
The YAP/TAZ Signaling Pathway in the Tumor Microenvironment and Carcinogenesis: Current Knowledge and Therapeutic Promises

 ,
,  , , , ,
, , , ,  and
and
Abstract
:1. Introduction
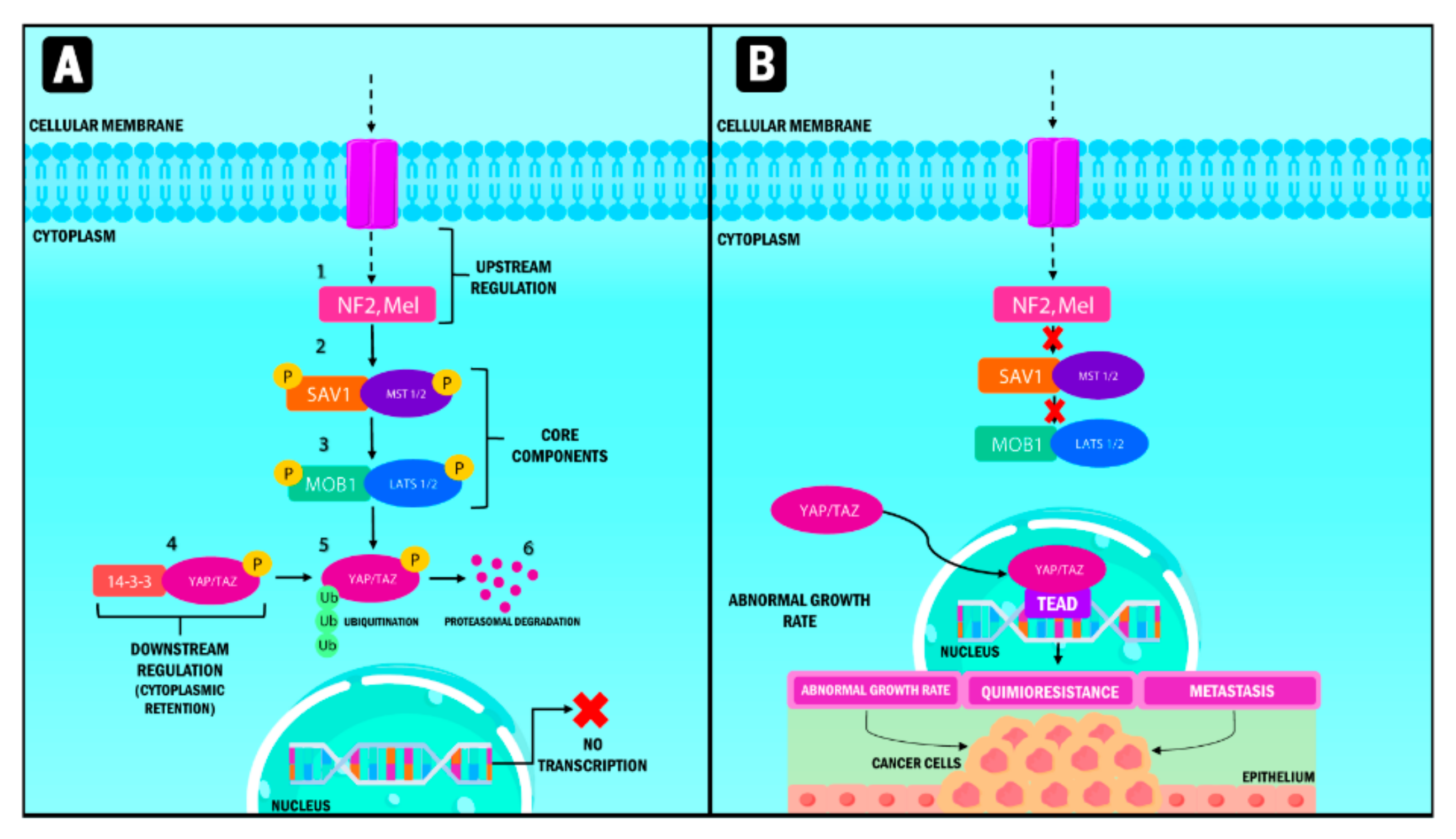
2. YAP/TAZ Structure and Function
3. Role of YAP/TAZ in Carcinogenesis
4. YAP/TAZ and the Tumor Microenvironment
5. Remodelling of the Tumor Microenvironment
5.1. Epithelial-Mesenchymal Transition
5.2. Angiogenesis
5.3. Mechanotransduction
5.4. Stromal Immunomodulatory Responses
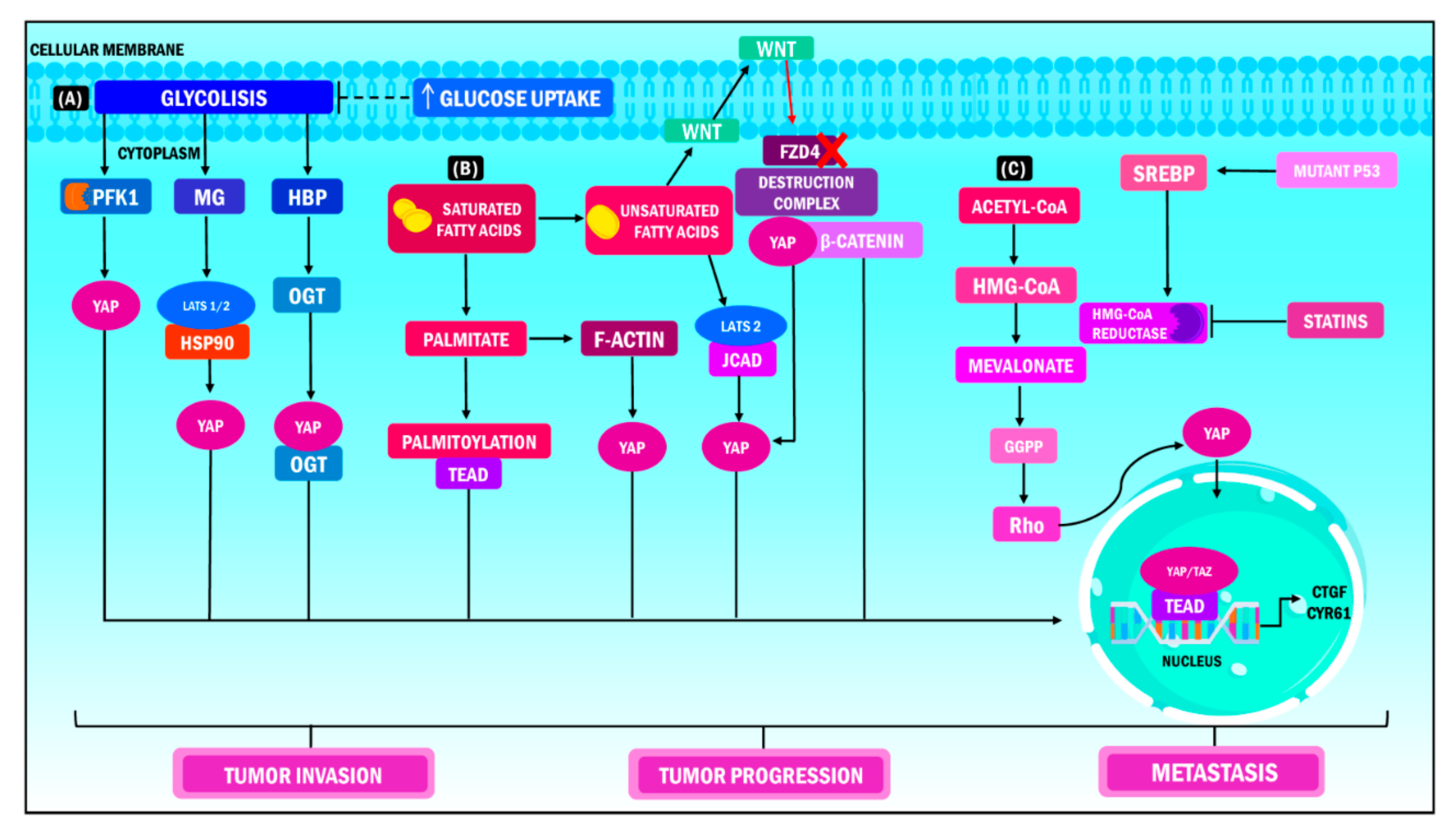
5.5. Metabolic Reprogramming
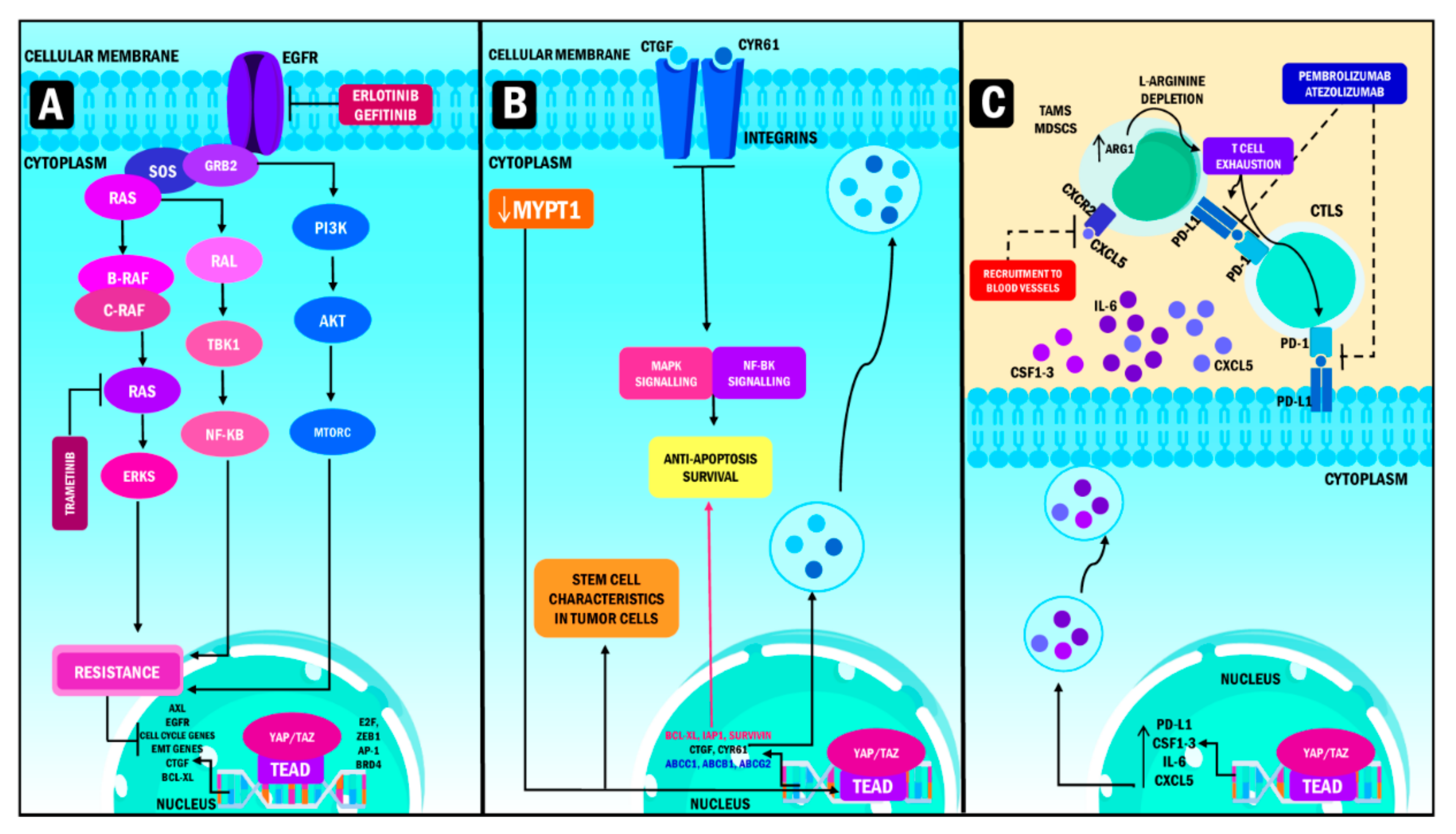
6. Resistance to Therapy: The Role of YAP and TAZ
7. YAP and TAZ as Therapeutic Targets
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- León, J.D.; Pareja, A. Inmunología del cáncer II: Bases moleculares y celulares de la carcinogénesis. Horiz. Médico (Lima) 2019, 19, 84–92. [Google Scholar] [CrossRef]
- Bizzarri, M.; Cucina, A. Tumor and the Microenvironment: A Chance to Reframe the Paradigm of Carcinogenesis? BioMed Res. Int. 2014, 2014, e934038. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sever, R.; Brugge, J.S. Signal Transduction in Cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.-X.; Zhao, B.; Guan, K.-L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Han, H.; Seo, G.; Vargas, R.E.; Yang, B.; Chuc, K.; Zhao, H.; Wang, W. Systematic Analysis of the Hippo Pathway Organization and Oncogenic Alteration in Evolution. Sci. Rep. 2020, 10, 3173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, C.G.; Moroishi, T.; Guan, K.-L. YAP and TAZ: A Nexus for Hippo Signaling and Beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Pobbati, A.V.; Hong, W. A Combat with the YAP/TAZ-TEAD Oncoproteins for Cancer Therapy. Theranostics 2020, 10, 3622–3635. [Google Scholar] [CrossRef]
- Omori, H.; Nishio, M.; Masuda, M.; Miyachi, Y.; Ueda, F.; Nakano, T.; Sato, K.; Mimori, K.; Taguchi, K.; Hikasa, H.; et al. YAP1 Is a Potent Driver of the Onset and Progression of Oral Squamous Cell Carcinoma. Sci. Adv. 2020, 6, eaay3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canu, V.; Donzelli, S.; Sacconi, A.; Lo Sardo, F.; Pulito, C.; Bossel, N.; Di Benedetto, A.; Muti, P.; Botti, C.; Domany, E.; et al. Aberrant Transcriptional and Post-Transcriptional Regulation of SPAG5, a YAP-TAZ-TEAD Downstream Effector, Fuels Breast Cancer Cell Proliferation. Cell Death Differ. 2021, 28, 1493–1511. [Google Scholar] [CrossRef] [PubMed]
- Shreberk-Shaked, M.; Dassa, B.; Sinha, S.; Di Agostino, S.; Azuri, I.; Mukherjee, S.; Aylon, Y.; Blandino, G.; Ruppin, E.; Oren, M. A Division of Labor between YAP and TAZ in Non-Small Cell Lung Cancer. Cancer Res. 2020, 80, 4145–4157. [Google Scholar] [CrossRef]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional Addiction in Cancer Cells Is Mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef]
- Ferrari, N.; Ranftl, R.; Chicherova, I.; Slaven, N.D.; Moeendarbary, E.; Farrugia, A.J.; Lam, M.; Semiannikova, M.; Westergaard, M.C.W.; Tchou, J.; et al. Dickkopf-3 Links HSF1 and YAP/TAZ Signalling to Control Aggressive Behaviours in Cancer-Associated Fibroblasts. Nat. Commun. 2019, 10, 130. [Google Scholar] [CrossRef]
- Lopez-Hernandez, A.; Sberna, S.; Campaner, S. Emerging Principles in the Transcriptional Control by YAP and TAZ. Cancers 2021, 13, 4242. [Google Scholar] [CrossRef]
- Varelas, X. The Hippo Pathway Effectors TAZ and YAP in Development, Homeostasis and Disease. Development 2014, 141, 1614–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plouffe, S.W.; Meng, Z.; Lin, K.C.; Lin, B.; Hong, A.W.; Chun, J.V.; Guan, K.-L. Characterization of Hippo Pathway Components by Gene Inactivation. Mol. Cell. 2016, 64, 993–1008. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Du, S.; Lei, T.; Wang, H.; He, X.; Tong, R.; Wang, Y. Multifaceted Regulation and Functions of YAP/TAZ in Tumors (Review). Oncol. Rep. 2018, 40, 16–28. [Google Scholar] [CrossRef]
- Fu, V.; Plouffe, S.W.; Guan, K.-L. The Hippo Pathway in Organ Development, Homeostasis, and Regeneration. Curr. Opin. Cell Biol. 2017, 49, 99–107. [Google Scholar] [CrossRef]
- Maugeri-Saccà, M.; De Maria, R. The Hippo Pathway in Normal Development and Cancer. Pharmacol. Ther. 2018, 186, 60–72. [Google Scholar] [CrossRef]
- Gumbiner, B.M.; Kim, N.-G. The Hippo-YAP Signaling Pathway and Contact Inhibition of Growth. J. Cell Sci. 2014, 127, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D. The Hippo Signaling Pathway in Development and Cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Jho, E.-H. Regulation of the Hippo Signaling Pathway by Ubiquitin Modification. BMB Rep. 2018, 51, 143–150. [Google Scholar] [CrossRef]
