
Pancreatic Cancer and Cellular Senescence: Tumor Microenvironment under the Spotlight

 ,
, 
 , , ,
, , ,
Abstract
:1. Introduction
2. Pancreatic Ductal Adenocarcinoma Heterogeneity
2.1. Genetic Heterogeneity
2.2. Tumor Microenvironment
2.2.1. Pancreatic Stellate Cells (PSCs)
2.2.2. Cancer-Associated Fibroblasts (CAFs)
2.2.3. Immune Cells
2.2.4. Other Cell Populations
2.2.5. Non-Cellular Components of the Stroma
3. Cellular Senescence: An Overview
3.1. Cellular Senescence
3.2. Markers and Molecular Mechanisms of Senescence
3.3. SASP
4. Dual Role of Senescence in PDAC
5. Senescence in Non-Tumor Cells
6. Senescence in Immune Cells
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Porciuncula, A.; Hajdu, C.; David, G. The Dual Role of Senescence in Pancreatic Ductal Adenocarcinoma. Adv. Cancer Res. 2016, 131, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Gheorghe, G.; Bungau, S.; Ilie, M.; Behl, T.; Vesa, C.M.; Brisc, C.; Bacalbasa, N.; Turi, V.; Costache, R.S.; Diaconu, C.C. Early Diagnosis of Pancreatic Cancer: The Key for Survival. Diagnostics 2020, 10, 869. [Google Scholar] [CrossRef]
- Gutiérrez, M.L.; Muñoz-Bellvís, L.; Orfao, A. Genomic Heterogeneity of Pancreatic Ductal Adenocarcinoma and Its Clinical Impact. Cancers 2021, 13, 4451. [Google Scholar] [CrossRef]
- Veenstra, V.L.; Garcia-Garijo, A.; Van Laarhoven, H.W.; Bijlsma, M.F. Extracellular Influences: Molecular Subclasses and the Microenvironment in Pancreatic Cancer. Cancers 2018, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Bernadotte, A.; Mikhelson, V.M.; Spivak, I.M. Markers of cellular senescence. Telomere shortening as a marker of cellular senescence. Aging 2016, 8, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Prasanna, P.G.; Citrin, D.E.; Hildesheim, J.; Ahmed, M.M.; Venkatachalam, S.; Riscuta, G.; Xi, D.; Zheng, G.; van Deursen, J.; Goronzy, J.; et al. Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J. Natl. Cancer Inst. 2021, 113, 1285–1298. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [Green Version]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Lau, L.; David, G. Pro- and anti-tumorigenic functions of the senescence-associated secretory phenotype. Expert Opin. Ther. Targets 2019, 23, 1041–1051. [Google Scholar] [CrossRef]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Fridman, J.S.; Yang, M.; Lee, S.; Baranov, E.; Hoffman, R.M.; Lowe, S.W. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002, 109, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Kanda, M.; Matthaei, H.; Wu, J.; Hong, S.M.; Yu, J.; Borges, M.; Hruban, R.H.; Maitra, A.; Kinzler, K.; Vogelstein, B.; et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology 2012, 142, 730–733. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Maitra, A. Precursor lesions of pancreatic cancer: Molecular pathology and clinical implications. Pancreatology 2007, 7, 9–19. [Google Scholar] [CrossRef]
- Hayashi, H.; Higashi, T.; Miyata, T.; Yamashita, Y.; Baba, H. Recent advances in precision medicine for pancreatic ductal adenocarcinoma. Ann. Gastroenterol. Surg. 2021, 5, 457–466. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Dardare, J.; Witz, A.; Merlin, J.L.; Gilson, P.; Harlé, A. SMAD4 and the TGFβ Pathway in Patients with Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 3534. [Google Scholar] [CrossRef]
- Waddell, N.N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Lemire, M.; Zhang, A.; Chan-Seng-Yue, M.; Wilson, G.; Grant, R.C.; Merico, D.; Lungu, I.; et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019, 35, 267.e7–282.e7. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef] [Green Version]
- Samuel, N.; Hudson, T.J. The molecular and cellular heterogeneity of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 9, 77–87. [Google Scholar] [CrossRef]
- Iguchi, E.; Safgren, S.L.; Marks, D.L.; Olson, R.L.; Fernandez-Zapico, M.E. Pancreatic cancer, a Mis-interpreter of the epigenetic language. Yale J. Biol. Med. 2016, 89, 575–590. [Google Scholar] [CrossRef]
