
CCN2 Increases TGF-β Receptor Type II Expression in Vascular Smooth Muscle Cells: Essential Role of CCN2 in the TGF-β Pathway Regulation

 , , , , ,
, , , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
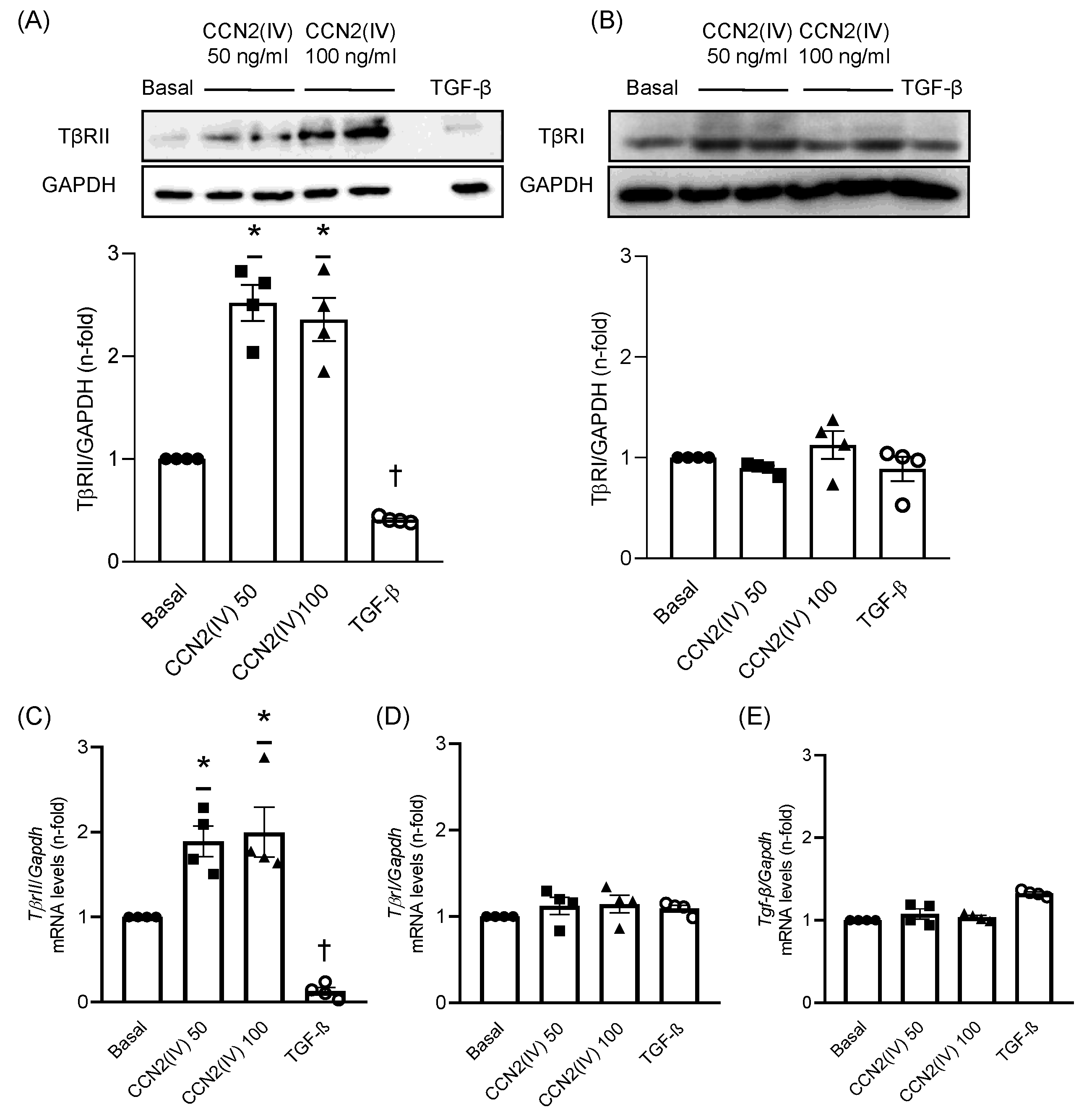
2.1. CCN2(IV) Increases TβRII Levels in Cultured VSMCs after 48 h
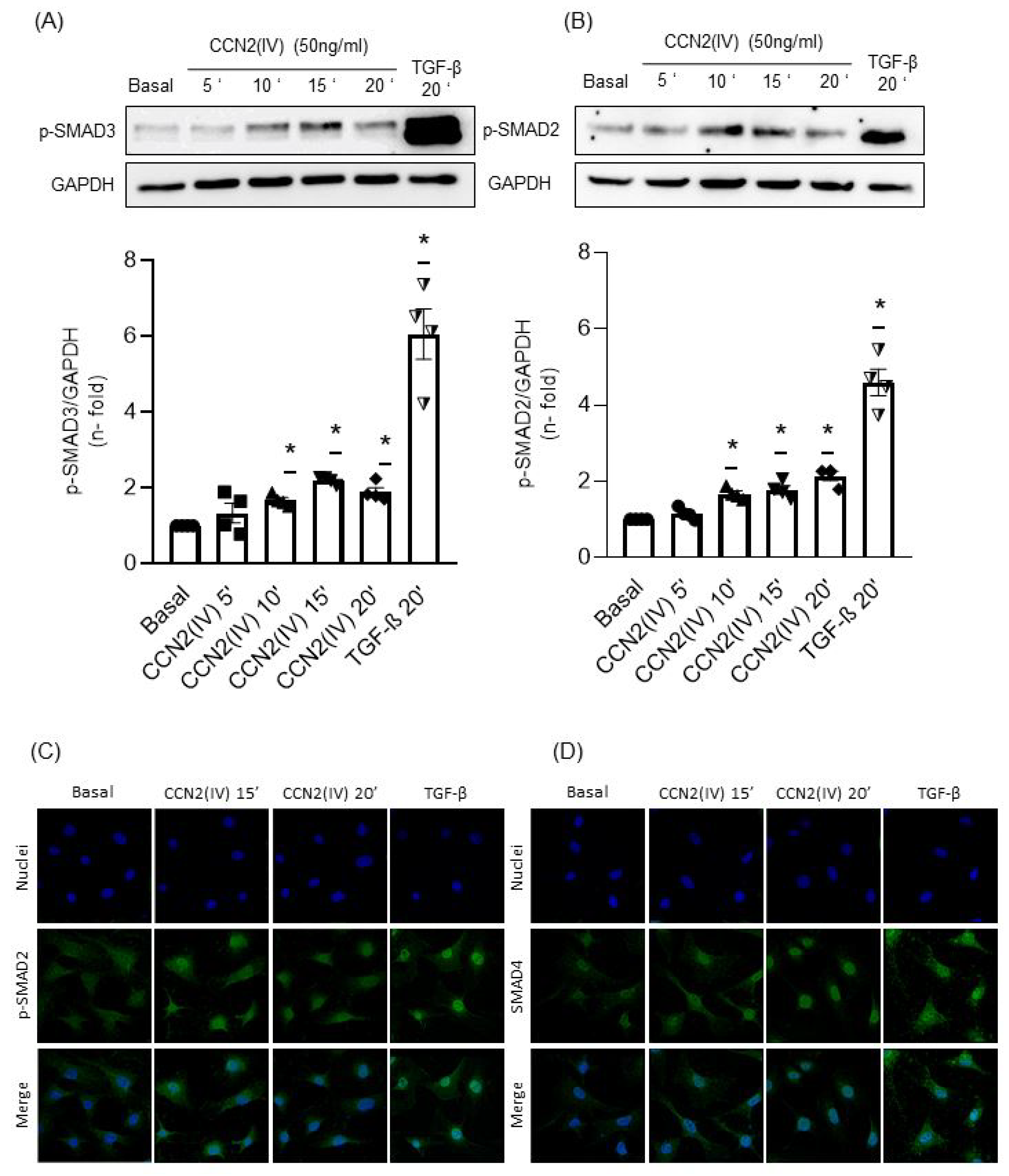
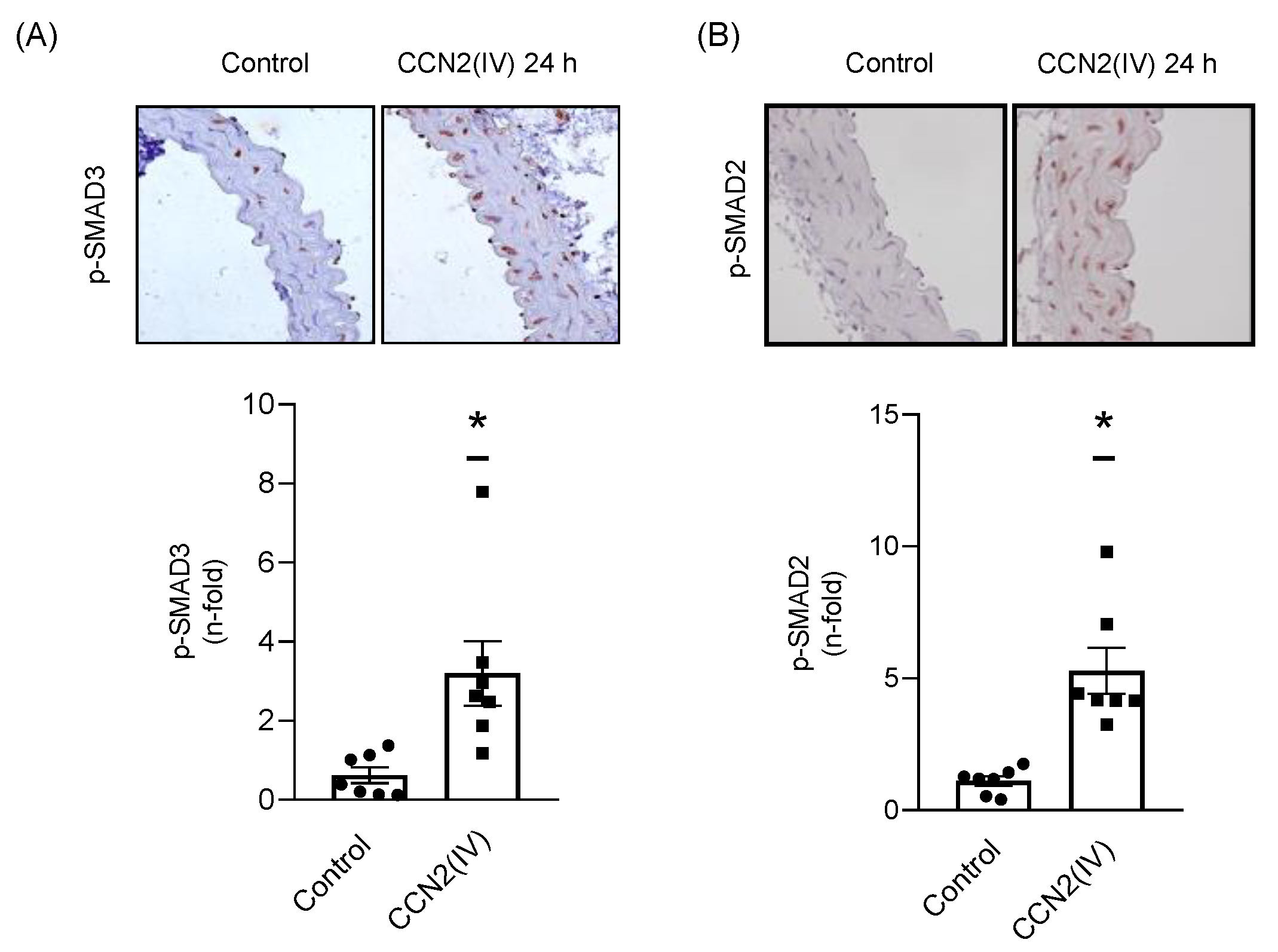
2.2. CCN2(IV) Activates the SMAD Pathway in Cultured and Aortic VSMCs at Short Times
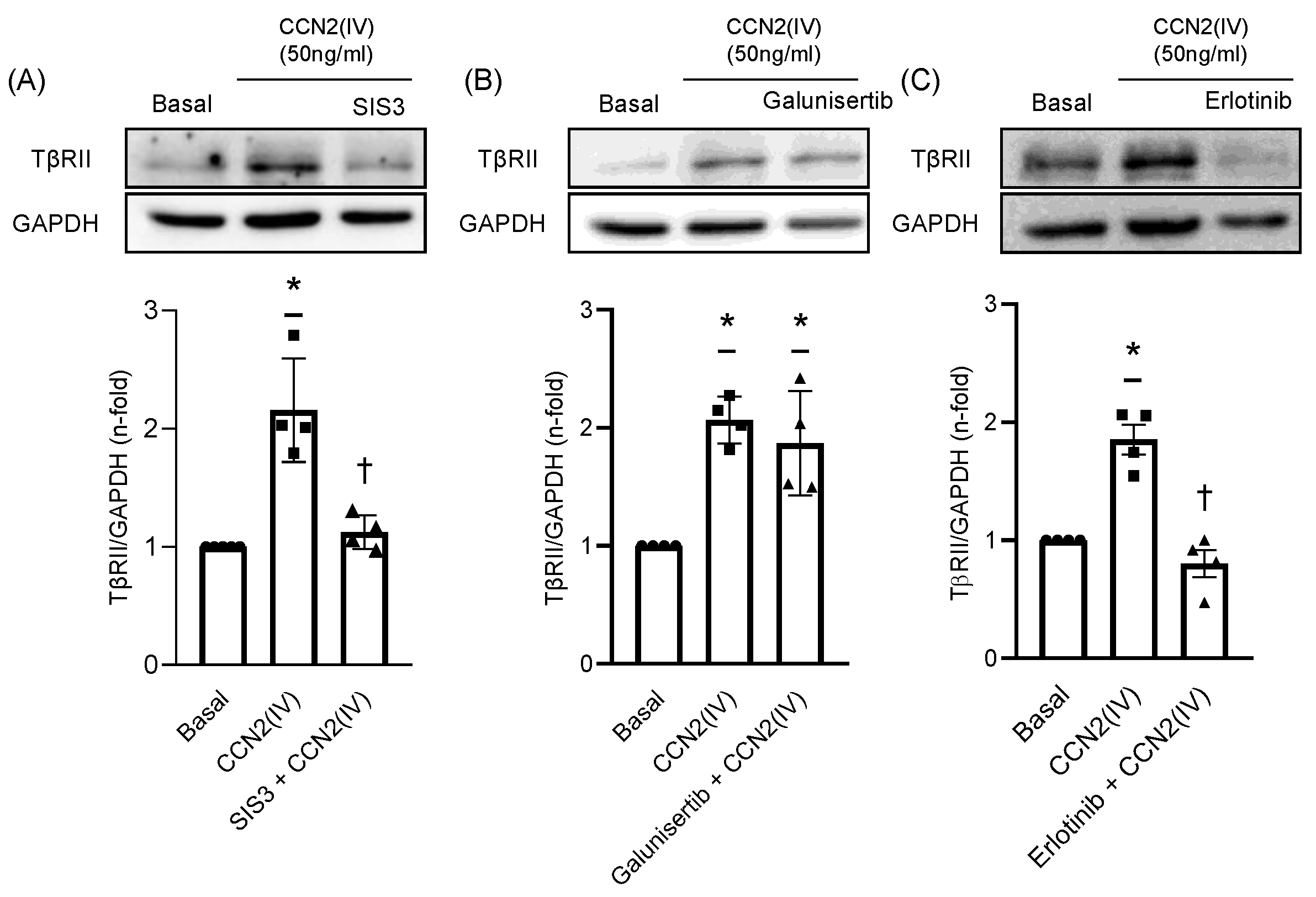
2.3. CCN2(IV) Increases TβRII Expression in VSMCs by TGF- β –Independent SMAD Activation
2.4. TβRII Expression Induced by CCN2(IV) in VSMCs Is Mediated by the EGF Receptor
3. Discussion
4. Materials and Methods
4.1. Experimental Mice Model
4.2. Histological Analysis
4.3. Cell Cultures
4.4. Immunofluorescence
4.5. qPCR Analysis
4.6. Western Blot
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Javelaud, D.; Mauviel, A. Mammalian transforming growth factor-betas: Smad signaling and physio-pathological roles. Int. J. Biochem. Cell Biol. 2004, 36, 1161–1165. [Google Scholar] [CrossRef]
- Massagué, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Pardali, K.; Gaal, A.; Heldin, C.H. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunol. Lett. 2002, 82, 85–91. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Goumans, M.J.; ten Dijke, P. TGF-β Signaling in Control of Cardiovascular Function. Cold Spring Harb. Perspect. Biol. 2018, 10, a022210. [Google Scholar] [CrossRef]
- Tzavlaki, K.; Moustakas, A. TGF-β Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef]
- Leask, A. Targeting the TGFβ, endothelin-1 and CCN2 axis to combat fibrosis in scleroderma. Cell. Signal. 2008, 20, 1409–1414. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Goumans, M.J.; Liu, Z.; Ten Dijke, P. TGF-beta signaling in vascular biology and dysfunction. Cell Res. 2009, 19, 116–127. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Rodriguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-beta signaling in vascular fibrosis. Cardiovasc. Res. 2007, 74, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Cichon, M.A.; Radisky, D.C. Extracellular matrix as a contextual determinant of transforming growth factor-β signaling in epithelial-mesenchymal transition and in cancer. Cell Adh. Migr. 2014, 8, 588–594. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.L.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Toma, I.; McCaffrey, T.A. Transforming growth factor-β and atherosclerosis: Interwoven atherogenic and atheroprotective aspects. Cell Tissue Res. 2012, 347, 155. [Google Scholar] [CrossRef]