- Misra, J.R.; Irvine, K.D. The Hippo Signaling Network and Its Biological Functions. Annu. Rev. Genet. 2018, 52, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-L.; Choi, S.-H.; Mo, J.-S. Role of the Hippo Pathway in Fibrosis and Cancer. Cells 2019, 8, E468. [Google Scholar] [CrossRef] [Green Version]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Santucci, M.; Vignudelli, T.; Ferrari, S.; Mor, M.; Scalvini, L.; Bolognesi, M.L.; Uliassi, E.; Costi, M.P. The Hippo Pathway and YAP/TAZ-TEAD Protein-Protein Interaction as Targets for Regenerative Medicine and Cancer Treatment. J. Med. Chem. 2015, 58, 4857–4873. [Google Scholar] [CrossRef]
- Chen, Y.-A.; Lu, C.-Y.; Cheng, T.-Y.; Pan, S.-H.; Chen, H.-F.; Chang, N.-S. WW Domain-Containing Proteins YAP and TAZ in the Hippo Pathway as Key Regulators in Stemness Maintenance, Tissue Homeostasis, and Tumorigenesis. Front. Oncol. 2019, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [Green Version]
- Debaugnies, M.; Sánchez-Danés, A.; Rorive, S.; Raphaël, M.; Liagre, M.; Parent, M.-A.; Brisebarre, A.; Salmon, I.; Blanpain, C. YAP and TAZ Are Essential for Basal and Squamous Cell Carcinoma Initiation. EMBO Rep. 2018, 19, e45809. [Google Scholar] [CrossRef]
- Hagenbeek, T.J.; Webster, J.D.; Kljavin, N.M.; Chang, M.T.; Pham, T.; Lee, H.-J.; Klijn, C.; Cai, A.G.; Totpal, K.; Ravishankar, B.; et al. The Hippo Pathway Effector TAZ Induces TEAD-Dependent Liver Inflammation and Tumors. Sci. Signal. 2018, 11, eaaj1757. [Google Scholar] [CrossRef]
- Zhou, X.; Lei, Q.-Y. Regulation of TAZ in Cancer. Protein Cell 2016, 7, 548–561. [Google Scholar] [CrossRef] [Green Version]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.-S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.-X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.K.; Lu, S.Y.; Kang, S.-A.; Tan, H.J.; Li, Z.; Wee, Z.N.A.; Guan, J.S.; Reddy Chichili, V.P.; Sivaraman, J.; Putti, T.; et al. Wnt Signaling Promotes Breast Cancer by Blocking ITCH-Mediated Degradation of YAP/TAZ Transcriptional Coactivator WBP2. Cancer Res. 2016, 76, 6278–6289. [Google Scholar] [CrossRef] [Green Version]
- Bisso, A.; Filipuzzi, M.; Gamarra Figueroa, G.P.; Brumana, G.; Biagioni, F.; Doni, M.; Ceccotti, G.; Tanaskovic, N.; Morelli, M.J.; Pendino, V.; et al. Cooperation Between MYC and β-Catenin in Liver Tumorigenesis Requires Yap/Taz. Hepatology 2020, 72, 1430–1443. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Shin, J.E.; Park, H.W. The Role of Hippo Pathway in Cancer Stem Cell Biology. Mol. Cells 2018, 41, 83–92. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A Signalling Hub of the Tumour Microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-Wide Association between YAP/TAZ/TEAD and AP-1 at Enhancers Drives Oncogenic Growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ Functions and Their Regulation at a Glance. J. Cell Sci. 2020, 133, jcs230425. [Google Scholar] [CrossRef]
- Gill, M.K.; Christova, T.; Zhang, Y.Y.; Gregorieff, A.; Zhang, L.; Narimatsu, M.; Song, S.; Xiong, S.; Couzens, A.L.; Tong, J.; et al. A Feed Forward Loop Enforces YAP/TAZ Signaling during Tumorigenesis. Nat. Commun. 2018, 9, 3510. [Google Scholar] [CrossRef]
- Koo, J.H.; Guan, K.-L. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018, 28, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva-Diz, V.; Lorenzo-Sanz, L.; Bernat-Peguera, A.; Lopez-Cerda, M.; Muñoz, P. Cancer Cell Plasticity: Impact on Tumor Progression and Therapy Response. Semin. Cancer Biol. 2018, 53, 48–58. [Google Scholar] [CrossRef]
- Merrell, A.J.; Stanger, B.Z. Adult Cell Plasticity in Vivo: De-Differentiation and Transdifferentiation Are Back in Style. Nat. Rev. Mol. Cell Biol. 2016, 17, 413–425. [Google Scholar] [CrossRef]
- Le Magnen, C.; Shen, M.M.; Abate-Shen, C. Lineage Plasticity in Cancer Progression and Treatment. Annu. Rev. Cancer Biol. 2018, 2, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Panciera, T.; Azzolin, L.; Fujimura, A.; Di Biagio, D.; Frasson, C.; Bresolin, S.; Soligo, S.; Basso, G.; Bicciato, S.; Rosato, A.; et al. Induction of Expandable Tissue-Specific Stem/Progenitor Cells through Transient Expression of YAP/TAZ. Cell Stem Cell 2016, 19, 725–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo Transducer TAZ Confers Cancer Stem Cell-Related Traits on Breast Cancer Cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Gil, J.; Rodriguez, T. Cancer: The Transforming Power of Cell Competition. Curr. Biol. 2016, 26, R164–R166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowling, S.; Lawlor, K.; Rodríguez, T.A. Cell Competition: The Winners and Losers of Fitness Selection. Development 2019, 146, dev167486. [Google Scholar] [CrossRef] [Green Version]
