
Antifungal Activity of N-(4-Halobenzyl)amides against Candida spp. and Molecular Modeling Studies

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
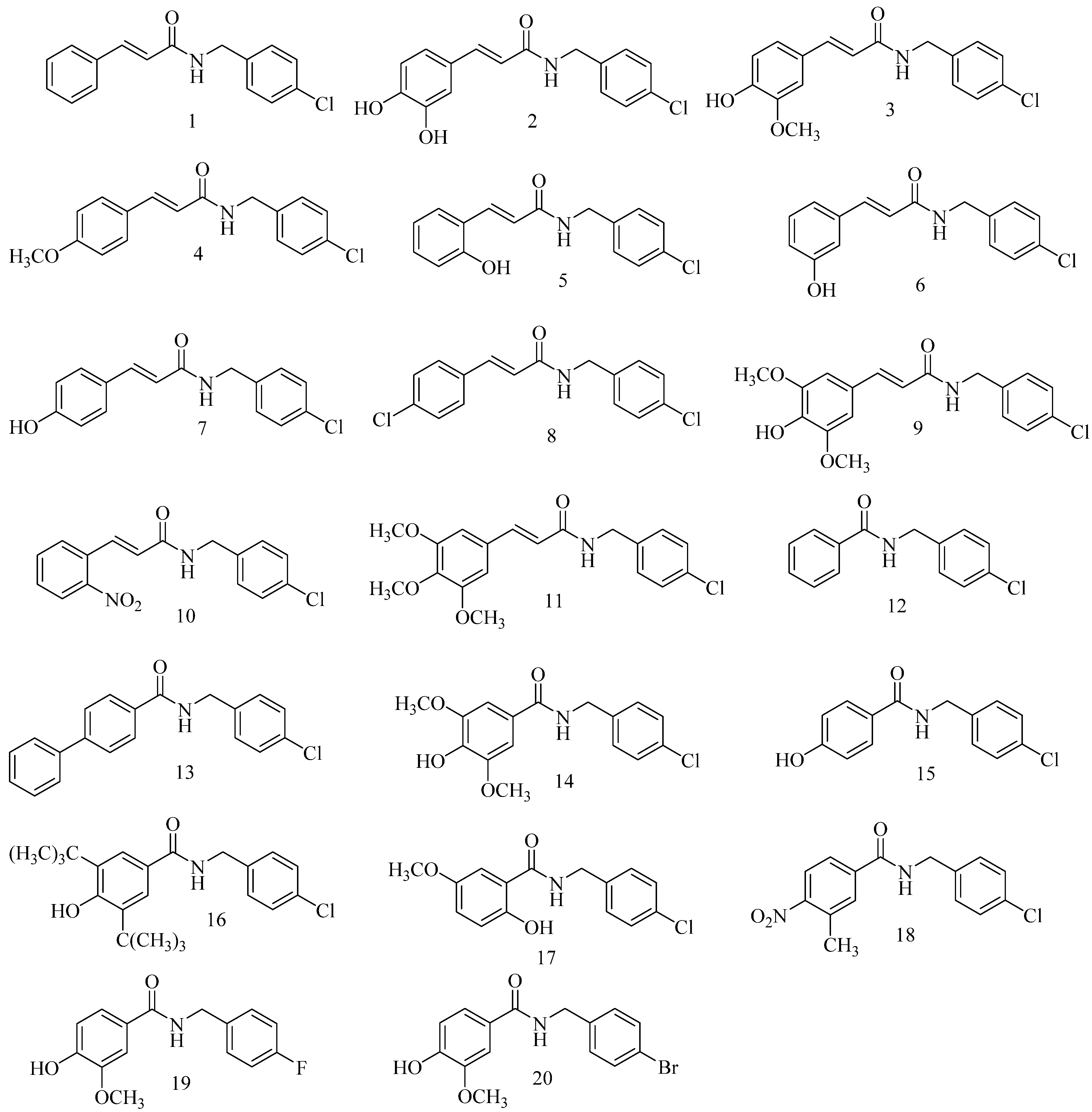
2.1. Chemistry
2.2. Antimicrobial Activity
2.3. Antifungal Activity of Compound 16
2.4. Molecular Modeling
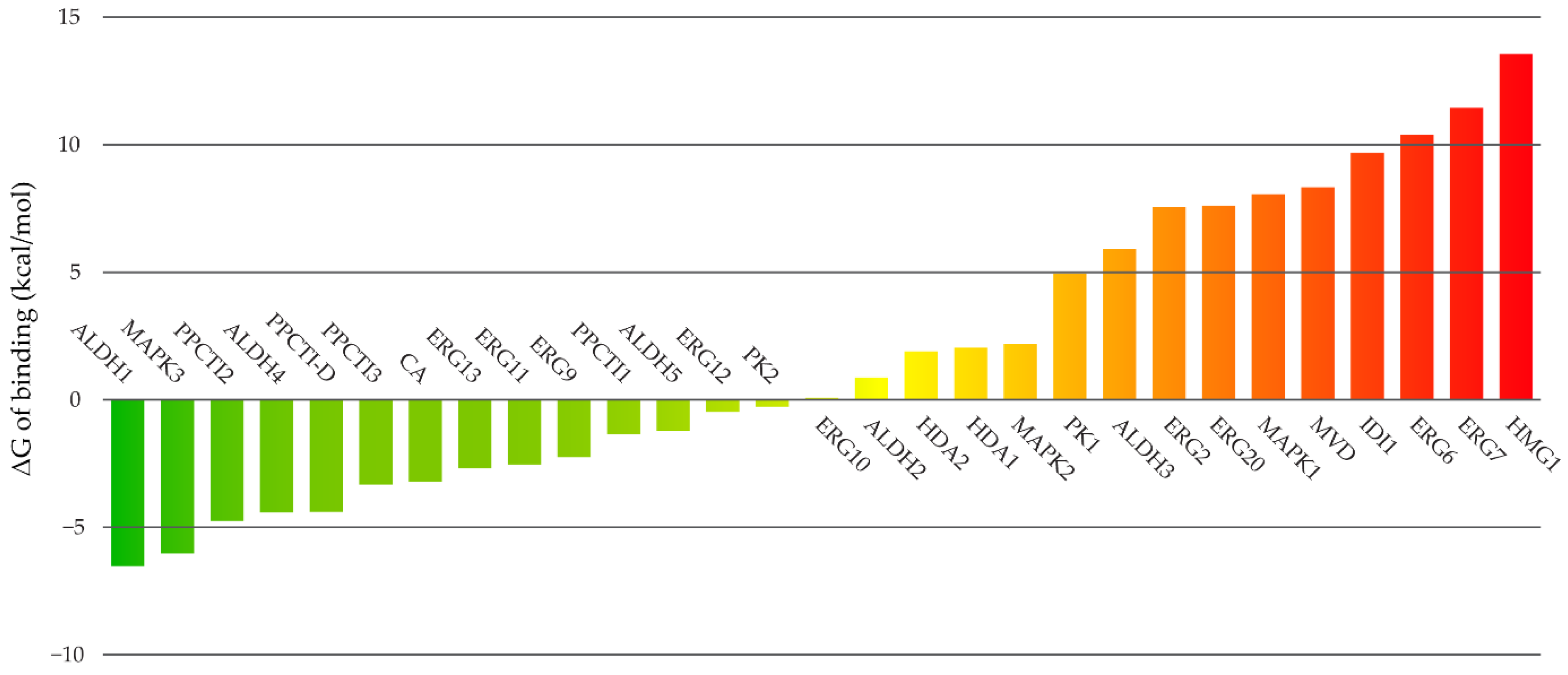
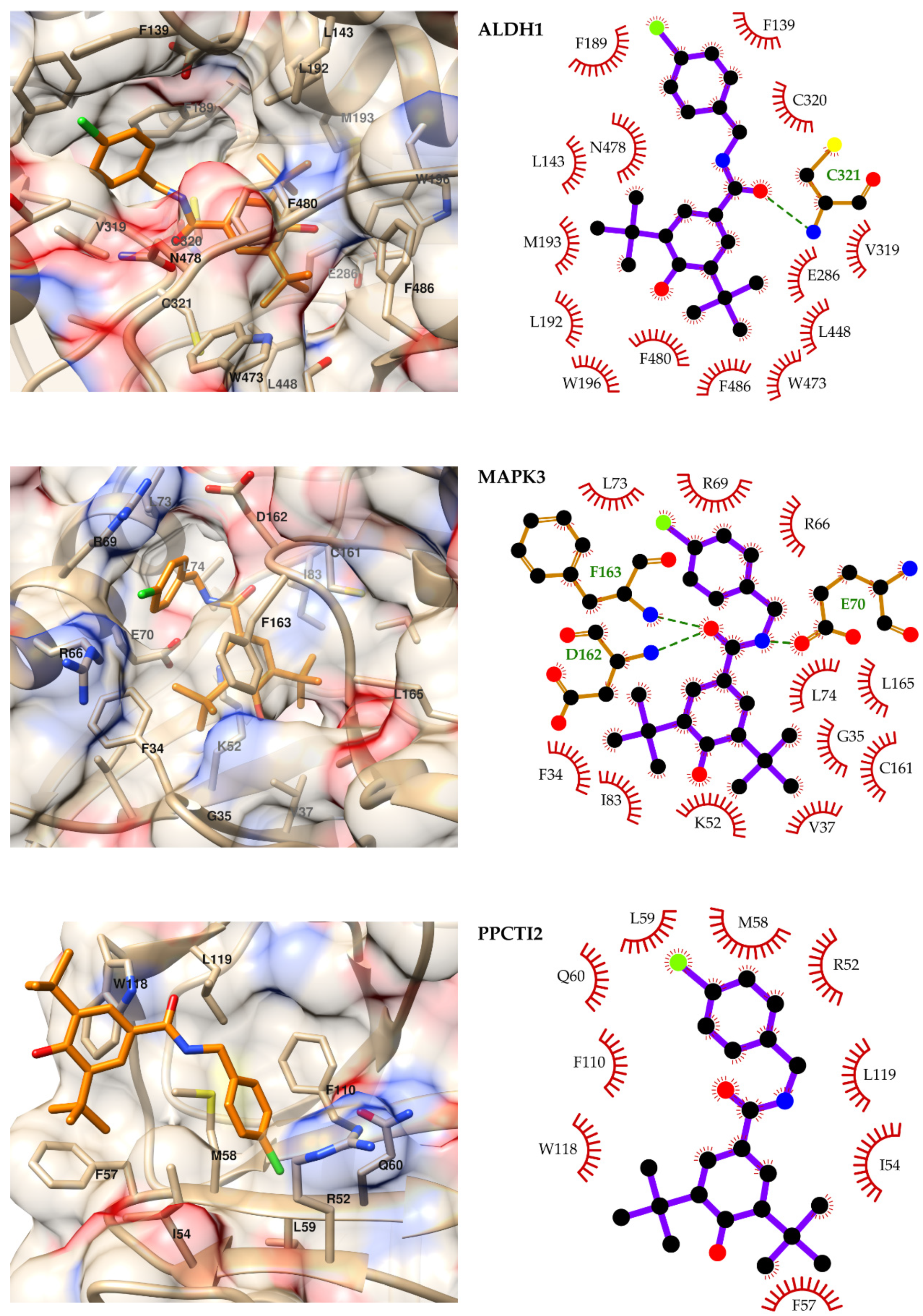

3. Discussion
4. Materials and Methods
4.1. General Information
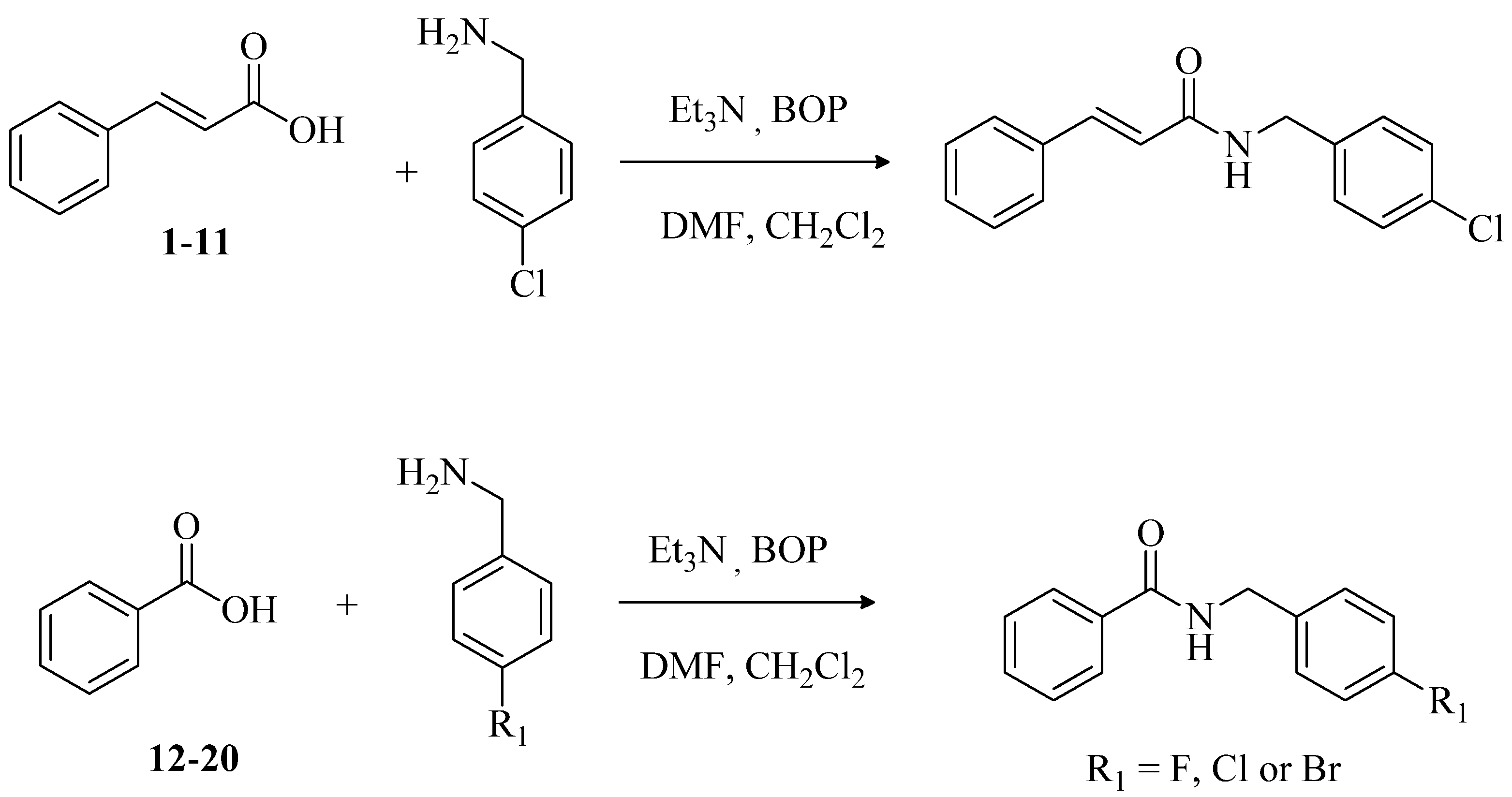
4.2. General Preparation of N-(4-Halobenzyl)amides
4.3. Evaluation of Antimicrobial Activity
4.3.1. Microbiological Strains and Culture Medium
4.3.2. Determination of the Minimum Inhibitory Concentration (MIC)
4.4. Evaluation of Antimicrobial Activity Compound 16
4.4.1. Microbiological Strains and Culture Medium
4.4.2. Determination of the Minimum Inhibitory Concentration (MIC)
4.5. Computational Methods
4.5.1. Molecular Modeling
4.5.2. Targets Selection
4.5.3. Molecular Docking
4.5.4. Molecular Dynamics Simulations and Prediction of the Free Energies of Binding
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sardi, J.C.O.; Scorzoni, L.; Bernardi, T.; Fusco-Almeida, A.M.; Mendes Giannini, M.J.S. Candida species: Current epidemiology, pathogenicity, biofilm formation, natural antifungal products and new therapeutic options. J. Med. Microbiol. 2013, 62, 10–24. [Google Scholar] [CrossRef]
- Ostrosky-Zeichner, L.; Kullberg, B.J.; Bow, E.J.; Hadley, S.; León, C.; Nucci, M.; Patterson, T.F.; Perfect, J.R. Early treatment of candidemia in adults: A review. Med. Mycol. 2011, 49, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Giri, S.; Kindo, A.J. A review of Candida species causing blood stream infection. Indian J. Med. Microbiol. 2012, 30, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Barbedo, L.S.; Sgarbi, D.B.G. Candidíase. DST J. Bras. Doenças Sex Transm. 2010, 22, 22–38. [Google Scholar]
- Lima, T.C.; Ferreira, A.R.; Silva, D.F.; Lima, E.O.; de Sousa, D.P. Antifungal activity of cinnamic acid and benzoic acid esters against Candida albicans strains. Nat. Prod. Res. 2018, 32, 572–575. [Google Scholar] [CrossRef]
- Lima, T.C.; Ferreira, A.R.; Barboza, J.N.; Filho, C.S.M.B.; Silva, D.F.; Lima, E.O.; de Sousa, D.P. Antimicrobial Activity of Cinnamic and Benzoic Methyl Esters. Lat. Am. J. Pharm. 2018, 37, 1011–1016. [Google Scholar]
- Korošec, B.; Sova, M.; Turk, S.; Kraševec, N.; Novak, M.; Lah, L.; Stojan, J.; Podobnik, B.; Berne, S.; Zupanec, N.; et al. Antifungal activity of cinnamic acid derivatives involves inhibition of benzoate 4-hydroxylase (CYP53). J. Appl. Microbiol. 2014, 116, 955–966. [Google Scholar] [CrossRef] [Green Version]