- Hung, Y.H.; Hsu, M.C.; Chen, L.T.; Hung, W.C.; Pan, M.R. Alteration of Epigenetic Modifiers in Pancreatic Cancer and Its Clinical Implication. J. Clin. Med. 2019, 8, 903. [Google Scholar] [CrossRef] [Green Version]
- Kadaba, R.; Birke, H.; Wang, J.; Hooper, S.; Andl, C.D.; Di Maggio, F.; Soylu, E.; Ghallab, M.; Bor, D.; Froeling, F.E.M.; et al. Imbalance of desmoplastic stromal cell numbers drives aggressive cancer processes. J. Pathol. 2013, 230, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Whatcott, C.J.; Diep, C.H.; Jiang, P.; Watanabe, A.; Lobello, J.; Sima, C.; Hostetter, G.; Shepard, H.M.; Von Hoff, D.D.; Han, H. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin. Cancer Res. 2015, 21, 3561–3568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res. 2008, 68, 2085–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, D.; Carvajo, M.; Bauden, M.; Andersson, R. Pancreatic cancer stroma: Controversies and current insights. Scand. J. Gastroenterol. 2017, 52, 641–646. [Google Scholar] [CrossRef]
- Allam, A.; Thomsen, A.R.; Gothwal, M.; Saha, D.; Maurer, J.; Brunner, T.B. Pancreatic stellate cells in pancreatic cancer: In focus. Pancreatology 2017, 17, 514–522. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Mol. Cancer 2018, 17, 62. [Google Scholar] [CrossRef] [Green Version]
- Schnittert, J.; Bansal, R.; Prakash, J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer 2019, 5, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Apte, M.V.; Wilson, J.S.; Lugea, A.; Pandol, S.J. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013, 144, 1210–1219. [Google Scholar] [CrossRef] [Green Version]
- Lugea, A.; Waldron, R.T. Exosome-Mediated Intercellular Communication Between Stellate Cells and Cancer Cells in Pancreatic Ductal Adenocarcinoma. Pancreas 2017, 46, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Kota, J.; Hancock, J.; Kwon, J.; Korc, M. Pancreatic cancer: Stroma and its current and emerging targeted therapies. Cancer Lett. 2017, 391, 38–49. [Google Scholar] [CrossRef]
- Nagathihalli, N.S.; Castellanos, J.A.; Van Saun, M.N.; Dai, X.; Ambrose, M.; Guo, Q.; Xiong, Y.; Merchant, N.B. Pancreatic stellate cell secreted IL-6 stimulates STAT3 dependent invasiveness of pancreatic intraepithelial neoplasia and cancer cells. Oncotarget 2016, 7, 65982–65992. [Google Scholar] [CrossRef] [Green Version]
- Berchtold, S.; Grünwald, B.; Krüger, A.; Reithmeier, A.; Hähl, T.; Cheng, T.; Feuchtinger, A.; Born, D.; Erkan, M.; Kleeff, J.; et al. Collagen type V promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Cancer Lett. 2015, 356, 721–732. [Google Scholar] [CrossRef]
- Di Maggio, F.; Arumugam, P.; Delvecchio, F.R.; Batista, S.; Lechertier, T.; Hodivala-Dilke, K.; Kocher, H.M. Pancreatic stellate cells regulate blood vessel density in the stroma of pancreatic ductal adenocarcinoma. Pancreatology 2016, 16, 995–1004. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, B.; Pignatelli, C.; Cossutta, M.; Citro, A.; Courty, J.; Piemonti, L. The Extracellular Matrix in Pancreatic Cancer: Description of a Complex Network and Promising Therapeutic Options. Cancers 2021, 13, 4442. [Google Scholar] [CrossRef]
- Tang, D.; Wang, D.; Yuan, Z.; Xue, X.; Zhang, Y.; An, Y.; Chen, J.; Tu, M.; Lu, Z.; Wei, J.; et al. Persistent activation of pancreatic stellate cells creates a microenvironment favorable for the malignant behavior of pancreatic ductal adenocarcinoma. Int. J. cancer 2013, 132, 993–1003. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. 2016, 381, 194–200. [Google Scholar] [CrossRef]
- Von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Viros, A.; Hooper, S.; Mitter, R.; Féral, C.C.; Cook, M.; et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef] [Green Version]
- Malik, R.; Luong, T.; Cao, X.; Han, B.; Shah, N.; Franco-Barraza, J.; Han, L.; Shenoy, V.B.; Lelkes, P.I.; Cukierman, E. Rigidity controls human desmoplastic matrix anisotropy to enable pancreatic cancer cell spread via extracellular signal-regulated kinase 2. Matrix Biol. 2019, 81, 50–69. [Google Scholar] [CrossRef]
- Avery, D.; Govindaraju, P.; Jacob, M.; Todd, L.; Monslow, J.; Puré, E. Extracellular matrix directs phenotypic heterogeneity of activated fibroblasts. Matrix Biol. 2018, 67, 90–106. [Google Scholar] [CrossRef]