- Suijkerbuijk, S.J.E.; Kolahgar, G.; Kucinski, I.; Piddini, E. Cell Competition Drives the Growth of Intestinal Adenomas in Drosophila. Curr. Biol. 2016, 26, 428–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Yee, P.P.; Wei, Y.; Liu, Z.; Kawasawa, Y.I.; Li, W. Differential YAP Expression in Glioma Cells Induces Cell Competition and Promotes Tumorigenesis. J. Cell Sci. 2019, 132, jcs225714. [Google Scholar] [CrossRef] [Green Version]
- Seager, R.J.; Hajal, C.; Spill, F.; Kamm, R.D.; Zaman, M.H. Dynamic Interplay between Tumour, Stroma and Immune System Can Drive or Prevent Tumour Progression. Converg. Sci. Phys. Oncol. 2017, 3, 034002. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The Tumor Microenvironment at a Glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, R.; Liu, S.; Zhang, S.; Min, L.; Zhu, S. Cellular and Extracellular Components in Tumor Microenvironment and Their Application in Early Diagnosis of Cancers. Anal. Cell. Pathol. 2020, 2020, 6283796. [Google Scholar] [CrossRef] [Green Version]
- Franco, P.I.R.; Rodrigues, A.P.; De Menezes, L.B.; Miguel, M.P. Tumor Microenvironment Components: Allies of Cancer Progression. Pathol. Res. Pract. 2020, 216, 152729. [Google Scholar] [CrossRef]
- Piccolo, S.; Cordenonsi, M.; Dupont, S. Molecular Pathways: YAP and TAZ Take Center Stage in Organ Growth and Tumorigenesis. Clin. Cancer Res. 2013, 19, 4925–4930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boopathy, G.T.K.; Hong, W. Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Front. Cell Dev. Biol. 2019, 7, 49. [Google Scholar] [CrossRef]
- White, S.M.; Murakami, S.; Yi, C. The Complex Entanglement of Hippo-Yap/Taz Signaling in Tumor Immunity. Oncogene 2019, 38, 2899–2909. [Google Scholar] [CrossRef]
- Pan, Z.; Tian, Y.; Cao, C.; Niu, G. The Emerging Role of YAP/TAZ in Tumor Immunity. Mol. Cancer Res. 2019, 17, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Horsman, M.R.; Vaupel, P. Pathophysiological Basis for the Formation of the Tumor Microenvironment. Front. Oncol. 2016, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Prieto-García, E.; Díaz-García, C.V.; García-Ruiz, I.; Agulló-Ortuño, M.T. Epithelial-to-Mesenchymal Transition in Tumor Progression. Med. Oncol. 2017, 34, 122. [Google Scholar] [CrossRef] [PubMed]
- Redfern, A.D.; Spalding, L.J.; Thompson, E.W. The Kraken Wakes: Induced EMT as a Driver of Tumour Aggression and Poor Outcome. Clin. Exp. Metastasis 2018, 35, 285–308. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Wang, P.; Toh, A.; Thompson, E.W. New Insights Into the Role of Phenotypic Plasticity and EMT in Driving Cancer Progression. Front. Mol. Biosci. 2020, 7, 71. [Google Scholar] [CrossRef]
- Pei, D.; Shu, X.; Gassama-Diagne, A.; Thiery, J.P. Mesenchymal-Epithelial Transition in Development and Reprogramming. Nat. Cell Biol. 2019, 21, 44–53. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Aiello, N.M.; Kang, Y. Context-Dependent EMT Programs in Cancer Metastasis. J. Exp. Med. 2019, 216, 1016–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Wang, Y.; Zhu, Y.; Yuan, C.; Wang, D.; Zhang, W.; Qi, B.; Qiu, J.; Song, X.; Ye, J.; et al. The Hippo Transducer TAZ Promotes Epithelial to Mesenchymal Transition and Cancer Stem Cell Maintenance in Oral Cancer. Mol. Oncol. 2015, 9, 1091–1105. [Google Scholar] [CrossRef] [Green Version]
- Ling, H.-H.; Kuo, C.-C.; Lin, B.-X.; Huang, Y.-H.; Lin, C.-W. Elevation of YAP Promotes the Epithelial-Mesenchymal Transition and Tumor Aggressiveness in Colorectal Cancer. Exp. Cell Res. 2017, 350, 218–225. [Google Scholar] [CrossRef]
- Diepenbruck, M.; Waldmeier, L.; Ivanek, R.; Berninger, P.; Arnold, P.; Van Nimwegen, E.; Christofori, G. Tead2 Expression Levels Control the Subcellular Distribution of Yap and Taz, Zyxin Expression and Epithelial-Mesenchymal Transition. J. Cell Sci. 2014, 127, 1523–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Li, G.; Huang, S.; Feng, Y.; Zhou, A. SOX9 Promotes Epithelial-Mesenchymal Transition via the Hippo-YAP Signaling Pathway in Gastric Carcinoma Cells. Oncol. Lett. 2019, 18, 599–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Xie, J.; Huang, P.; Yang, Z. Overexpression of TAZ Promotes Cell Proliferation, Migration and Epithelial-Mesenchymal Transition in Ovarian Cancer. Oncol. Lett. 2016, 12, 1821–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Xu, Z.; An, Q.; Jiang, D.; Wang, L.; Liang, B.; Li, Z. TAZ Promotes Epithelial to Mesenchymal Transition via the Upregulation of Connective Tissue Growth Factor Expression in Neuroblastoma Cells. Mol Med. Rep. 2015, 11, 982–988. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Jin, L.; Chen, Y.; Xi, X.; Guo, Y. YAP Promotes Epithelial Mesenchymal Transition by Upregulating Slug Expression in Human Colorectal Cancer Cells. Int. J. Clin. Exp. Pathol. 2020, 13, 701–710. [Google Scholar]