- Chhillar, R.; Dhingra, D. Antidepressant-like activity of gallic acid in mice subjected to unpredictable chronic mild stress. Fundam. Clin. Pharmacol. 2013, 27, 409–418. [Google Scholar] [CrossRef]
- Dhingra, D.; Chhillar, R.; Gupta, A. Antianxiety-like activity of gallic acid in unstressed and stressed mice: Possible involvement of nitriergic system. Neurochem. Res. 2012, 37, 487–494. [Google Scholar] [CrossRef]
- Kroes, B.H.; van den Berg, A.J.; van Ufford, H.C.Q.; van Dijk, H.; Labadie, R.P. Anti-inflammatory activity of gallic acid. Planta Med. 1992, 58, 499–504. [Google Scholar] [CrossRef]
- Lorusso, L.; Mikhaylova, S.V.; Capelli, E.; Ferrari, D.; Ngonga, G.K.; Ricevuti, G. Immunological aspects of chronic fatigue syndrome. Autoimmun. Rev. 2009, 8, 287–291. [Google Scholar] [CrossRef]
- Kim, K.D.; Song, M.H.; Yum, E.K.; Jeon, O.S.; Ju, Y.W.; Chang, M.S. Melanogenesis inhibition by mono-hydroxycinnamic ester derivatives in B16 melanoma cells. Bull. Korean Chem. Soc. 2010, 31, 181–184. [Google Scholar] [CrossRef] [Green Version]
- Guzman, J.D. Natural cinnamic acids, synthetic derivatives and hybrids with antimicrobial activity. Molecules 2014, 19, 19292–19349. [Google Scholar] [CrossRef]
- Ueda, S.; Okada, T.; Nagasawa, H. Oxindole synthesis by palladium-catalysed aromatic C-H alkenylation. Chem. Commun. 2010, 46, 2462–2464. [Google Scholar] [CrossRef] [Green Version]
- Watson, A.J.; Maxwell, A.C.; Williams, J.M. Ruthenium-catalyzed oxidation of alcohols into amides. Org. Lett. 2009, 11, 2667–2670. [Google Scholar] [CrossRef]
- Foo, S.W.; Oishi, S.; Saito, S. Aldol condensation of amides using phosphazene-based catalysis. Tetrahedron lett. 2012, 53, 5445–5448. [Google Scholar] [CrossRef]
- Wu, J.W.; Wu, Y.D.; Dai, J.J.; Xu, H.J. Benzoic Acid-Catalyzed Transamidation Reactions of Carboxamides, Phthalimide, Ureas and Thioamide with Amines. Adv. Synth. Catal. 2014, 356, 2429–2436. [Google Scholar] [CrossRef]
- Shen, X.X.; Liu, Q.; Xing, R.G.; Zhou, B. Reduction of N-(alkoxy (aryl) methyl) benzamide Compounds by a Hantzsch Ester 1, 4-Dihydropyridine Using Pd/C as a Catalyst. Catal. Lett. 2008, 126, 361–366. [Google Scholar] [CrossRef]
- Zhang, M.; Lu, X.; Zhang, H.J.; Li, N.; Xiao, Y.; Zhu, H.L.; Ye, Y.H. Synthesis, structure, and biological assay of cinnamic amides as potential EGFR kinase inhibitors. Med. Chem. Res. 2013, 22, 986–994. [Google Scholar] [CrossRef]