- Chen, P.Y.; Qin, L.; Li, G.; Wang, Z.; Dahlman, J.E.; Malagon-Lopez, J.; Gujja, S.; Cilfone, N.A.; Kauffman, K.J.; Sun, L.; et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat. Metab. 2019, 1, 912–926. [Google Scholar] [CrossRef] [PubMed]
- Perbal, B.; Tweedie, S.; Bruford, E. The official unified nomenclature adopted by the HGNC calls for the use of the acronyms, CCN1–6, and discontinuation in the use of CYR61, CTGF, NOV and WISP 1–3 respectively. J. Cell Commun. Signal. 2018, 12, 625–629. [Google Scholar] [CrossRef]
- Perbal, A.; Perbal, B. The CCN family of proteins: A 25th anniversary picture. J. Cell Commun. Signal. 2016, 10, 177–190. [Google Scholar] [CrossRef]
- De Winter, P.; Leoni, P.; Abraham, D. Connective tissue growth factor: Structure-function relationships of a mosaic, multifunctional protein. Growth Factors 2008, 26, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Perbal, B. CCN proteins: Multifunctional signalling regulators. Lancet 2004, 363, 62–64. [Google Scholar] [CrossRef]
- Ponticos, M. Connective tissue growth factor (CCN2) in blood vessels. Vascul. Pharmacol. 2013, 58, 189–193. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. All in the CCN family: Essential matricellular signaling modulators emerge from the bunker. J. Cell Sci. 2006, 119, 4803–4810. [Google Scholar] [CrossRef] [PubMed]
- Kaasbøll, O.J.; Gadicherla, A.K.; Wang, J.H.; Monsen, V.T.; Hagelin, E.M.V.; Dong, M.Q.; Attramadal, H. Connective tissue growth factor (CCN2) is a matricellular preproprotein controlled by proteolytic activation. J. Biol. Chem. 2018, 293, 17953–17970. [Google Scholar] [CrossRef] [PubMed]
- Cicha, I.; Yilmaz, A.; Klein, M.; Raithel, D.; Brigstock, D.R.; Daniel, W.G.; Goppelt-Struebe, M.; Garlichs, C.D. Connective tissue growth factor is overexpressed in complicated atherosclerotic plaques and induces mononuclear cell chemotaxis in vitro. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1008–1013. [Google Scholar] [CrossRef]
- Kundi, R.; Hollenbeck, S.T.; Yamanouchi, D.; Herman, B.C.; Edlin, R.; Ryer, E.J.; Wang, C.; Tsai, S.; Liu, B.; Kent, K.C. Arterial gene transfer of the TGF-beta signalling protein Smad3 induces adaptive remodelling following angioplasty: A role for CTGF. Cardiovasc. Res. 2009, 84, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Oemar, B.S.; Werner, A.; Garnier, J.M.; Do, D.D.; Godoy, N.; Nauck, M.; März, W.; Rupp, J.; Pech, M.; Lüscher, T.F. Human connective tissue growth factor is expressed in advanced atherosclerotic lesions. Circulation 1997, 95, 831–839. [Google Scholar] [CrossRef]