- Franzetti, G.-A.; Laud-Duval, K.; Van der Ent, W.; Brisac, A.; Irondelle, M.; Aubert, S.; Dirksen, U.; Bouvier, C.; De Pinieux, G.; Snaar-Jagalska, E.; et al. Cell-to-Cell Heterogeneity of EWSR1-FLI1 Activity Determines Proliferation/Migration Choices in Ewing Sarcoma Cells. Oncogene 2017, 36, 3505–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovar, H.; Bierbaumer, L.; Radic-Sarikas, B. The YAP/TAZ Pathway in Osteogenesis and Bone Sarcoma Pathogenesis. Cells 2020, 9, E972. [Google Scholar] [CrossRef] [Green Version]
- Piccolo, S.; Cordenonsi, M. Regulation of YAP and TAZ by Epithelial Plasticity. In The Hippo Signaling Pathway and Cancer; Oren, M., Aylon, Y., Eds.; Springer: New York, NY, USA, 2013; pp. 89–113. ISBN 978-1-4614-6219-4. [Google Scholar]
- Yamaguchi, H.; Taouk, G.M. A Potential Role of YAP/TAZ in the Interplay Between Metastasis and Metabolic Alterations. Front. Oncol. 2020, 10, 928. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivée, B. Tumor Angiogenesis and Vascular Normalization: Alternative Therapeutic Targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Zuazo-Gaztelu, I.; Casanovas, O. Unraveling the Role of Angiogenesis in Cancer Ecosystems. Front. Oncol. 2018, 8, 248. [Google Scholar] [CrossRef]
- Azad, T.; Ghahremani, M.; Yang, X. The Role of YAP and TAZ in Angiogenesis and Vascular Mimicry. Cells 2019, 8, E407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elaimy, A.L.; Mercurio, A.M. Convergence of VEGF and YAP/TAZ Signaling: Implications for Angiogenesis and Cancer Biology. Sci. Signal. 2018, 11, eaau1165. [Google Scholar] [CrossRef] [Green Version]
- Frezzetti, D.; Gallo, M.; Maiello, M.R.; D’Alessio, A.; Esposito, C.; Chicchinelli, N.; Normanno, N.; De Luca, A. VEGF as a Potential Target in Lung Cancer. Expert Opin. Ther. Targets 2017, 21, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Elaimy, A.L.; Guru, S.; Chang, C.; Ou, J.; Amante, J.J.; Zhu, L.J.; Goel, H.L.; Mercurio, A.M. VEGF-Neuropilin-2 Signaling Promotes Stem-like Traits in Breast Cancer Cells by TAZ-Mediated Repression of the Rac GAP Β2-Chimaerin. Sci. Signal. 2018, 11, eaao6897. [Google Scholar] [CrossRef] [Green Version]
- Elaimy, A.L.; Amante, J.J.; Zhu, L.J.; Wang, M.; Walmsley, C.S.; FitzGerald, T.J.; Goel, H.L.; Mercurio, A.M. The VEGF Receptor Neuropilin 2 Promotes Homologous Recombination by Stimulating YAP/TAZ-Mediated Rad51 Expression. Proc. Natl. Acad. Sci. USA 2019, 116, 14174–14180. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Mao, L.; Xiong, J.; Wen, J.; Wang, Y.; Geng, D.; Liu, Y. TAZ Expression on Endothelial Cells Is Closely Related to Blood Vascular Density and VEGFR2 Expression in Astrocytomas. J. Neuropathol. Exp. Neurol. 2019, 78, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Teleanu, R.I.; Chircov, C.; Grumezescu, A.M.; Teleanu, D.M. Tumor Angiogenesis and Anti-Angiogenic Strategies for Cancer Treatment. J. Clin. Med. 2019, 9, 84. [Google Scholar] [CrossRef] [Green Version]
- Lobov, I.; Mikhailova, N. The Role of Dll4/Notch Signaling in Normal and Pathological Ocular Angiogenesis: Dll4 Controls Blood Vessel Sprouting and Vessel Remodeling in Normal and Pathological Conditions. J. Ophthalmol. 2018, 2018, 3565292. [Google Scholar] [CrossRef]
- Olsen, J.J.; Pohl, S.Ö.-G.; Deshmukh, A.; Visweswaran, M.; Ward, N.C.; Arfuso, F.; Agostino, M.; Dharmarajan, A. The Role of Wnt Signalling in Angiogenesis. Clin. Biochem. Rev. 2017, 38, 131–142. [Google Scholar] [PubMed]
- Manzat Saplacan, R.M.; Balacescu, L.; Gherman, C.; Chira, R.I.; Craiu, A.; Mircea, P.A.; Lisencu, C.; Balacescu, O. The Role of PDGFs and PDGFRs in Colorectal Cancer. Mediat. Inflamm. 2017, 2017, 4708076. [Google Scholar] [CrossRef] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in Mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Cobbaut, M.; Karagil, S.; Bruno, L.; Diaz de la Loza, M.D.C.; Mackenzie, F.E.; Stolinski, M.; Elbediwy, A. Dysfunctional Mechanotransduction through the YAP/TAZ/Hippo Pathway as a Feature of Chronic Disease. Cells 2020, 9, 151. [Google Scholar] [CrossRef] [Green Version]