- Nomura, E.; Kashiwada, A.; Hosoda, A.; Nakamura, K.; Morishita, H.; Tsuno, T.; Taniguchi, H. Synthesis of amide compounds of ferulic acid, and their stimulatory effects on insulin secretion in vitro. Bioorg. Med. Chem. 2003, 11, 3807–3813. [Google Scholar] [CrossRef]
- Fu, J.; Cheng, K.; Zhang, Z.M.; Fang, R.Q.; Zhu, H.L. Synthesis, structure and structure-activity relationship analysis of caffeic acid amides as potential antimicrobials. Eur. J. Med. Chem. 2010, 45, 2638–2643. [Google Scholar] [CrossRef]
- Waisser, K.; Pesina, M.; Holý, P.; Pour, M.; Bures, O.; Kunes, J.; Klimesová, V.; Buchta, V.; Kubanová, P.; Kaustová, J. Antimycobacterial and antifungal isosters of salicylamides. Arch. Pharm. 2003, 336, 322–335. [Google Scholar] [CrossRef]
- Rognan, D. Structure-Based Approaches to Target Fishing and Ligand Profiling. Mol. Inform. 2010, 29, 176–187. [Google Scholar] [CrossRef]
- Schomburg, K.T.; Rarey, M. What is the potential of structure-based target prediction methods? Future Med. Chem. 2014, 6, 1987–1999. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Kurgan, L. Survey of Similarity-Based Prediction of Drug-Protein Interactions. Curr. Med. Chem. 2020, 27, 5856–5886. [Google Scholar] [CrossRef]
- Wang, C.; Kurgan, L. Review and comparative assessment of similarity-based methods for prediction of drug-protein interactions in the druggable human proteome. Brief Bioinform. 2019, 20, 2066–2087. [Google Scholar] [CrossRef]
- Sydow, D.; Burggraaff, L.; Szengel, A.; van Vlijmen, H.W.T.; IJzerman, A.P.; van Westen, G.J.P.; Volkamer, A. Advances and Challenges in Computational Target Prediction. J. Chem. Inf. Model. 2019, 59, 1728–1742. [Google Scholar] [CrossRef] [Green Version]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.L.; Lavan, P.; Weber, E.; Doak, A.K.; Côté, S.; et al. Large-scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361–367. [Google Scholar] [CrossRef]
- Liu, B.; Fu, X.Q.; Li, T.; Su, T.; Guo, H.; Zhu, P.L.; Tse, A.K.; Liu, S.M.; Yu, Z.L. Computational and experimental prediction of molecules involved in the anti-melanoma action of berberine. J. Ethnopharmacol. 2017, 208, 225–235. [Google Scholar] [CrossRef]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA Methods in Virtual Screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef] [Green Version]
- Tutone, M.; Virzì, A.; Almerico, A.M. Reverse screening on indicaxanthin from Opuntia ficus-indica as natural chemoactive and chemopreventive agent. J. Theor. Biol. 2018, 455, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Turkez, H.; Nóbrega, F.R.D.; Ozdemir, O.; Filho, C.S.M.B.; Almeida, R.N.; Tejera, E.; Perez-Castillo, Y.; de Sousa, D.P. NFBTA: A Potent Cytotoxic Agent against Glioblastoma. Molecules 2019, 24, 2411. [Google Scholar] [CrossRef] [Green Version]