- Rodriguez-Vita, J.; Ruiz-Ortega, M.; Rupérez, M.; Esteban, V.; Sanchez-López, E.; Plaza, J.J.; Egido, J. Endothelin-1, via ETA receptor and independently of transforming growth factor-β, increases the connective tissue growth factor in vascular smooth muscle cells. Circ. Res. 2005, 97, 125–134. [Google Scholar] [CrossRef]
- Rupérez, M.; Lorenzo, Ó.; Blanco-Colio, L.M.; Esteban, V.; Egido, J.; Ruiz-Ortega, M. Connective tissue growth factor is a mediator of angiotensin II-induced fibrosis. Circulation 2003, 108, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Diez, R.R.; Garcia-Redondo, A.B.; Orejudo, M.; Rodrigues-Diez, R.; Briones, A.M.; Bosch-Panadero, E.; Kery, G.; Pato, J.; Ortiz, A.; Salaices, M.; et al. The C-terminal module IV of connective tissue growth factor, through EGFR/Nox1 signaling, activates the NF-κB pathway and proinflammatory factors in vascular smooth muscle cells. Antioxid. Redox Signal. 2015, 22, 29–47. [Google Scholar] [CrossRef]
- Sánchez-López, E.; Rayego, S.; Rodrigues-Díez, R.; Rodriguez, J.S.; Rodrigues-Díez, R.; Rodríguez-Vita, J.; Carvajal, G.; Aroeira, L.S.; Selgas, R.; Mezzano, S.A.; et al. CTGF promotes inflammatory cell infiltration of the renal interstitium by activating NF-κB. J. Am. Soc. Nephrol. 2009, 20, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Charrier, A.; Chen, R.; Kemper, S.; Brigstock, D.R. Regulation of pancreatic inflammation by connective tissue growth factor (CTGF/CCN2). Immunology 2014, 141, 564–576. [Google Scholar] [CrossRef]
- Rodrigues-Díez, R.; Rodrigues-Díez, R.R.; Rayego-Mateos, S.; Suarez-Alvarez, B.; Lavoz, C.; Stark Aroeira, L.; Sánchez-López, E.; Orejudo, M.; Alique, M.; Lopez-Larrea, C.; et al. The C-terminal module IV of connective tissue growth factor is a novel immune modulator of the Th17 response. Lab. Investig. 2013, 93, 812–824. [Google Scholar] [CrossRef]
- Koitabashi, N.; Arai, M.; Niwano, K.; Watanabe, A.; Endoh, M.; Suguta, M.; Yokoyama, T.; Tada, H.; Toyama, T.; Adachi, H.; et al. Plasma connective tissue growth factor is a novel potential biomarker of cardiac dysfunction in patients with chronic heart failure. Eur. J. Heart Fail. 2008, 10, 373–379. [Google Scholar] [CrossRef]