- Low, B.C.; Pan, C.Q.; Shivashankar, G.V.; Bershadsky, A.; Sudol, M.; Sheetz, M. YAP/TAZ as Mechanosensors and Mechanotransducers in Regulating Organ Size and Tumor Growth. FEBS Lett. 2014, 588, 2663–2670. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.; Kim, J. Regulation of Hippo Signaling by Actin Remodeling. BMB Rep. 2018, 51, 151–156. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, F.; Gao, Z.; Zhang, P.; Gao, J.; Wu, X. Regulation of Hippo Signaling by Mechanical Signals and the Cytoskeleton. DNA Cell Biol. 2020, 39, 159–166. [Google Scholar] [CrossRef]
- Pocaterra, A.; Santinon, G.; Romani, P.; Brian, I.; Dimitracopoulos, A.; Ghisleni, A.; Carnicer-Lombarte, A.; Forcato, M.; Braghetta, P.; Montagner, M.; et al. F-Actin Dynamics Regulates Mammalian Organ Growth and Cell Fate Maintenance. J. Hepatol. 2019, 71, 130–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, Y.; Lai, Z.-C. Mutual Regulation between Hippo Signaling and Actin Cytoskeleton. Protein Cell 2013, 4, 904–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, I.; McCollum, D. Control of Cellular Responses to Mechanical Cues through YAP/TAZ Regulation. J. Biol. Chem. 2019, 294, 17693–17706. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Goel, H.L.; Gao, H.; Pursell, B.; Shultz, L.D.; Greiner, D.L.; Ingerpuu, S.; Patarroyo, M.; Cao, S.; Lim, E.; et al. A Laminin 511 Matrix Is Regulated by TAZ and Functions as the Ligand for the A6Bβ1 Integrin to Sustain Breast Cancer Stem Cells. Genes Dev. 2015, 29, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-Dependent Matrix Remodelling Is Required for the Generation and Maintenance of Cancer-Associated Fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Hong, S.-H. Hippo Pathway as Another Oncogenic Mediator to Promote Immune Evasion by PD-L1 Signaling. J. Thorac. Dis. 2019, 11, S318–S321. [Google Scholar] [CrossRef]
- Ju, X.; Zhang, H.; Zhou, Z.; Wang, Q. Regulation of PD-L1 Expression in Cancer and Clinical Implications in Immunotherapy. Am. J. Cancer Res. 2020, 10, 1–11. [Google Scholar]
- Janse van Rensburg, H.J.; Azad, T.; Ling, M.; Hao, Y.; Snetsinger, B.; Khanal, P.; Minassian, L.M.; Graham, C.H.; Rauh, M.J.; Yang, X. The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res. 2018, 78, 1457–1470. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.S.; Park, D.I.; Lee, D.H.; Lee, J.E.; Yeo, M.-K.; Park, Y.H.; Lim, D.S.; Choi, W.; Lee, D.H.; Yoo, G.; et al. Hippo Effector YAP Directly Regulates the Expression of PD-L1 Transcripts in EGFR-TKI-Resistant Lung Adenocarcinoma. Biochem. Biophys. Res. Commun. 2017, 491, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.-C.; Yang, C.-T.; Jablons, D.M.; You, L. The Role of Yes-Associated Protein (YAP) in Regulating Programmed Death-Ligand 1 (PD-L1) in Thoracic Cancer. Biomedicines 2018, 6, E114. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Yang, S.; Zhang, F.; Cheng, F.; Wang, X.; Rao, J. Influence of the Hippo-YAP Signalling Pathway on Tumor Associated Macrophages (TAMs) and Its Implications on Cancer Immunosuppressive Microenvironment. Ann. Transl. Med. 2020, 8, 399. [Google Scholar] [CrossRef]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2019, 9, 1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Wang, X.; You, Y.; Wen, D.; Feng, Z.; Zhou, Y.; Que, K.; Gong, J.; Liu, Z. Nogo-B Fosters HCC Progression by Enhancing Yap/Taz-Mediated Tumor-Associated Macrophages M2 Polarization. Exp. Cell Res. 2020, 391, 111979. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Yang, C.-K.; Wei, P.-L.; Huynh, T.-T.; Whang-Peng, J.; Meng, T.-C.; Hsiao, M.; Tzeng, Y.-M.; Wu, A.T.; Yen, Y. Ovatodiolide Suppresses Colon Tumorigenesis and Prevents Polarization of M2 Tumor-Associated Macrophages through YAP Oncogenic Pathways. J. Hematol. Oncol. 2017, 10, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-de-Frutos, K.; Segrelles, C.; Lorz, C. Hippo Pathway and YAP Signaling Alterations in Squamous Cancer of the Head and Neck. J. Clin. Med. 2019, 8, 2131. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [Green Version]
- Stampouloglou, E.; Cheng, N.; Federico, A.; Slaby, E.; Monti, S.; Szeto, G.L.; Varelas, X. Yap Suppresses T-Cell Function and Infiltration in the Tumor Microenvironment. PLoS Biol. 2020, 18, e3000591. [Google Scholar] [CrossRef]
- Ni, X.; Tao, J.; Barbi, J.; Chen, Q.; Park, B.V.; Li, Z.; Zhang, N.; Lebid, A.; Ramaswamy, A.; Wei, P.; et al. YAP Is Essential for Treg-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2018, 8, 1026–1043. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Gao, Y.; Rao, J.; Wang, K.; Zhang, F.; Zhang, C. YAP-1 Promotes Tregs Differentiation in Hepatocellular Carcinoma by Enhancing TGFBR2 Transcription. Cell Physiol. Biochem. 2017, 41, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, H.; Li, Y.; Xia, D.; Yang, L.; Ma, Y.; Li, H. The Role of YAP/TAZ Activity in Cancer Metabolic Reprogramming. Mol. Cancer 2018, 17, 134. [Google Scholar] [CrossRef] [PubMed]