- Montes, R.C.; de Freitas, T.S.; Costa, M.S.; Oliveira, F.S.; Campina, F.F.; Ferreira, A.R.; Ferreira, S.O.; Melo, H.D.; Dias, C.S.; de Sousa, D.P. Antimicrobial evaluation of cinnamic and benzoic haloamides. J. Chem. Pharm. Res. 2016, 8, 311–320. [Google Scholar]
- Montes, R.C.; Perez, A.L.; Medeiros, C.I.; Araújo, M.O.; Lima, E.O.; Scotti, M.T.; de Sousa, D.P. Synthesis, Antifungal Evaluation and In Silico Study of N-(4-Halobenzyl)amides. Molecules 2016, 21, 1716. [Google Scholar] [CrossRef] [Green Version]
- Joullié, M.M.; Lassen, K.M. Evolution of amide bond formation. Arkivoc 2010, 8, 189–250. [Google Scholar] [CrossRef] [Green Version]
- Montalbetti, C.A.; Falque, V. Amide bond formation and peptide coupling. Tetrahedron 2005, 61, 10827–10852. [Google Scholar] [CrossRef]
- Malz, F.; Jancke, H. Validation of quantitative NMR. J. Pharm. Biomed. Anal. 2005, 38, 813–823. [Google Scholar] [CrossRef]
- Rastelli, G.; Pinzi, L. Refinement and Rescoring of Virtual Screening Results. Front. Chem. 2019, 7, 498. [Google Scholar] [CrossRef] [Green Version]
- Santos Oliveira, A.J.M.; de Castro, R.D.; Pessôa, H.L.F.; Wadood, A.; de Sousa, D.P. Amides Derived from Vanillic Acid: Coupling Reactions, Antimicrobial Evaluation, and Molecular Docking. Biomed. Res. Int. 2019, 2019, 9209676. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Garcia-Bustos, V.; Cabanero-Navalon, M.D.; Ruiz-Saurí, A.; Ruiz-Gaitán, A.C.; Salavert, M.; Tormo, M.Á.; Pemán, J. What Do We Know about Candida auris? State of the Art, Knowledge Gaps, and Future Directions. Microorganisms 2021, 9, 2177. [Google Scholar] [CrossRef] [PubMed]
- Dejani, N.N.; Elshabrawy, H.A.; Bezerra Filho, C.d.S.M.; de Sousa, D.P. Anticoronavirus and Immunomodulatory Phenolic Compounds: Opportunities and Pharmacotherapeutic Perspectives. Biomolecules 2021, 11, 1254. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Xia, B.; Li, W.; Wu, J.; Zhang, H. Two novel phenolic compounds from Stenoloma chusanum and their antifungal activity. Chem. Nat. Compd. 2009, 45, 182–186. [Google Scholar] [CrossRef]
- Alves, D.D.N.; Ferreira, A.R.; Duarte, A.B.S.; Melo, A.K.V.; de Sousa, D.P.; de Castro, R.D. Breakpoints for the Classification of Anti-Candida Compounds in Antifungal Screening. Biomed Res. Int. 2021, 2021, 6653311. [Google Scholar] [CrossRef]