- Wang, X.; McLennan, S.V.; Allen, T.J.; Twigg, S.M. Regulation of pro-inflammatory and pro-fibrotic factors by CCN2/CTGF in H9c2 cardiomyocytes. J. Cell Commun. Signal. 2010, 4, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Xie, Y.; Peng, M.; Ma, L.; Zhou, Y.; Zhang, Y.; Kang, W.; Wang, J.; Bai, X.; Wang, P.; et al. Inhibition of connective tissue growth factor suppresses hepatic stellate cell activation in vitro and prevents liver fibrosis in vivo. Clin. Exp. Med. 2014, 14, 141–150. [Google Scholar] [CrossRef]
- Phanish, M.K.; Winn, S.K.; Dockrell, M.E.C. Connective tissue growth factor-(CTGF, CCN2)-A marker, mediator and therapeutic target for renal fibrosis. Nephron-Exp. Nephrol. 2010, 114, e83–e92. [Google Scholar] [CrossRef]
- Ponticos, M.; Holmes, A.M.; Shi-wen, X.; Leoni, P.; Khan, K.; Rajkumar, V.S.; Hoyles, R.K.; Bou-Gharios, G.; Black, C.M.; Denton, C.P.; et al. Pivotal role of connective tissue growth factor in lung fibrosis: MAPK-dependent transcriptional activation of type I collagen. Arthritis Rheum. 2009, 60, 2142–2155. [Google Scholar] [CrossRef]
- Shakil Ahmed, M.; Gravning, J.; Martinov, V.N.; von Lueder, T.G.; Edvardsen, T.; Czibik, G.; Moe, I.T.; Vinge, L.E.; Øie, E.; Valen, G.; et al. Mechanisms of novel cardioprotective functions of CCN2/CTGF in myocardial ischemia-reperfusion injury. Am. J. Physiol.-Hear. Circ. Physiol. 2011, 300, H1291–H1302. [Google Scholar] [CrossRef]
- Gravning, J.; Ahmed, M.S.; Von Lueder, T.G.; Edvardsen, T.; Attramadal, H. CCN2/CTGF attenuates myocardial hypertrophy and cardiac dysfunction upon chronic pressure-overload. Int. J. Cardiol. 2013, 168, 2049–2056. [Google Scholar] [CrossRef]
- Moe, I.T.; Ahmed, M.S.; Stang, E.; Hagelin, E.M.V.; Attramadal, H. CTGF/CCN2 postconditioning increases tolerance of murine hearts towards ischemia-reperfusion injury 1ole jørgen kaasbøll. PLoS ONE 2016, 11, e0149000. [Google Scholar] [CrossRef]
- Panek, A.N.; Posch, M.G.; Alenina, N.; Ghadge, S.K.; Erdmann, B.; Popova, E.; Perrot, A.; Geier, C.; Morano, R.D.I.; Bader, M.; et al. Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertrophy and protection against pressure overload. PLoS ONE 2009, 4, e6743. [Google Scholar] [CrossRef]
- Moe, I.T.; Pham, T.A.; Hagelin, E.M.V.; Ahmed, M.S.; Attramadal, H. CCN2 exerts direct cytoprotective actions in adult cardiac myocytes by activation of the PI3-kinase/Akt/GSK-3β signaling pathway. J. Cell Commun. Signal. 2013, 7, 31–47. [Google Scholar] [CrossRef]
