0.4526 | |||||||
| 4 | 0.6002 | 0.0993 | 0.4469 | 0.1017 | 0.4398 | 0.1064 | 0.4426 | |||||
| 5 | 0.5990 | 0.1058 | 0.4373 | 0.1042 | 0.4343 | 0.1052 | 0.4333 | 0.1079 | 0.4409 | |||
| 6 | 0.5986 | 0.1091 | 0.4320 | 0.1145 | 0.4208 | 0.1039 | 0.4320 | 0.1043 | 0.4336 | 0.1063 | 0.442 | |
| V | 1 | 0.6577 | ||||||||||
| 2 | 0.6535 | 0.0701 | 0.55 | |||||||||
| 3 | 0.6522 | 0.072 | 0.5425 | 0.0722 | 0.5444 | |||||||
| 4 | 0.6517 | 0.0741 | 0.5391 | 0.0594 | 0.5556 | 0.0739 | 0.5397 | |||||
| 5 | 0.6516 | 0.0735 | 0.5394 | 0.0626 | 0.5502 | 0.062 | 0.5499 | 0.0741 | 0.5395 | |||
| 6 | 0.6521 | 0.0732 | 0.5395 | 0.0701 | 0.5404 | 0.0579 | 0.5581 | 0.0624 | 0.5499 | 0.0728 | 0.5423 | |
| #n | 1–2 | 2–3 | 3–4 | 4–5 | 5–6 | |
|---|---|---|---|---|---|---|
| I | 1 | 0.2845 | ||||
| 2 | 0.2854 | 0.2884 | ||||
| 3 | 0.2858 | 0.2897 | 0.2894 | |||
| 4 | 0.2860 | 0.2902 | 0.2906 | 0.2898 | ||
| 5 | 0.2861 | 0.2903 | 0.2912 | 0.2910 | 0.2899 | |
| II | 1 | 0.2524 | ||||
| 2 | 0.2537 | 0.2564 | ||||
| 3 | 0.2541 | 0.258 | 0.2574 | |||
| 4 | 0.2543 | 0.2585 | 0.259 | 0.2515 | ||
| 5 | 0.2543 | 0.2587 | 0.2595 | 0.2593 | 0.2578 | |
| III | 1 | 0.3056 | ||||
| 2 | 0.3064 | 0.3092 | ||||
| 3 | 0.3071 | 0.3106 | 0.3105 | |||
| 4 | 0.3072 | 0.3115 | 0.3110 | 0.3110 | ||
| 5 | 0.3075 | 0.3116 | 0.3129 | 0.3124 | 0.3112 | |
| IV | 1 | 0.2279 | ||||
| 2 | 0.2418 | 0.2449 | ||||
| 3 | 0.2461 | 0.2623 | 0.2573 | |||
| 4 | 0.2559 | 0.2697 | 0.2717 | 0.2612 | ||
| 5 | 0.2609 | 0.2823 | 0.2752 | 0.2712 | 0.2585 | |
| V | 1 | 0.1658 | ||||
| 2 | 0.1692 | 0.1721 | ||||
| 3 | 0.1745 | 0.1589 | 0.1717 | |||
| 4 | 0.1727 | 0.1635 | 0.1609 | 0.1719 | ||
| 5 | 0.1702 | 0.1705 | 0.1647 | 0.1658 | 0.1735 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domagała, M.; Simon, S.; Palusiak, M. Resonance-Assisted Hydrogen Bond—Revisiting the Original Concept in the Context of Its Criticism in the Literature. Int. J. Mol. Sci. 2022, 23, 233. https://doi.org/10.3390/ijms23010233
Domagała M, Simon S, Palusiak M. Resonance-Assisted Hydrogen Bond—Revisiting the Original Concept in the Context of Its Criticism in the Literature. International Journal of Molecular Sciences. 2022; 23(1):233. https://doi.org/10.3390/ijms23010233
Chicago/Turabian StyleDomagała, Małgorzata, Sílvia Simon, and Marcin Palusiak. 2022. "Resonance-Assisted Hydrogen Bond—Revisiting the Original Concept in the Context of Its Criticism in the Literature" International Journal of Molecular Sciences 23, no. 1: 233. https://doi.org/10.3390/ijms23010233
APA StyleDomagała, M., Simon, S., & Palusiak, M. (2022). Resonance-Assisted Hydrogen Bond—Revisiting the Original Concept in the Context of Its Criticism in the Literature. International Journal of Molecular Sciences, 23(1), 233. https://doi.org/10.3390/ijms23010233