- Singh, S.; Brocker, C.; Koppaka, V.; Chen, Y.; Jackson, B.C.; Matsumoto, A.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic. Biol. Med. 2013, 56, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Schmid, F.X.; Mayr, L.M.; Mücke, M.; Schönbrunner, E.R. Prolyl isomerases: Role in protein folding. Adv. Protein Chem. 1993, 44, 25–66. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, H.; Devasahayam, G.; Gemmill, T.; Chaturvedi, V.; Hanes, S.D.; van Roey, P. The structure of the Candida albicans Ess1 prolyl isomerase reveals a well-ordered linker that restricts domain mobility. Biochemistry 2005, 44, 6180–6189. [Google Scholar] [CrossRef] [Green Version]
- Samaranayake, D.; Atencio, D.; Morse, R.; Wade, J.T.; Chaturvedi, V.; Hanes, S.D. Role of Ess1 in growth, morphogenetic switching, and RNA polymerase II transcription in Candida albicans. PLoS ONE 2013, 8, e59094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, J.M.; Kauffman, S.J.; Hauser, M.; Huang, L.; Lin, M.; Sillaots, S.; Jiang, B.; Xu, D.; Roemer, T. Pathway analysis of Candida albicans survival and virulence determinants in a murine infection model. Proc. Natl. Acad. Sci. USA 2010, 107, 22044–22049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Hou, Y.; Yue, L.; Liu, S.; Du, J.; Sun, S. Potential Targets for Antifungal Drug Discovery Based on Growth and Virulence in Candida albicans. Antimicrob. Agents Chemother. 2015, 59, 5885–5891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, M.W.; Kontoyiannis, D.P.; Cornely, O.A.; Perfect, J.R.; Walsh, T.J. Novel Agents and Drug Targets to Meet the Challenges of Resistant Fungi. J. Infect Dis. 2017, 216, S474–S483. [Google Scholar] [CrossRef] [Green Version]
- Ahmad Khan, M.S.; Alshehrei, F.; Al-Ghamdi, S.B.; Bamaga, M.A.; Al-Thubiani, A.S.; Alam, M.Z. Virulence and biofilms as promising targets in developing antipathogenic drugs against candidiasis. Future Sci. OA 2020, 6, FSO440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso-Monge, R.; Navarro-García, F.; Román, E.; Negredo, A.I.; Eisman, B.; Nombela, C.; Pla, J. The Hog1 mitogen-activated protein kinase is essential in the oxidative stress response and chlamydospore formation in Candida albicans. Eukaryot. Cell 2003, 2, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Neto, J.B.A.; da Silva, C.R.; Nascimento, F.B.S.A.; Sampaio, L.S.; da Silva, A.R.; de Freitas, D.D.; Campos, R.S.; de Andrade, L.N.D.; Gonçalves, T.B.; Nagao-Dias, A.T.; et al. Screening of Antimicrobial Metabolite Yeast Isolates Derived Biome Ceará against Pathogenic Bacteria, Including MRSA: Antibacterial Activity and mode of Action Evaluated by Flow Cytometry. Int. J. Curr. Microbiol. App. Sci. 2015, 4, 459–472. [Google Scholar]