- Mori, T.; Kawara, S.; Shinozaki, M.; Hayashi, N.; Kakinuma, T.; Igarashi, A.; Takigawa, M.; Nakanishi, T.; Takehara, K. Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: A mouse fibrosis model. J. Cell. Physiol. 1999, 181, 153–159. [Google Scholar] [CrossRef]
- Lasky, J.; Ortiz, L.A.; Tonthat, B.; Hoyle, G.W.; Corti, M.; Athas, G.; Lungarella, G.; Brody, A.; Friedman, M. Connective tissue growth factor mRNA expression is upregulated in bleomycin-induced lung fibrosis. Am. J. Physiol. 1998, 275, L365–L371. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F. Signaling in fibrosis: Targeting the TGF beta, endothelin-1 and CCN2 axis in scleroderma. Front. Biosci. (Elite Ed.) 2009, 1, 125–131. [Google Scholar] [CrossRef]
- Bonniaud, P.; Martin, G.; Margetts, P.J.; Ask, K.; Robertson, J.; Gauldie, J.; Kolb, M. Connective tissue growth factor is crucial to inducing a profibrotic environment in “fibrosis-resistant” BALB/c mouse lungs. Am. J. Respir. Cell Mol. Biol. 2004, 31, 510–516. [Google Scholar] [CrossRef]
- Bonniaud, P.; Margetts, P.J.; Kolb, M.; Haberberger, T.; Kelly, M.; Robertson, J.; Gauldie, J. Adenoviral gene transfer of connective tissue growth factor in the lung induces transient fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 770–778. [Google Scholar] [CrossRef]
- Rojas, A.; Padidam, M.; Cress, D.; Grady, W.M. TGF-beta receptor levels regulate the specificity of signaling pathway activation and biological effects of TGF-beta. Biochim. Biophys. Acta 2009, 1793, 1165–1173. [Google Scholar] [CrossRef]
- Lönn, P.; Morén, A.; Raja, E.; Dahl, M.; Moustakas, A. Regulating the stability of TGFbeta receptors and Smads. Cell Res. 2009, 19, 21–35. [Google Scholar] [CrossRef]
- Kang, J.S.; Liu, C.; Derynck, R. New regulatory mechanisms of TGF-beta receptor function. Trends Cell Biol. 2009, 19, 385–394. [Google Scholar] [CrossRef]
- Rayego-Mateos, S.; Rodrigues-Díez, R.; Morgado-Pascual, J.L.; Rodrigues Díez, R.R.; Mas, S.; Lavoz, C.; Alique, M.; Pato, J.; Keri, G.; Ortiz, A.; et al. Connective tissue growth factor is a new ligand of epidermal growth factor receptor. J. Mol. Cell Biol. 2013, 5, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef] [PubMed]