- Strell, C.; Paulsson, J.; Jin, S.B.; Tobin, N.P.; Mezheyeuski, A.; Roswall, P.; Mutgan, C.; Mitsios, N.; Johansson, H.; Wickberg, S.M.; et al. Impact of Epithelial-Stromal Interactions on Peritumoral Fibroblasts in Ductal Carcinoma in Situ. J. Natl. Cancer Inst. 2019, 111, 983–995. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Shindo, K.; Aishima, S.; Ohuchida, K.; Fujiwara, K.; Fujino, M.; Mizuuchi, Y.; Hattori, M.; Mizumoto, K.; Tanaka, M.; Oda, Y. Podoplanin expression in cancer-associated fibroblasts enhances tumor progression of invasive ductal carcinoma of the pancreas. Mol. Cancer 2013, 12, 168. [Google Scholar] [CrossRef] [Green Version]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernández-Porras, I.; Cañamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Xu, X.; Guo, S.; Zhang, C.; Tang, Y.; Tian, Y.; Ni, B.; Lu, B.; Wang, H. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS ONE 2014, 9, e91551. [Google Scholar] [CrossRef] [Green Version]
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 2015, 3, 51. [Google Scholar] [CrossRef] [Green Version]
- Bulle, A.; Lim, K.H. Beyond just a tight fortress: Contribution of stroma to epithelial-mesenchymal transition in pancreatic cancer. Signal Transduct. Target. Ther. 2020, 5, 249. [Google Scholar] [CrossRef]
- Markowitz, J.; Brooks, T.R.; Duggan, M.C.; Paul, B.K.; Pan, X.; Wei, L.; Abrams, Z.; Luedke, E.; Lesinski, G.B.; Mundy-Bosse, B.; et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease. Cancer Immunol. Immunother. 2015, 64, 149–159. [Google Scholar] [CrossRef]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Otsuji, M.; Kimura, Y.; Aoe, T.; Okamoto, Y.; Saito, T. Oxidative stress by tumor-derived macrophages suppresses the expression of CD3 zeta chain of T-cell receptor complex and antigen-specific T-cell responses. Proc. Natl. Acad. Sci. USA 1996, 93, 13119–13124. [Google Scholar] [CrossRef] [Green Version]
- Stromnes, I.M.; Brockenbrough, J.S.; Izeradjene, K.; Carlson, M.A.; Cuevas, C.; Simmons, R.M.; Greenberg, P.D.; Hingorani, S.R. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014, 63, 1769–1781. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Jeong, H.; Bae, Y.; Shin, K.; Kang, S.; Kim, H.; Oh, J.; Bae, H. Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. J. Immunother. Cancer 2019, 7, 147. [Google Scholar] [CrossRef] [Green Version]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [Green Version]
- Hao, N.B.; Lü, M.H.; Fan, Y.H.; Cao, Y.L.; Zhang, Z.R.; Yang, S.M. Macrophages in tumor microenvironments and the progression of tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat. Commun. 2015, 6, 7158. [Google Scholar] [CrossRef] [Green Version]
- Elaileh, A.; Saharia, A.; Potter, L.; Baio, F.; Ghafel, A.; Abdelrahim, M.; Heyne, K. Promising new treatments for pancreatic cancer in the era of targeted and immune therapies. Am. J. Cancer Res. 2019, 9, 1871–1888. [Google Scholar]
- Qian, J.; Olbrecht, S.; Boeckx, B.; Vos, H.; Laoui, D.; Etlioglu, E.; Wauters, E.; Pomella, V.; Verbandt, S.; Busschaert, P.; et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020, 30, 745–762. [Google Scholar] [CrossRef]
- Büchler, P.; Reber, H.A.; Büchler, M.; Shrinkante, S.; Büchler, M.W.; Friess, H.; Semenza, G.L.; Hines, O.J. Hypoxia-inducible factor 1 regulates vascular endothelial growth factor expression in human pancreatic cancer. Pancreas 2003, 26, 56–64. [Google Scholar] [CrossRef]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Deucker, S.; Sauliunaite, D.; Streit, S.; Esposito, I.; Friess, H.; Kleeff, J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 2009, 11, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Korc, M. Pathways for aberrant angiogenesis in pancreatic cancer. Mol. Cancer 2003, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, E.; Hermann, P.C.; Mueller, M.T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Hou, Y.C.; Chao, Y.J.; Tung, H.L.; Wang, H.C.; Shan, Y.S. Coexpression of CD44-positive/CD133-positive cancer stem cells and CD204-positive tumor-associated macrophages is a predictor of survival in pancreatic ductal adenocarcinoma. Cancer 2014, 120, 2766–2777. [Google Scholar] [CrossRef] [Green Version]