- Clinical and Laboratory Standards Institute. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; CLSI: Wayne, PA, USA, 2012. [Google Scholar]
- Araújo, M.O.; Pérez-Castillo, Y.; Oliveira, L.H.G.; Nunes, F.C.; de Sousa, D.P. Larvicidal Activity of Cinnamic Acid Derivatives: Investigating Alternative Products for Aedes aegypti L. Control. Molecules 2020, 26, 61. [Google Scholar] [CrossRef] [PubMed]
- Lopes, S.P.; Castillo, Y.P.; Monteiro, M.L.; Menezes, R.R.P.P.B.; Almeida, R.N.; Martins, A.M.C.; de Sousa, D.P. Trypanocidal Mechanism of Action and in silico Studies of p-Coumaric Acid Derivatives. Int. J. Mol. Sci. 2019, 20, 5916. [Google Scholar] [CrossRef] [Green Version]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Skrzypek, M.S.; Binkley, J.; Binkley, G.; Miyasato, S.R.; Simison, M.; Sherlock, G. The Candida Genome Database (CGD): Incorporation of Assembly 22, systematic identifiers and visualization of high throughput sequencing data. Nucleic Acids Res. 2017, 45, D592–D596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Castillo, Y.; Lima, T.C.; Ferreira, A.R.; Silva, C.R.; Campos, R.S.; Neto, J.B.A.; Magalhães, H.I.F.; Cavalcanti, B.C.; Júnior, H.V.N.; de Sousa, D.P. Bioactivity and Molecular Docking Studies of Derivatives from Cinnamic and Benzoic Acids. Biomed Res. Int. 2020, 2020, 6345429. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. OMEGA. Santa Fe, NM: OpenEye Scientific Software. 2021. Available online: http://www.eyesopen.com (accessed on 15 September 2021).
- QUACPAC. OpenEye Scientific Software, Santa Fe, NM. Available online: http://www.eyesopen.com (accessed on 16 September 2021).
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Giambasu, G.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Pang, Y.P. Successful molecular dynamics simulation of two zinc complexes bridged by a hydroxide in phosphotriesterase using the cationic dummy atom method. Proteins 2001, 45, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.C.; de Souza, M.M.; Mercer, I.P.; Gould, I.R.; Klug, D.R. Large and fast relaxations inside a protein: Calculation and measurement of reorganization energies in alcohol dehydrogenase. J. Phys. Chem. B 2002, 106, 11658–11665. [Google Scholar] [CrossRef] [Green Version]
- Pavelites, J.J.; Gao, J.; Bash, P.A.; Mackerell, A.D., Jr. A molecular mechanics force field for NAD+ NADH, and the pyrophosphate groups of nucleotides. J. Comput. Chem. 1998, 18, 221–239. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
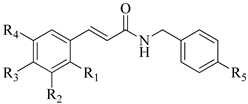 1–11 | 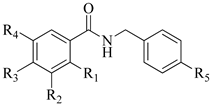 12–20 | ||||||
