
Role of Macrophages and Plasminogen Activator Inhibitor-1 in Delayed Bone Repair Induced by Glucocorticoids in Mice


Abstract
:1. Introduction
2. Results
2.1. Effects of Dex and PAI-1 Deficiency on Macrophage Number after Femoral Bone Injury
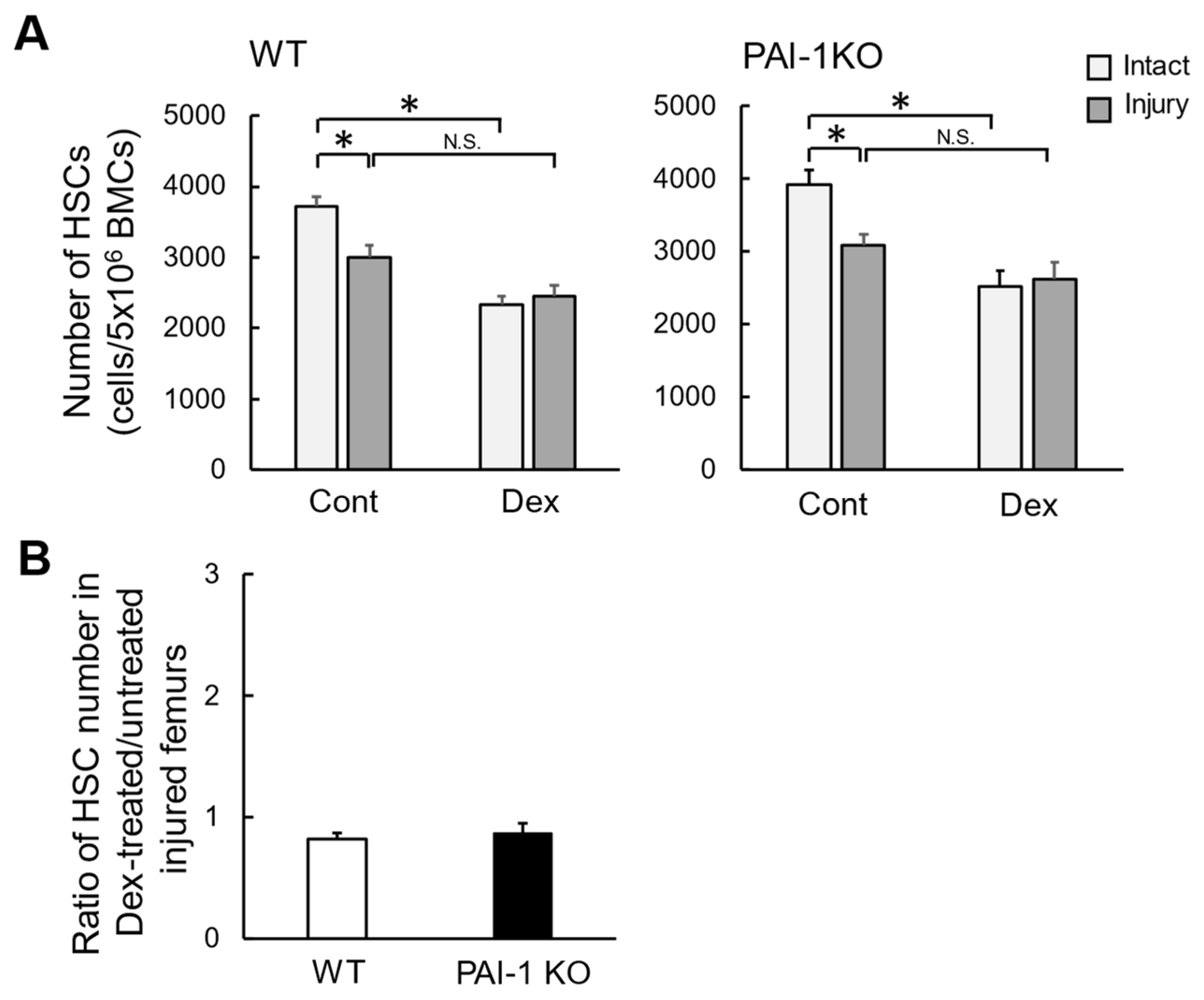
2.2. Effects of Dex on the Number of HSCs in Bone Marrow after Femoral Bone Injury
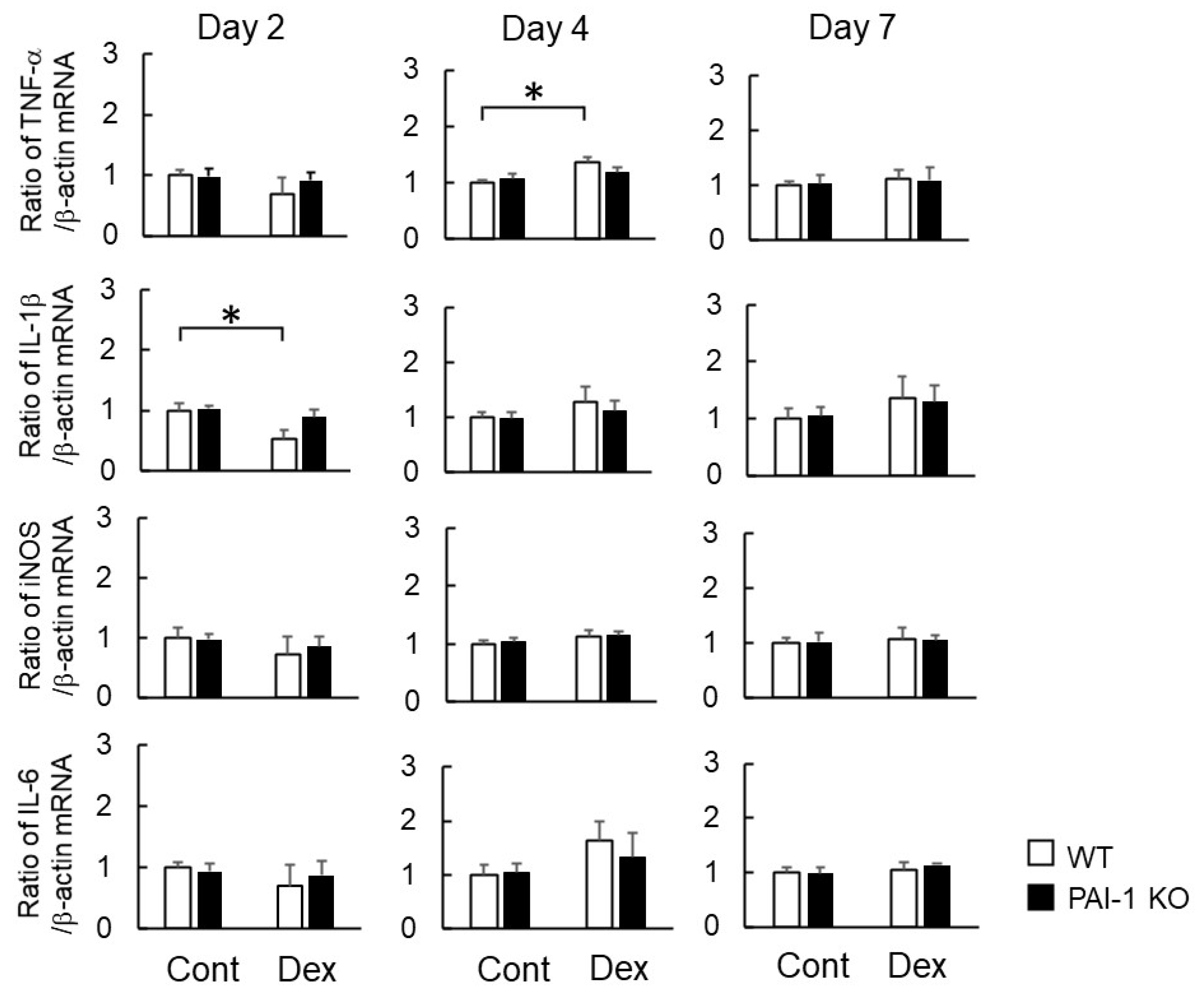
2.3. Effects of Dex on Macrophage-Related Gene Expression at the Damaged Site after Bone Injury
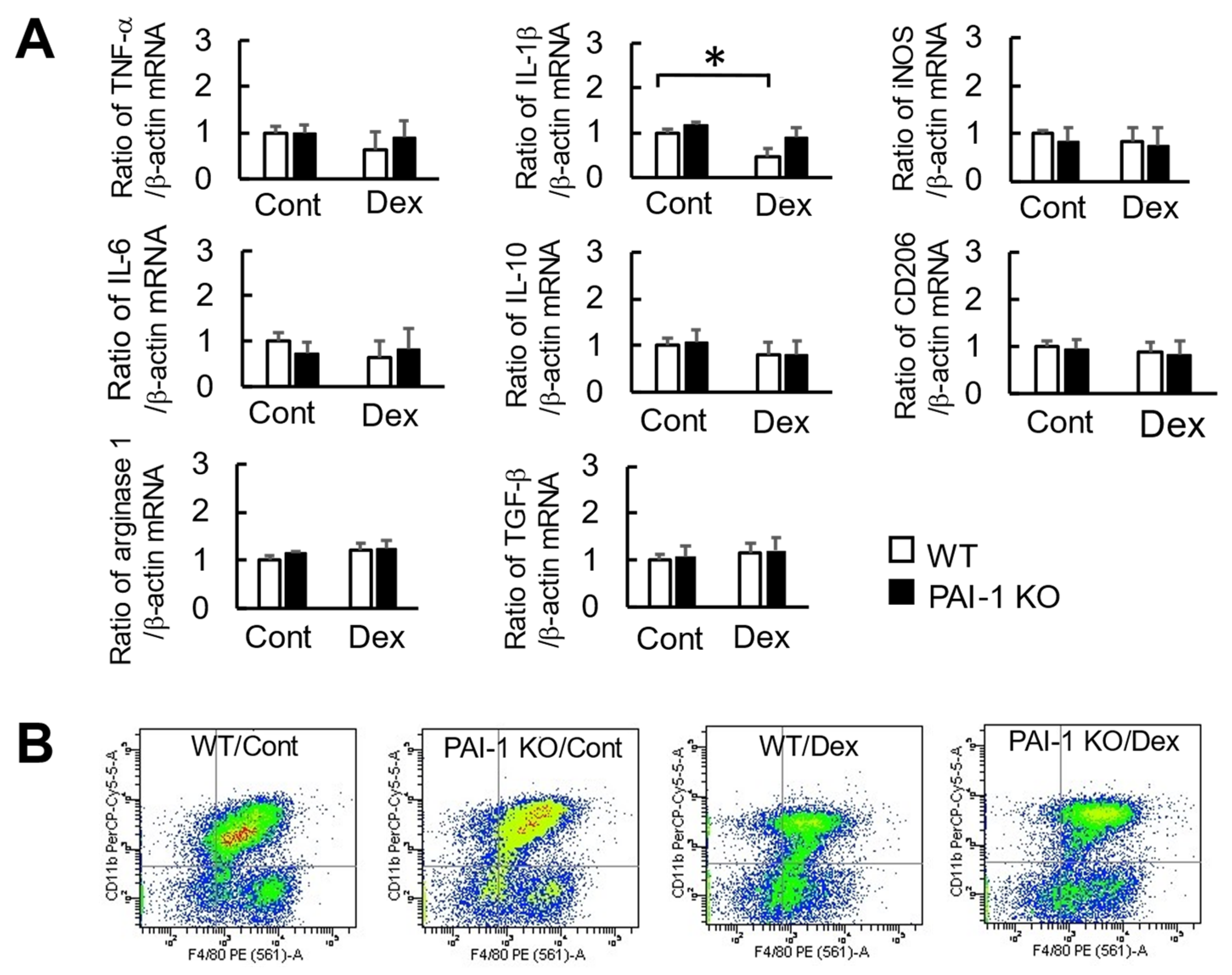
2.4. Effects of Dex and PAI-1 Deficiency on Factors Produced from Macrophages Derived from the Bone Marrow of Damaged Femurs
2.5. Effects of Dex and PAI-1 Deficiency on the Phagocytosis of Macrophages at Damaged Femurs
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Murine Bone Injury Model
4.3. Histological Analysis
4.4. Flow Cytometric Analysis
4.5. Quantitative Real-Time Polymerase Chain Reaction (PCR)
4.6. Isolation of F4/80 and CD11b Double-Positive Cells from the Femur
4.7. Transmission Electron Microscopy