- Onishi, H.; Katano, M. Hedgehog signaling pathway as a new therapeutic target in pancreatic cancer. World J. Gastroenterol. 2014, 20, 2335–2342. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Clauser, K.R.; Öhlund, D.; Rickelt, S.; Huang, Y.; Gupta, M.; Mani, D.R.; Carr, S.A.; Tuveson, D.A.; Hynes, R.O. Proteomic analyses of ECM during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc. Natl. Acad. Sci. USA 2019, 116, 19609–19618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.; Giancotti, F.G. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell 2019, 35, 347–367. [Google Scholar] [CrossRef]
- Schaffner, F.; Ray, A.M.; Dontenwill, M. Integrin α5β1, the Fibronectin Receptor, as a Pertinent Therapeutic Target in Solid Tumors. Cancers 2013, 5, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [Green Version]
- Abbadie, C.; Pluquet, O.; Pourtier, A. Epithelial cell senescence: An adaptive response to pre-carcinogenic stresses? Cell. Mol. Life Sci. 2017, 74, 4471–4509. [Google Scholar] [CrossRef]
- Erusalimsky, J.D.; Kurz, D.J. Endothelial cell senescence. Handb. Exp. Pharmacol. 2006, 176, 213–248. [Google Scholar] [CrossRef]
- Liu, J.; Ding, Y.; Liu, Z.; Liang, X. Senescence in Mesenchymal Stem Cells: Functional Alterations, Molecular Mechanisms, and Rejuvenation Strategies. Front. Cell Dev. Biol. 2020, 8, 258. [Google Scholar] [CrossRef]
- Zeng, S.; Shen, W.H.; Liu, L. Senescence and Cancer. Cancer Transl. Med. 2018, 4, 70. [Google Scholar] [CrossRef]
- Paez-Ribes, M.; González-Gualda, E.; Doherty, G.J.; Muñoz-Espín, D. Targeting senescent cells in translational medicine. EMBO Mol. Med. 2019, 11, e10234. [Google Scholar] [CrossRef]
- Rhinn, M.; Ritschka, B.; Keyes, W.M. Cellular senescence in development, regeneration and disease. Development 2019, 146, dev151837. [Google Scholar] [CrossRef] [Green Version]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [Green Version]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garré, M.; Giovanni Nuciforo, P.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Nakamura, A.J.; Chiang, Y.J.; Hathcock, K.S.; Horikawa, I.; Sedelnikova, O.A.; Hodes, R.J.; Bonner, W.M. Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenet. Chromatin 2008, 1, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin remodeling underlies the senescence-associated secretory phenotype of tumor stromal fibroblasts that supports cancer progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef] [Green Version]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A review in the theme: Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am. J. Physiol. Cell Physiol. 2015, 308, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Campisi, J. Cancer, aging and cellular senescence. Vivo 2000, 14, 183–188. [Google Scholar]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Martínez-Zamudio, R.I.; Robinson, L.; Roux, P.F.; Bischof, O. SnapShot: Cellular Senescence Pathways. Cell 2017, 170, 816–816.e1. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrillon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring tumorigenesis and senescence in vivo with a p16INK4a-luciferase model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.; Gong, Y.; Yan, G.; Wang, D.; Wang, Q.; Qiao, Y.; Hou, J.; Liu, B.; Tang, C. Atorvastatin treatment modulates p16 promoter methylation to regulate p16 expression. FEBS J. 2017, 284, 1868–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Poi, M.J.; Tsai, M.D. Regulatory mechanisms of tumor suppressor P16INK4A and their relevance to cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, B.H.; Kim, Y.; Je Cho, Y.; Kim, D.S. Distinct roles of transforming growth factor-β signaling and transforming growth factor-β receptor inhibitor SB431542 in the regulation of p21 expression. Eur. J. Pharmacol. 2015, 764, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell. Signal. 2010, 22, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Al-Khalaf, H.H.; Aboussekhra, A. p16INK4A positively regulates p21(WAF1) expression by suppressing AUF1-dependent mRNA decay. PLoS ONE 2013, 8, e70133. [Google Scholar] [CrossRef] [Green Version]