|---|---|---|---|---|---|---|---|
| Compounds | R1 | R2 | R3 | R4 | R5 | MIC 1 | |
| C. parapsilosis (ATCC 22019) | C. krusei (ATCC 14243) | ||||||
| 1 | - | - | - | - | Cl | + | + |
| 2 | - | OH | OH | - | Cl | 31.25 | 250 |
| 3 | - | OCH3 | OH | - | Cl | + | + |
| 4 | - | - | OCH3 | - | Cl | + | + |
| 5 | OH | - | - | - | Cl | + | + |
| 6 | - | OH | - | - | Cl | + | + |
| 7 | - | - | OH | - | Cl | + | + |
| 8 | - | - | Cl | - | Cl | + | + |
| 9 | - | OCH3 | OH | OCH3 | Cl | 250 | 150 |
| 10 | NO2 | - | - | - | Cl | + | + |
| 11 | - | OCH3 | OCH3 | OCH3 | Cl | + | + |
| 12 | - | - | - | - | Cl | 125 | 250 |
| 13 | - | - | C6H5 | - | Cl | + | + |
| 14 | - | OCH3 | OH | OCH3 | Cl | 31.25 | 31.25 |
| 15 | - | - | OH | - | Cl | 250 | 250 |
| 16 | - | C(CH3)3 | OH | C(CH3)3 | Cl | 125 | 7.8 |
| 17 | - | OH | - | OCH3 | Cl | + | + |
| 18 | - | CH3 | - | NO2 | Cl | + | + |
| 19 | - | OCH3 | OH | - | F | + | + |
| 20 | - | OCH3 | OH | - | Br | + | + |
| Fluconazole | - | - | - | - | 2 | 16 | |
| Voriconazole | - | - | - | - | 0.125 | 0.25 | |
| MIC50 | |
|---|---|
| Strains a | Compound 16 b |
| C. parapsilosis ATCC 22019 | 106.6 µg/mL |
| C. krusei ATCC 6258 | 85.3 µg/mL |
| C. auris 01256P CDC | 85.3 µg/mL |
| C. albicans 1 | 341.3 µg/mL |
| C. albicans 2 | 128 µg/mL |
| C. tropicalis | 106.6 µg/mL |
| C. parapsilosis | 128 µg/mL |
| C. glabrata | 128 µg/mL |
| UniProt Accession | ID a | Description |
|---|---|---|
| A0A099P395 b | ALDH5 | Aldehyde dehydrogenase 5, mitochondrial |
| A0A2U9R6B5 b | ALDH1 | Aldedh domain-containing protein |
| A0A099P647 b | ALDH3 | Aldedh domain-containing protein |
| A0A2U9R5Y1 b | ALDH2 | Aldedh domain-containing protein |
| A0A2U9R723 b | ALDH4 | Aldedh domain-containing protein |
| A0A099P843 b | MAPK1 | Mitogen-activated protein kinase CEK1 |
| A0A099NVZ1 b | MAPK2 | Mitogen-activated protein kinase |
| A0A1V2LG11 b | MAPK3 | Mitogen-activated protein kinase;uncharacterized protein HOG1 |
| A0A1Z8JVT5 b | PK1 | Protein kinase domain-containing protein |
| A0A2U9R7Y2 b | PK2 | Protein kinase domain-containing protein |
| A0A099P0U0 b | HDA1 | Histone deacetylase RPD3 |
| A0A2U9RAK0 b | HDA2 | Histone deacetylase |
| A0A1V2LMB8 b | PPCTI-D | Peptidyl-prolyl cis-trans isomerase D |
| A0A2U9QXT4 b | PPCTI1 | Peptidyl-prolyl cis-trans isomerase |
| A0A099P2W3 b | PPCTI2 | Peptidyl-prolyl cis-trans isomerase |
| A0A099P773 b | PPCTI3 | Peptidyl-prolyl cis-trans isomerase |
| A0A1Z8JS57 b | CA | Carbonic anhydrase |
| A0A2U9RBI7 c | IDI1 | Isopentenyl-diphosphate Delta-isomerase |