- Rayego-Mateos, S.; Campillo, S.; Rodrigues-Diez, R.R.; Tejera-Muñoz, A.; Marquez-Exposito, L.; Goldschmeding, R.; Rodríguez-Puyol, D.; Calleros, L.; Ruiz-Ortega, M. Interplay between extracellular matrix components and cellular and molecular mechanisms in kidney fibrosis. Clin. Sci. (Lond.) 2021, 135, 1999–2029. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, N.; Chu, H.Y.; Yu, Y.; Zhang, Z.K.; Zhang, G.; Zhang, B.T. Connective Tissue Growth Factor: From Molecular Understandings to Drug Discovery. Front. Cell Dev. Biol. 2020, 8, 1239. [Google Scholar] [CrossRef]
- Di Guglielmo, G.M.; Le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat. Cell Biol. 2003, 5, 410–421. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, T.A.; Consigli, S.; Du, B.; Falcone, D.J.; Sanborn, T.A.; Spokojny, A.M.; Bush, H.L. Decreased type II/type I TGF-beta receptor ratio in cells derived from human atherosclerotic lesions. Conversion from an antiproliferative to profibrotic response to TGF-beta1. J. Clin. Investig. 1995, 96, 2667–2675. [Google Scholar] [CrossRef] [PubMed]
- McCaffrey, T.A.; Du, B.; Fu, C.; Bray, P.J.; Sanborn, T.A.; Deutsch, E.; Tarazona, N.; Shaknovitch, A.; Newman, G.; Patterson, C.; et al. The expression of TGF-beta receptors in human atherosclerosis: Evidence for acquired resistance to apoptosis due to receptor imbalance. J. Mol. Cell. Cardiol. 1999, 31, 1627–1642. [Google Scholar] [CrossRef]
- McCaffrey, T.A.; Du, B.; Consigli, S.; Szabo, P.; Bray, P.J.; Hartner, L.; Weksler, B.B.; Sanborn, T.A.; Bergman, G.; Bush, H.L. Genomic instability in the type II TGF-beta1 receptor gene in atherosclerotic and restenotic vascular cells. J. Clin. Investig. 1997, 100, 2182–2188. [Google Scholar] [CrossRef]
- Rodríguez-Vita, J.; Sínchez-Galín, E.; Santamaría, B.; Sánchez-López, E.; Rodrigues-Díez, R.; Blanco-Colio, L.M.; Egido, J.; Ortiz, A.; Ruiz-Ortega, M. Essential role of TGF-beta/Smad pathway on statin dependent vascular smooth muscle cell regulation. PLoS ONE 2008, 3, e3959. [Google Scholar] [CrossRef] [PubMed]
- Chaqour, B. Caught between a “Rho” and a hard place: Are CCN1/CYR61 and CCN2/CTGF the arbiters of microvascular stiffness? J. Cell Commun. Signal. 2020, 14, 21–29. [Google Scholar] [CrossRef]
- Kubota, S.; Takigawa, M. Cellular and molecular actions of CCN2/CTGF and its role under physiological and pathological conditions. Clin. Sci. (Lond.) 2015, 128, 181–196. [Google Scholar] [CrossRef]
- Wang, R.; Xu, Y.J.; Liu, X.S.; Zeng, D.X.; Xiang, M. Knockdown of connective tissue growth factor by plasmid-based short hairpin RNA prevented pulmonary vascular remodeling in cigarette smoke-exposed rats. Arch. Biochem. Biophys. 2011, 508, 93–100. [Google Scholar] [CrossRef]