- Zou, L. Single- and double-stranded DNA: Building a trigger of ATR-mediated DNA damage response. Genes Dev. 2007, 21, 879–885. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, Y. The ATM-mediated DNA-damage response: Taking shape. Trends Biochem. Sci. 2006, 31, 402–410. [Google Scholar] [CrossRef]
- Ayrapetov, M.K.; Gursoy-Yuzugullu, O.; Xu, C.; Xu, Y.; Price, B.D. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. USA 2014, 111, 9169–9174. [Google Scholar] [CrossRef] [Green Version]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef]
- Summer, R.; Shaghaghi, H.; Schriner, D.L.; Roque, W.; Sales, D.; Cuevas-Mora, K.; Desai, V.; Bhushan, A.; Ramirez, M.I.; Romero, F. Activation of the mTORC1/PGC-1 axis promotes mitochondrial biogenesis and induces cellular senescence in the lung epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L1049–L1060. [Google Scholar] [CrossRef]
- Sun, Y.; Coppé, J.P.; Lam, E.W.F. Cellular Senescence: The Sought or the Unwanted? Trends Mol. Med. 2018, 24, 871–885. [Google Scholar] [CrossRef]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef]
- Tanaka, Y.; Takahashi, A. Senescence-associated extracellular vesicle release plays a role in senescence-associated secretory phenotype (SASP) in age-associated diseases. J. Biochem. 2021, 169, 147–153. [Google Scholar] [CrossRef]
- Coppé, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Maciel-Barón, L.A.; Morales-Rosales, S.L.; Aquino-Cruz, A.A.; Triana-Martínez, F.; Galván-Arzate, S.; Luna-López, A.; González-Puertos, V.Y.; López-Díazguerrero, N.E.; Torres, C.; Königsberg, M. Senescence associated secretory phenotype profile from primary lung mice fibroblasts depends on the senescence induction stimuli. Age 2016, 38, 26. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Kale, A.; Sharma, A.; Stolzing, A.; Stolzing, A.; Desprez, P.Y.; Desprez, P.Y.; Campisi, J.; Campisi, J. Role of immune cells in the removal of deleterious senescent cells. Immun. Ageing 2020, 17, 16. [Google Scholar] [CrossRef]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; Van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Montero, P.; Londoño-Vallejo, A.; Vernot, J.P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal. 2017, 15, 17. [Google Scholar] [CrossRef] [Green Version]
- Gasek, N.S.; Kuchel, G.A.; Kirkland, J.L.; Xu, M. Strategies for targeting senescent cells in human disease. Nat. Aging 2021, 1, 870–879. [Google Scholar] [CrossRef]
- Rübe, C.E.; Bäumert, C.; Schuler, N.; Isermann, A.; Schmal, Z.; Glanemann, M.; Mann, C.; Scherthan, H. Human skin aging is associated with increased expression of the histone variant H2A.J in the epidermis. NPJ Aging Mech. Dis. 2021, 7, 7. [Google Scholar] [CrossRef]
- Campisi, J.; D’Adda Di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Funayama, R.; Ishikawa, F. Cellular senescence and chromatin structure. Chromosoma 2007, 116, 431–440. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [Green Version]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Gallage, S.; Gil, J. Mitochondrial Dysfunction Meets Senescence. Trends Biochem. Sci. 2016, 41, 207–209. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, M.E.; Denicola, G.M.; Martins, C.P.; Jacobetz, M.A.; Maitra, A.; Hruban, R.H.; Tuveson, D.A. Cellular features of senescence during the evolution of human and murine ductal pancreatic cancer. Oncogene 2012, 31, 1599–1608. [Google Scholar] [CrossRef] [Green Version]
- Frey, N.; Venturelli, S.; Zender, L.; Bitzer, M. Cellular senescence in gastrointestinal diseases: From pathogenesis to therapeutics. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16Ink4a and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansel, D.E.; Kern, S.E.; Hruban, R.H. Molecular pathogenesis of pancreatic cancer. Annu. Rev. Genomics Hum. Genet. 2003, 4, 237–256. [Google Scholar] [CrossRef] [PubMed]