| A0A1Z8JJU5 c | MVD | Diphosphomevalonate decarboxylase |
| A0A2U9R9Y8 c,d | ERG8 | Phosphomevalonate kinase |
| A0A2U9R5L9 c | ERG12 | Mevalonate kinase |
| A0A2U9R6J4 c | HMG1 | 3-hydroxy-3-methylglutaryl coenzyme A reductase |
| A0A099P154 c | ERG13 | 3-hydroxy-3-methylglutaryl coenzyme A synthase |
| A0A099P5C0 c | ERG10 | Acetyl-CoA C-acetyltransferase IA |
| A0A099P078 c,d | ERG26 | Sterol-4-alpha-carboxylate 3-dehydrogenase |
| A0A1Z8JS11 c,d | ERG27 | 3-keto-steroid reductase |
| A0A2U9RAJ8 c,d | ERG4 | Delta(24(24(1)))-sterol reductase |
| A0A099P3X9 c,d | ERG5 | C-22 sterol desaturase |
| A0A099P0V3 c,d | ERG3 | C-5 sterol desaturase |
| A0A2U9QYN8 c | ERG2 | C-8 sterol isomerase |
| A0A1V2LUI2 c | ERG6 | Sterol 24-C-methyltransferase |
| A0A099NZM6 c,d | ERG25 | Methylsterol monooxygenase |
| A0A099P4I0 c,d | ERG24 | Delta(14)-sterol reductase |
| A9YUC7 c | ERG11 | Lanosterol 14-alpha demethylase |
| A0A2U9R113 c | ERG9 | Squalene synthase |
| A0A2U9R171 c | ERG7 | Lanosterol synthase |
| A0A099NX62 c | ERG1 | Squalene monooxygenase |
| A0A099P5M1 c | ERG20 | (2E,6E)-farnesyl diphosphate synthase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez-Castillo, Y.; Montes, R.C.; da Silva, C.R.; Neto, J.B.d.A.; Dias, C.d.S.; Brunna Sucupira Duarte, A.; Júnior, H.V.N.; de Sousa, D.P. Antifungal Activity of N-(4-Halobenzyl)amides against Candida spp. and Molecular Modeling Studies. Int. J. Mol. Sci. 2022, 23, 419. https://doi.org/10.3390/ijms23010419
Perez-Castillo Y, Montes RC, da Silva CR, Neto JBdA, Dias CdS, Brunna Sucupira Duarte A, Júnior HVN, de Sousa DP. Antifungal Activity of N-(4-Halobenzyl)amides against Candida spp. and Molecular Modeling Studies. International Journal of Molecular Sciences. 2022; 23(1):419. https://doi.org/10.3390/ijms23010419
Chicago/Turabian StylePerez-Castillo, Yunierkis, Ricardo Carneiro Montes, Cecília Rocha da Silva, João Batista de Andrade Neto, Celidarque da Silva Dias, Allana Brunna Sucupira Duarte, Hélio Vitoriano Nobre Júnior, and Damião Pergentino de Sousa. 2022. "Antifungal Activity of N-(4-Halobenzyl)amides against Candida spp. and Molecular Modeling Studies" International Journal of Molecular Sciences 23, no. 1: 419. https://doi.org/10.3390/ijms23010419
APA StylePerez-Castillo, Y., Montes, R. C., da Silva, C. R., Neto, J. B. d. A., Dias, C. d. S., Brunna Sucupira Duarte, A., Júnior, H. V. N., & de Sousa, D. P. (2022). Antifungal Activity of N-(4-Halobenzyl)amides against Candida spp. and Molecular Modeling Studies. International Journal of Molecular Sciences, 23(1), 419. https://doi.org/10.3390/ijms23010419