- Szabó, Z.; Magga, J.; Alakoski, T.; Ulvila, J.; Piuhola, J.; Vainio, L.; Kivirikko, K.I.; Vuolteenaho, O.; Ruskoaho, H.; Lipson, K.E.; et al. Connective tissue growth factor inhibition attenuates left ventricular remodeling and dysfunction in pressure overload-induced heart failure. Hypertension 2014, 63, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Gravning, J.; Ørn, S.; Kaasbøll, O.J.; Martinov, V.N.; Manhenke, C.; Dickstein, K.; Edvardsen, T.; Attramadal, H.; Ahmed, M.S. Myocardial connective tissue growth factor (CCN2/CTGF) attenuates left ventricular remodeling after myocardial infarction. PLoS ONE 2012, 7, e52120. [Google Scholar] [CrossRef] [PubMed]
- Ivkovic, S.; Yoon, B.S.; Popoff, S.N.; Safadi, F.F.; Libuda, D.E.; Stephenson, R.C.; Daluiski, A.; Lyons, K.M. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development 2003, 130, 2779–2791. [Google Scholar] [CrossRef] [PubMed]
- Rayego-Mateos, S.; Morgado-Pascual, J.L.; Rodrigues-Diez, R.R.; Rodrigues-Diez, R.; Falke, L.L.; Mezzano, S.; Ortiz, A.; Egido, J.; Goldschmeding, R.; Ruiz-Ortega, M. Connective tissue growth factor induces renal fibrosis via epidermal growth factor receptor activation. J. Pathol. 2018, 244, 227–241. [Google Scholar] [CrossRef]
- Fontes, M.S.C.; Kessler, E.L.; van Stuijvenberg, L.; Brans, M.A.; Falke, L.L.; Kok, B.; Leask, A.; van Rijen, H.V.M.; Vos, M.A.; Goldschmeding, R.; et al. CTGF knockout does not affect cardiac hypertrophy and fibrosis formation upon chronic pressure overload. J. Mol. Cell. Cardiol. 2015, 88, 82–90. [Google Scholar] [CrossRef]
- Rodrigues-Díez, R.R.; Tejera-Muñoz, A.; Esteban, V. CCN2 (Cellular Communication Network Factor 2) Deletion Alters Vascular Integrity and Function Predisposing to Aneurysm Formation. Hypertension, 2022; in press. [Google Scholar] [CrossRef]
- Chen, X.; Rateri, D.L.; Howatt, D.A.; Balakrishnan, A.; Moorleghen, J.J.; Cassis, L.A.; Daugherty, A. TGF-β neutralization enhances angii-induced aortic rupture and aneurysm in both thoracic and abdominal regions. PLoS ONE 2016, 11, e0153811. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Ait-Oufella, H.; Tedgui, A. The Pathogenic Transforming Growth Factor-β Overdrive Hypothesis in Aortic Aneurysms and Dissections: A Mirage? Circ. Res. 2017, 120, 1718–1720. [Google Scholar] [CrossRef] [PubMed]