- Ag Moir, J.; A. White, S.; Mann, J. Arrested development and the great escape--the role of cellular senescence in pancreatic cancer. Int. J. Biochem. Cell Biol. 2014, 57, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.; Iacobuzio-Donahue, C.A. Pancreatic cancer biology and genetics from an evolutionary perspective. Nat. Rev. Cancer 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Ellenrieder, V. Senescence in pancreatic carcinogenesis: From signalling to chromatin remodelling and epigenetics. Gut 2013, 62, 1364–1372. [Google Scholar] [CrossRef]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Peeper, D.S.; Dannenberg, J.H.; Douma, S.; Te Riele, H.; Bernards, R. Escape from premature senescence is not sufficient for oncogenic transformation by Ras. Nat. Cell Biol. 2001, 3, 198–203. [Google Scholar] [CrossRef]
- Lowenfels, A.B.; Maisonneuve, P. Epidemiology and risk factors for pancreatic cancer. Best Pract. Res. Clin. Gastroenterol. 2006, 20, 197–209. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Morton, J.P.; Manoharan, I.; Nelson, D.M.; Jamieson, N.B.; Pawlikowski, J.S.; McBryan, T.; Doyle, B.; McKay, C.; Oien, K.A.; et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol. Cell 2011, 42, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.; Yoshizuka, N.; New, L.; Moser, B.A.; Li, Y.; Liao, R.; Xie, C.; Chen, J.; Deng, Q.; Yamout, M.; et al. PRAK is essential for ras-induced senescence and tumor suppression. Cell 2007, 128, 295–308. [Google Scholar] [CrossRef]
- Wang, J.; Kobayashi, T.; Floc’h, N.; Kinkade, C.W.; Aytes, A.; Dankort, D.; Lefebvre, C.; Mitrofanova, A.; Cardiff, R.D.; McMahon, M.; et al. B-Raf activation cooperates with PTEN loss to drive c-Myc expression in advanced prostate cancer. Cancer Res. 2012, 72, 4765–4776. [Google Scholar] [CrossRef] [Green Version]
- Tront, J.S.; Hoffman, B.; Liebermann, D.A. Gadd45a suppresses Ras-driven mammary tumorigenesis by activation of c-Jun NH2-terminal kinase and p38 stress signaling resulting in apoptosis and senescence. Cancer Res. 2006, 66, 8448–8454. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, P.; Seidler, B.; Schüler, S.; Schnieke, A.; Göttlicher, M.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009, 58, 1399–1409. [Google Scholar] [CrossRef] [Green Version]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef] [Green Version]
- Jochems, F.; Thijssen, B.; De Conti, G.; Jansen, R.; Pogacar, Z.; Groot, K.; Wang, L.; Schepers, A.; Wang, C.; Jin, H.; et al. The Cancer SENESCopedia: A delineation of cancer cell senescence. Cell Rep. 2021, 36, 109441. [Google Scholar] [CrossRef]
- Holmer, R.; Goumas, F.A.; Waetzig, G.H.; Rose-John, S.; Kalthoff, H. Interleukin-6: A villain in the drama of pancreatic cancer development and progression. Hepatobiliary Pancreat. Dis. Int 2014, 13, 371–380. [Google Scholar] [CrossRef]
- Slapak, E.J.; Duitman, J.; Tekin, C.; Bijlsma, M.F.; Spek, C.A. Matrix Metalloproteases in Pancreatic Ductal Adenocarcinoma: Key Drivers of Disease Progression? Biology 2020, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Yako, Y.Y.; Kruger, D.; Smith, M.; Brand, M. Cytokines as Biomarkers of Pancreatic Ductal Adenocarcinoma: A Systematic Review. PLoS ONE 2016, 11, e0154016. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Qi, Q.; Wang, P.; Chen, H.; Chen, Z.; Meng, Z.; Liu, L. Serum levels of IL-6, IL-8, and IL-10 are indicators of prognosis in pancreatic cancer. J. Int. Med. Res. 2018, 46, 5228–5236. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.M.; Johansen, A.Z.; Dehlendorff, C.; Jensen, B.V.; Bojesen, S.E.; Pfeiffer, P.; Bjerregaard, J.K.; Nielsen, S.E.; Andersen, F.; Holländer, N.H.; et al. Prognostic Value of Combined Detection of Serum IL6, YKL-40, and C-reactive Protein in Patients with Unresectable Pancreatic Cancer. Cancer Epidemiol. Biomark. Prev. 2020, 29, 176–184. [Google Scholar] [CrossRef]
- Lesina, M.; Wörmann, S.M.; Neuhöfer, P.; Song, L.; Algül, H. Interleukin-6 in inflammatory and malignant diseases of the pancreas. Semin. Immunol. 2014, 26, 80–87. [Google Scholar] [CrossRef]