- Santinon, G.; Enzo, E.; Dupont, S. The Sweet Side of YAP/TAZ. Cell Cycle 2015, 14, 2543–2544. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Chen, X.; Sun, X.; Wang, L.; Chen, S. The Glycolytic Switch in Tumors: How Many Players Are Involved? J. Cancer 2017, 8, 3430–3440. [Google Scholar] [CrossRef]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic Glycolysis Tunes YAP/TAZ Transcriptional Activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef]
- Kocemba, K.A.; Dulińska-Litewka, J.; Wojdyła, K.L.; Pękala, P.A. The Role of 6-Phosphofructo-2-Kinase (PFK-2)/Fructose 2,6-Bisphosphatase (FBPase-2) in Metabolic Reprogramming of Cancer Cells. Postepy Hig. I Med. Dosw. (Online) 2016, 70, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Xu, X. YAP1-TEAD1-Glut1 Axis Dictates the Oncogenic Phenotypes of Breast Cancer Cells by Modulating Glycolysis. Biomed. Pharmacother. 2017, 95, 789–794. [Google Scholar] [CrossRef]
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of YAP/TAZ Activity by Metabolic and Nutrient-Sensing Pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef]
- Ye, J.; Li, T.-S.; Xu, G.; Zhao, Y.-M.; Zhang, N.-P.; Fan, J.; Wu, J. JCAD Promotes Progression of Nonalcoholic Steatohepatitis to Liver Cancer by Inhibiting LATS2 Kinase Activity. Cancer Res. 2017, 77, 5287–5300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Frangou, C.; Zhang, J. Fatty Acid Oxidation (FAO) Metabolic Switch: Metastasis in Lymph Nodes Driven by Yes-Associated Protein (YAP) Activation. Biotarget 2019, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Bathaie, S.Z.; Ashrafi, M.; Azizian, M.; Tamanoi, F. Mevalonate Pathway and Human Cancers. Curr. Mol. Pharmacol. 2017, 10, 77–85. [Google Scholar] [CrossRef]
- Mullen, P.J.; Yu, R.; Longo, J.; Archer, M.C.; Penn, L.Z. The Interplay between Cell Signalling and the Mevalonate Pathway in Cancer. Nat. Rev. Cancer 2016, 16, 718–731. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic Control of YAP and TAZ by the Mevalonate Pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Zanconato, F.; Battilana, G.; Cordenonsi, M.; Piccolo, S. YAP/TAZ as Therapeutic Targets in Cancer. Curr. Opin. Pharmacol. 2016, 29, 26–33. [Google Scholar] [CrossRef]
- Kim, M.H.; Kim, J. Role of YAP/TAZ Transcriptional Regulators in Resistance to Anti-Cancer Therapies. Cell. Mol. Life Sci. 2017, 74, 1457–1474. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-B.; Myung, S.-J. Clinical Implications of the Hippo-YAP Pathway in Multiple Cancer Contexts. BMB Rep. 2018, 51, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular Targeted Therapy: Treating Cancer with Specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Ghiso, E.; Migliore, C.; Ciciriello, V.; Morando, E.; Petrelli, A.; Corso, S.; De Luca, E.; Gatti, G.; Volante, M.; Giordano, S. YAP-Dependent AXL Overexpression Mediates Resistance to EGFR Inhibitors in NSCLC. Neoplasia 2017, 19, 1012–1021. [Google Scholar] [CrossRef]
- Coggins, G.E.; Farrel, A.; Rathi, K.S.; Hayes, C.M.; Scolaro, L.; Rokita, J.L.; Maris, J.M. YAP1 Mediates Resistance to MEK1/2 Inhibition in Neuroblastomas with Hyperactivated RAS Signaling. Cancer Res. 2019, 79, 6204–6214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, D.; Ho, K.C.; Hao, Y.; Yang, X. Taxol Resistance in Breast Cancer Cells Is Mediated by the Hippo Pathway Component TAZ and Its Downstream Transcriptional Targets Cyr61 and CTGF. Cancer Res. 2011, 71, 2728–2738. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Galván, S.; Felipe-Abrio, B.; Verdugo-Sivianes, E.M.; Perez, M.; Jiménez-García, M.P.; Suarez-Martinez, E.; Estevez-Garcia, P.; Carnero, A. Downregulation of MYPT1 Increases Tumor Resistance in Ovarian Cancer by Targeting the Hippo Pathway and Increasing the Stemness. Mol. Cancer 2020, 19, 7. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kim, C.G.; Kim, S.-K.; Shin, S.J.; Choe, E.A.; Park, S.-H.; Shin, E.-C.; Kim, J. YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol. Res. 2018, 6, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Gong, R.; Yu, F.-X. Targeting the Hippo Pathway for Anti-Cancer Therapies. Curr. Med. Chem. 2015, 22, 4104–4117. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. Bioessays 2020, 42, e1900162. [Google Scholar] [CrossRef] [Green Version]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small Molecules Inhibiting the Nuclear Localization of YAP/TAZ for Chemotherapeutics and Chemosensitizers against Breast Cancers. FEBS Open Bio. 2015, 5, 542–549. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Gou, J.; Jia, J.; Yi, T.; Cui, T.; Li, Z. Verteporfin, a Suppressor of YAP-TEAD Complex, Presents Promising Antitumor Properties on Ovarian Cancer. OncoTargets Ther. 2016, 9, 5371–5381. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Wang, X.; Tang, B.; Liu, H.; Zhang, M.; Wang, Y.; Ping, F.; Ding, J.; Shen, A.; Geng, M. A Tightly Controlled Src-YAP Signaling Axis Determines Therapeutic Response to Dasatinib in Renal Cell Carcinoma. Theranostics 2018, 8, 3256–3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, F.; Xu, Q.; Wang, J.; Yu, S.; Chang, H.-H.; Sinnett-Smith, J.; Eibl, G.; Rozengurt, E. Lipophilic Statins Inhibit YAP Nuclear Localization, Co-Activator Activity and Colony Formation in Pancreatic Cancer Cells and Prevent the Initial Stages of Pancreatic Ductal Adenocarcinoma in KrasG12D Mice. PLoS ONE 2019, 14, e0216603. [Google Scholar] [CrossRef]
- Zhao, W.; Liu, H.; Wang, J.; Wang, M.; Shao, R. Cyclizing-Berberine A35 Induces G2/M Arrest and Apoptosis by Activating YAP Phosphorylation (Ser127). J. Exp. Clin. Cancer Res. 2018, 37, 98. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A Peptide Mimicking VGLL4 Function Acts as a YAP Antagonist Therapy against Gastric Cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lui, J.W.; Xiao, S.; Ogomori, K.; Hammarstedt, J.E.; Little, E.C.; Lang, D. The Efficiency of Verteporfin as a Therapeutic Option in Pre-Clinical Models of Melanoma. J. Cancer 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gibault, F.; Corvaisier, M.; Bailly, F.; Huet, G.; Melnyk, P.; Cotelle, P. Non-Photoinduced Biological Properties of Verteporfin. Curr. Med. Chem. 2016, 23, 1171–1184. [Google Scholar] [CrossRef]
- Lu, J.; Roy, B.; Anderson, M.; Leggett, C.L.; Levy, M.J.; Pogue, B.; Hasan, T.; Wang, K.K. Verteporfin- and Sodium Porfimer-Mediated Photodynamic Therapy Enhances Pancreatic Cancer Cell Death without Activating Stromal Cells in the Microenvironment. J. Biomed. Opt. 2019, 24, 118001. [Google Scholar] [CrossRef] [Green Version]
- Gibault, F.; Bailly, F.; Corvaisier, M.; Coevoet, M.; Huet, G.; Melnyk, P.; Cotelle, P. Molecular Features of the YAP Inhibitor Verteporfin: Synthesis of Hexasubstituted Dipyrrins as Potential Inhibitors of YAP/TAZ, the Downstream Effectors of the Hippo Pathway. ChemMedChem 2017, 12, 954–961. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, X.; Feng, W.; Yu, Y.; Jeong, K.; Guo, W.; Lu, Y.; Mills, G.B. Verteporfin Inhibits YAP Function through Up-Regulating 14-3-3σ Sequestering YAP in the Cytoplasm. Am. J. Cancer Res. 2016, 6, 27–37. [Google Scholar]
- Shi, G.; Wang, H.; Han, H.; Gan, J.; Wang, H. Verteporfin Enhances the Sensitivity of LOVO/TAX Cells to Taxol via YAP Inhibition. Exp. Ther. Med. 2018, 16, 2751–2755. [Google Scholar] [CrossRef] [Green Version]
- Keskin, D.; Sadri, S.; Eskazan, A.E. Dasatinib for the Treatment of Chronic Myeloid Leukemia: Patient Selection and Special Considerations. Drug Des. Dev. Ther. 2016, 10, 3355–3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, A.; Zheng, J.; Yalamanchili, S.; Conley, A.P.; Ratan, R.; Somaiah, N.; Livingston, J.A.; Zarzour, M.A.; Araujo, D.M.; Benjamin, R.S.; et al. Modulation of YAP/TAZ by Statins to Improve Survival in Epithelioid Hemangioendothelioma (EHE). J. Clin. Oncol. 2020, 38, e23527. [Google Scholar] [CrossRef]
- Santos, D.M.; Pantano, L.; Pronzati, G.; Grasberger, P.; Probst, C.K.; Black, K.E.; Spinney, J.J.; Hariri, L.P.; Nichols, R.; Lin, Y.; et al. Screening for YAP Inhibitors Identifies Statins as Modulators of Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Ma, Q.; Wang, L.; Liu, C.; Gao, H.; Yang, Z.; Liu, Z.; Zhang, H.; Ji, L.; Jiang, G. A Brief Review: Some Compounds Targeting YAP against Malignancies. Future Oncol. 2019, 15, 1535–1543. [Google Scholar] [CrossRef]
- Morice, S.; Danieau, G.; Rédini, F.; Brounais-Le-Royer, B.; Verrecchia, F. Hippo/YAP Signaling Pathway: A Promising Therapeutic Target in Bone Paediatric Cancers? Cancers 2020, 12, E645. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Fang, L. VGLL4 Is a Transcriptional Cofactor Acting as a Novel Tumor Suppressor via Interacting with TEADs. Am. J. Cancer Res. 2018, 8, 932–943. [Google Scholar]