- Lareyre, F.; Clment, M.; Raffort, J.; Pohlod, S.; Patel, M.; Esposito, B.; Master, L.; Finigan, A.; Vandestienne, M.; Stergiopulos, N.; et al. TGFβ (transforming growth factor-β) blockade induces a human-like disease in a nondissecting mouse model of abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2171–2181. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Vita, J.; Sanchez-Lopez, E.; Esteban, V.; Ruperez, M.; Egido, J.; Ruiz-Ortega, M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-beta-independent mechanism. Circulation 2005, 111, 2509–2517. [Google Scholar] [CrossRef] [PubMed]
- Jinnin, M.; Ihn, H.; Tamaki, K. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-beta1-induced extracellular matrix expression. Mol. Pharmacol. 2006, 69, 597–607. [Google Scholar] [CrossRef]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Devel. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tejera-Muñoz, A.; Marquez-Exposito, L.; Tejedor-Santamaría, L.; Rayego-Mateos, S.; Orejudo, M.; Suarez-Álvarez, B.; López-Larrea, C.; Ruíz-Ortega, M.; Rodrigues-Díez, R.R. CCN2 Increases TGF-β Receptor Type II Expression in Vascular Smooth Muscle Cells: Essential Role of CCN2 in the TGF-β Pathway Regulation. Int. J. Mol. Sci. 2022, 23, 375. https://doi.org/10.3390/ijms23010375
Tejera-Muñoz A, Marquez-Exposito L, Tejedor-Santamaría L, Rayego-Mateos S, Orejudo M, Suarez-Álvarez B, López-Larrea C, Ruíz-Ortega M, Rodrigues-Díez RR. CCN2 Increases TGF-β Receptor Type II Expression in Vascular Smooth Muscle Cells: Essential Role of CCN2 in the TGF-β Pathway Regulation. International Journal of Molecular Sciences. 2022; 23(1):375. https://doi.org/10.3390/ijms23010375
Chicago/Turabian StyleTejera-Muñoz, Antonio, Laura Marquez-Exposito, Lucía Tejedor-Santamaría, Sandra Rayego-Mateos, Macarena Orejudo, Beatriz Suarez-Álvarez, Carlos López-Larrea, Marta Ruíz-Ortega, and Raúl R. Rodrigues-Díez. 2022. "CCN2 Increases TGF-β Receptor Type II Expression in Vascular Smooth Muscle Cells: Essential Role of CCN2 in the TGF-β Pathway Regulation" International Journal of Molecular Sciences 23, no. 1: 375. https://doi.org/10.3390/ijms23010375
APA StyleTejera-Muñoz, A., Marquez-Exposito, L., Tejedor-Santamaría, L., Rayego-Mateos, S., Orejudo, M., Suarez-Álvarez, B., López-Larrea, C., Ruíz-Ortega, M., & Rodrigues-Díez, R. R. (2022). CCN2 Increases TGF-β Receptor Type II Expression in Vascular Smooth Muscle Cells: Essential Role of CCN2 in the TGF-β Pathway Regulation. International Journal of Molecular Sciences, 23(1), 375. https://doi.org/10.3390/ijms23010375