- Fitzner, B.; Müller, S.; Walther, M.; Fischer, M.; Engelmann, R.; Müller-Hilke, B.; Pützer, B.M.; Kreutzer, M.; Nizze, H.; Jaster, R. Senescence determines the fate of activated rat pancreatic stellate cells. J. Cell. Mol. Med. 2012, 16, 2620–2630. [Google Scholar] [CrossRef]
- Shao, C.; Tu, C.; Cheng, X.; Xu, Z.; Wang, X.; Shen, J.; Chai, K.; Chen, W. Inflammatory and Senescent Phenotype of Pancreatic Stellate Cells Induced by Sqstm1 Downregulation Facilitates Pancreatic Cancer Progression. Int. J. Biol. Sci. 2019, 15, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Straub, J.M.; New, J.; Hamilton, C.D.; Lominska, C.; Shnayder, Y.; Thomas, S.M. Radiation-induced fibrosis: Mechanisms and implications for therapy. J. Cancer Res. Clin. Oncol. 2015, 141, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Ragunathan, K.; Upfold, N.L.E.; Oksenych, V. Interaction between Fibroblasts and Immune Cells Following DNA Damage Induced by Ionizing Radiation. Int. J. Mol. Sci. 2020, 21, 8635. [Google Scholar] [CrossRef] [PubMed]
- Krisnawan, V.E.; Stanley, J.A.; Schwarz, J.K.; Denardo, D.G. Tumor Microenvironment as a Regulator of Radiation Therapy: New Insights into Stromal-Mediated Radioresistance. Cancers 2020, 12, 2916. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Notta, F.; Navab, R.; Joseph, J.; Ibrahimov, E.; Xu, J.; Zhu, C.Q.; Borgida, A.; Gallinger, S.; Tsao, M.S. Senescent Carcinoma-Associated Fibroblasts Upregulate IL8 to Enhance Prometastatic Phenotypes. Mol. Cancer Res. 2017, 15, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansems, M.; Span, P.N. The tumor microenvironment and radiotherapy response; a central role for cancer-associated fibroblasts. Clin. Transl. Radiat. Oncol. 2020, 22, 90–97. [Google Scholar] [CrossRef]
- Levi, N.; Papismadov, N.; Solomonov, I.; Sagi, I.; Krizhanovsky, V. The ECM path of senescence in aging: Components and modifiers. FEBS J. 2020, 287, 2636–2646. [Google Scholar] [CrossRef]
- Blokland, K.E.C.; Pouwels, S.D.; Schuliga, M.; Knight, D.A.; Burgess, J.K. Regulation of cellular senescence by extracellular matrix during chronic fibrotic diseases. Clin. Sci. 2020, 134, 2681–2706. [Google Scholar] [CrossRef]
- Hwang, H.J.; Lee, Y.R.; Kang, D.; Lee, H.C.; Seo, H.R.; Ryu, J.K.; Kim, Y.N.; Ko, Y.G.; Park, H.J.; Lee, J.S. Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett. 2020, 490, 100–110. [Google Scholar] [CrossRef]
- Wang, D.; Xiao, F.; Feng, Z.; Li, M.; Kong, L.; Huang, L.; Wei, Y.; Li, H.; Liu, F.; Zhang, H.; et al. Sunitinib facilitates metastatic breast cancer spreading by inducing endothelial cell senescence. Breast Cancer Res. 2020, 22, 103. [Google Scholar] [CrossRef]
- Ruscetti, M.; Morris, J.P.; Mezzadra, R.; Russell, J.; Leibold, J.; Romesser, P.B.; Simon, J.; Kulick, A.; Ho, Y.jui; Fennell, M.; et al. Senescence-Induced Vascular Remodeling Creates Therapeutic Vulnerabilities in Pancreas Cancer. Cell 2021, 184, 4838–4839. [Google Scholar] [CrossRef]
- Lian, J.; Yue, Y.; Yu, W.; Zhang, Y. Immunosenescence: A key player in cancer development. J. Hematol. Oncol. 2020, 13, 151. [Google Scholar] [CrossRef]
- Prata, L.G.P.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef]
- Song, P.; An, J.; Zou, M.H. Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases. Cells 2020, 9, 671. [Google Scholar] [CrossRef] [Green Version]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Ye, J.; Huang, X.; Hsueh, E.C.; Zhang, Q.; Ma, C.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Human regulatory T cells induce T-lymphocyte senescence. Blood 2012, 120, 2021–2031. [Google Scholar] [CrossRef]
- Ye, J.; Ma, C.; Hsueh, E.C.; Eickhoff, C.S.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Tumor-derived γδ regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J. Immunol. 2013, 190, 2403–2414. [Google Scholar] [CrossRef]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef]
- Etxebeste-Mitxeltorena, M.; del Rincón-Loza, I.; Martín-Antonio, B. Tumor Secretome to Adoptive Cellular Immunotherapy: Reduce Me Before I Make You My Partner. Front. Immunol. 2021, 12, 3201. [Google Scholar] [CrossRef]