- Guo, L.; Teng, L. YAP/TAZ for Cancer Therapy: Opportunities and Challenges (Review). Int. J. Oncol. 2015, 46, 1444–1452. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hu, G.; Liu, F.; Wang, X.; Wu, M.; Schwarz, J.J.; Zhou, J. Deletion of Yes-Associated Protein (YAP) Specifically in Cardiac and Vascular Smooth Muscle Cells Reveals a Crucial Role for YAP in Mouse Cardiovascular Development. Circ. Res. 2014, 114, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Daoud, F.; Holmberg, J.; Alajbegovic, A.; Grossi, M.; Rippe, C.; Swärd, K.; Albinsson, S. Inducible Deletion of YAP and TAZ in Adult Mouse Smooth Muscle Causes Rapid and Lethal Colonic Pseudo-Obstruction. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Schwartzman, M.; Reginensi, A.; Wong, J.S.; Basgen, J.M.; Meliambro, K.; Nicholas, S.B.; D’Agati, V.; McNeill, H.; Campbell, K.N. Podocyte-Specific Deletion of Yes-Associated Protein Causes FSGS and Progressive Renal Failure. J. Am. Soc. Nephrol. 2016, 27, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Levasseur, A.; St-Jean, G.; Paquet, M.; Boerboom, D.; Boyer, A. Targeted Disruption of YAP and TAZ Impairs the Maintenance of the Adrenal Cortex. Endocrinology 2017, 158, 3738–3753. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Jablons, D.; You, L. Hippo Pathway in Lung Development. J. Thorac. Dis. 2017, 9, 2246–2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Finegold, M.J.; Johnson, R.L. Hippo Pathway Coactivators Yap and Taz Are Required to Coordinate Mammalian Liver Regeneration. Exp. Mol. Med. 2018, 50, e423. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | Exp. Model | Duration | Relevant Results | References |
|---|---|---|---|---|
| Verteporfin | OVCAR8 xenograft mice. | 3 weeks | In vivo, VP significantly affected tumor growth in OVCAR8 xenograft mice, resulting in tumor nodules with lower average weight and reduced volume of gross ascites. | [142] |
| Dasatinib | Four-to six-week-old nu/nu athymic BALB/c female mice. | 3 weeks | Dasatinib can impair renal carcinoma cell viability in vitro and decrease tumor growth in vivo. | [143] |
| Lipophilic statins | PANC-1 and MiaPaCa-2LSL-human pancreatic cancer cell lines, and KrasG12D/+ and p48-Cre+/− mice. | 2 weeks | Lipophilic statins limit YAP activity and proliferation in pancreatic cancer cell models in vitro and they attenuate early lesions that lead to pancreatic ductal adenocarcinoma in vivo. | [144] |
| Dasatinib, statins and pazopanib | MDA-MB-231, MDA-MB-453, HBC-4, HBC-5, MCF-7, BSY-1, ZR-75-1, and SKBR-3 breast cancer cell lines. | 2 weeks | All drugs induced phosphorylation of YAP and TAZ, and pazopanib induced proteasomal degradation of YAP/TAZ. | [141] |
| A35 | Human K562, HepG2, Raji, HCT116 and HCT116-KO cancer cells. | 1 week | A35 decreased YAP1 nuclear localization by activating YAP phosphorylation (Ser127), which subsequently regulated the transcription of YAP target genes associated with growth and cycle regulation to induce G2/M arrest and growth inhibition. | [145] |
| Super-TDU | Human gastric tumor clinical specimens. | - | Super-TDU inhibited cell viability and colony formation of GC cell lines MGC-803, BGC-823, and HGC27. This peptide downregulated the expression of YAP-TEADs target genes CTGF, CYR61, and CDX2. | [146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega, Á.; Vera, I.; Diaz, M.P.; Navarro, C.; Rojas, M.; Torres, W.; Parra, H.; Salazar, J.; De Sanctis, J.B.; Bermúdez, V. The YAP/TAZ Signaling Pathway in the Tumor Microenvironment and Carcinogenesis: Current Knowledge and Therapeutic Promises. Int. J. Mol. Sci. 2022, 23, 430. https://doi.org/10.3390/ijms23010430
Ortega Á, Vera I, Diaz MP, Navarro C, Rojas M, Torres W, Parra H, Salazar J, De Sanctis JB, Bermúdez V. The YAP/TAZ Signaling Pathway in the Tumor Microenvironment and Carcinogenesis: Current Knowledge and Therapeutic Promises. International Journal of Molecular Sciences. 2022; 23(1):430. https://doi.org/10.3390/ijms23010430
Chicago/Turabian StyleOrtega, Ángel, Ivana Vera, Maria P. Diaz, Carla Navarro, Milagros Rojas, Wheeler Torres, Heliana Parra, Juan Salazar, Juan B. De Sanctis, and Valmore Bermúdez. 2022. "The YAP/TAZ Signaling Pathway in the Tumor Microenvironment and Carcinogenesis: Current Knowledge and Therapeutic Promises" International Journal of Molecular Sciences 23, no. 1: 430. https://doi.org/10.3390/ijms23010430
APA StyleOrtega, Á., Vera, I., Diaz, M. P., Navarro, C., Rojas, M., Torres, W., Parra, H., Salazar, J., De Sanctis, J. B., & Bermúdez, V. (2022). The YAP/TAZ Signaling Pathway in the Tumor Microenvironment and Carcinogenesis: Current Knowledge and Therapeutic Promises. International Journal of Molecular Sciences, 23(1), 430. https://doi.org/10.3390/ijms23010430