- Elkabets, M.; Ribeiro, V.S.G.; Dinarello, C.A.; Ostrand-Rosenberg, S.; Di Santo, J.P.; Apte, R.N.; Vosshenrich, C.A.J. IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur. J. Immunol. 2010, 40, 3347–3357. [Google Scholar] [CrossRef]
- Wu, J.; Gao, F.X.; Wang, C.; Qin, M.; Han, F.; Xu, T.; Hu, Z.; Long, Y.; He, X.M.; Deng, X.; et al. IL-6 and IL-8 secreted by tumour cells impair the function of NK cells via the STAT3 pathway in oesophageal squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 321. [Google Scholar] [CrossRef]
- Behmoaras, J.; Gil, J. Similarities and interplay between senescent cells and macrophages. J. Cell Biol. 2021, 220, e202010162. [Google Scholar] [CrossRef]
- Ogata, Y.; Yamada, T.; Hasegawa, S.; Sanada, A.; Iwata, Y.; Arima, M.; Nakata, S.; Sugiura, K.; Akamatsu, H. SASP-induced macrophage dysfunction may contribute to accelerated senescent fibroblast accumulation in the dermis. Exp. Dermatol. 2021, 30, 84–91. [Google Scholar] [CrossRef]
- Lesina, M.; Wörmann, S.M.; Morton, J.; Diakopoulos, K.N.; Korneeva, O.; Wimmer, M.; Einwächter, H.; Sperveslage, J.; Demir, I.E.; Kehl, T.; et al. RelA regulates CXCL1/CXCR2-dependent oncogene-induced senescence in murine Kras-driven pancreatic carcinogenesis. J. Clin. Investig. 2016, 126, 2919–2932. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Demoor-Goldschmidt, C.; De Vathaire, F. Review of risk factors of secondary cancers among cancer survivors. Br. J. Radiol. 2019, 92, 1093. [Google Scholar] [CrossRef] [PubMed]
- Ducreux, M.; Sa Cuhna, A.; Caramella, C.; Hollebecque, A.; Burtin, P.; Goéré, D.; Seufferlein, T.; Haustermans, K.; Van Laethem, J.L.; Brousse, P. Cancer of the pancreas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. ESMO Updat. Clin. Pract. Guidel. 2015, 26, v56–v68. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Kapahi, P.; Lithgow, G.J.; Melov, S.; Newman, J.C.; Verdin, E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019, 571, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
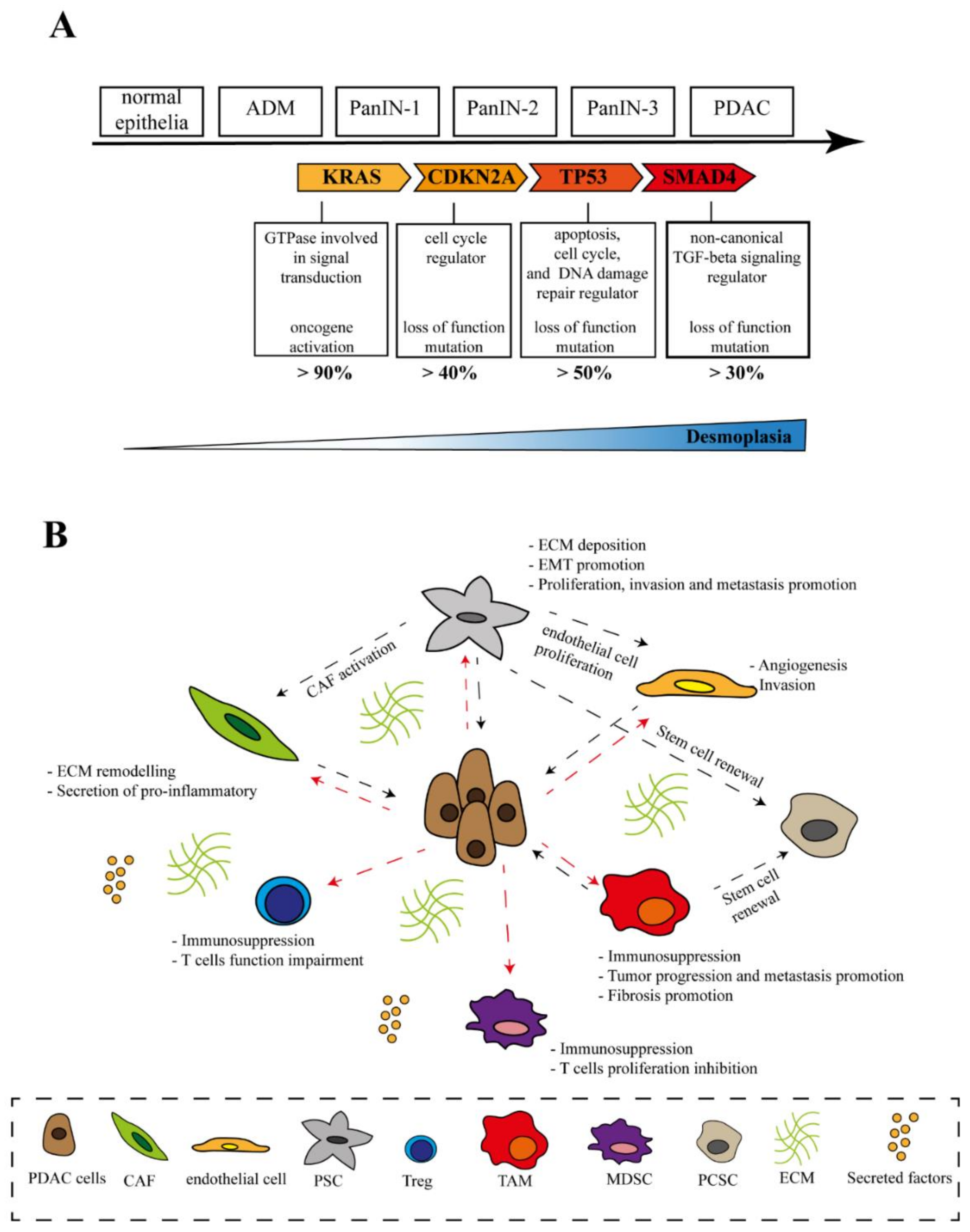
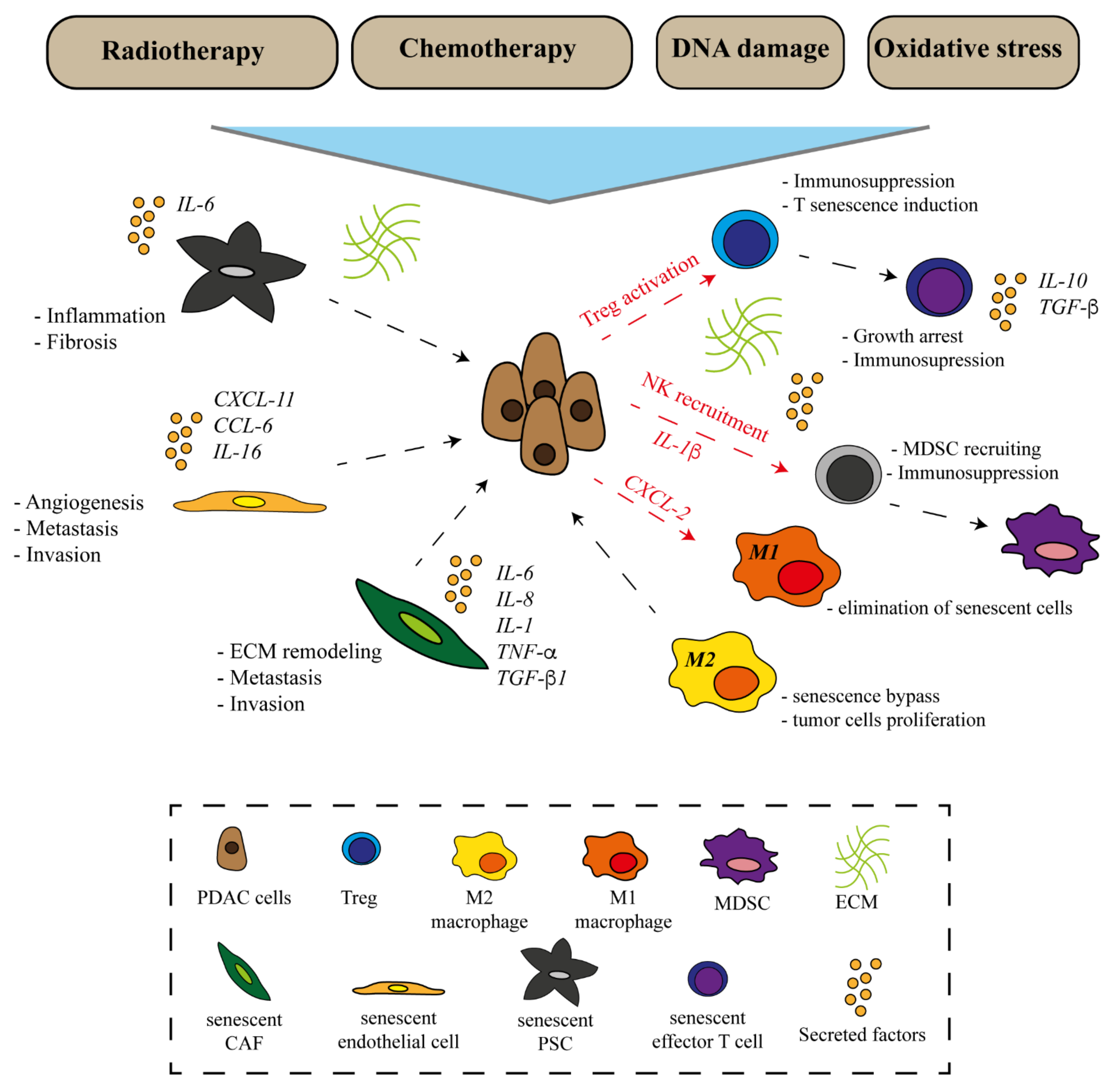

{kind=link}
{kind=link}
| Characteristics | Markers | Ref. |
|---|---|---|
| Proliferative arrest | BrDU + Ki-67 - pRB inactivation | Campisi et al. [133] |
| Persistent DDR activation | p53, p16, p21 up-regulation Lamin B1 down-regulation | Gorgoulis et al. [106] Freund et al. [134] |
| Heterochromatic foci | gamma-H2AX + DAPI staining | Funayama et al. [135] |
| Cells flattened and enlarged | Microscopy visible changes | Hernandez-Segura et al. [107] |
| Apoptosis resistance | Bcl-2; Bcl-XL, Bcl-W expression increase | Childs et al. [136] |
| Altered metabolism | SA-β-gal + Lipofuscin + | Debacq-Chainaux et al. [137] |
| Organelle dysfunction | Increased number of mitochondria Morphological changes Increased ROS levels | Gallage et al. [138] |
| SASP | IL-6; IL-8; MMPs; PAI-1 | Coppe et al. [124] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortesi, M.; Zanoni, M.; Pirini, F.; Tumedei, M.M.; Ravaioli, S.; Rapposelli, I.G.; Frassineti, G.L.; Bravaccini, S. Pancreatic Cancer and Cellular Senescence: Tumor Microenvironment under the Spotlight. Int. J. Mol. Sci. 2022, 23, 254. https://doi.org/10.3390/ijms23010254
Cortesi M, Zanoni M, Pirini F, Tumedei MM, Ravaioli S, Rapposelli IG, Frassineti GL, Bravaccini S. Pancreatic Cancer and Cellular Senescence: Tumor Microenvironment under the Spotlight. International Journal of Molecular Sciences. 2022; 23(1):254. https://doi.org/10.3390/ijms23010254
Chicago/Turabian StyleCortesi, Michela, Michele Zanoni, Francesca Pirini, Maria Maddalena Tumedei, Sara Ravaioli, Ilario Giovanni Rapposelli, Giovanni Luca Frassineti, and Sara Bravaccini. 2022. "Pancreatic Cancer and Cellular Senescence: Tumor Microenvironment under the Spotlight" International Journal of Molecular Sciences 23, no. 1: 254. https://doi.org/10.3390/ijms23010254
APA StyleCortesi, M., Zanoni, M., Pirini, F., Tumedei, M. M., Ravaioli, S., Rapposelli, I. G., Frassineti, G. L., & Bravaccini, S. (2022). Pancreatic Cancer and Cellular Senescence: Tumor Microenvironment under the Spotlight. International Journal of Molecular Sciences, 23(1), 254. https://doi.org/10.3390/ijms23010254