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hofbauer, L.C.; Rauner, M. Minireview: Live and let die: Molecular effects of glucocorticoids on bone cells. Mol Endocrinol. 2009, 10, 1525–1531. [Google Scholar] [CrossRef] [Green Version]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids—New mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [Green Version]
- Strehl, C.; Buttgereit, F. Optimized glucocorticoid therapy: Teaching old drugs new tricks. Mol. Cell. Endocrinol. 2013, 380, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Hachemi, Y.; Rapp, A.E.; Picke, A.K.; Weidinger, G.; Ignatius, A.; Tuckermann, J. Molecular mechanisms of glucocorticoids on skeletal and bone regeneration after fracture. J. Mol. Endocrinol. 2018, 61, R75–R90. [Google Scholar] [CrossRef]
- Henneicke, H.; Gasparini, S.J.; Brennan-Speranza, T.C.; Zhou, H.; Seibel, M.J. Glucocorticoids and bone: Local effects and systemic implications. Trends Endocrinol. Metab. 2014, 25, 197–211. [Google Scholar] [CrossRef]
- Moutsatsou, P.; Kassi, E.; Papavassiliou, A.G. Glucocorticoid receptor signaling in bone cells. Trends Mol. Med. 2012, 18, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Okamoto, T.; Okumoto, K.; Takafuji, Y.; Ishida, M.; Kawao, N.; Matsuo, O.; Kaji, H. PAI-1 is involved in delayed bone repair induced by glucocorticoids in mice. Bone 2020, 134, 115310. [Google Scholar] [CrossRef] [PubMed]
- Claes, L.; Recknagel, S.; Ignatius, A. Fracture healing under healthy and inflammatory conditions. Nat. Rev. Rheumatol. 2012, 8, 133–143. [Google Scholar] [CrossRef]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Alexander, K.A.; Chang, M.K.; Maylin, E.R.; Kohler, T.; Muller, R.; Wu, A.C.; Van Rooijen, N.; Sweet, M.J.; Hume, D.A.; Raggatt, L.J.; et al. Osteal macrophages promote in vivo intramembranous bone healing in a mouse tibial injury model. J. Bone Miner. Res. 2011, 26, 1517–1532. [Google Scholar] [CrossRef] [PubMed]
- Sinder, B.P.; Pettit, A.R.; McCauley, L.K. Macrophages: Their emerging roles in bone. J Bone Miner. Res. 2015, 30, 2140–2149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vi, L.; Baht, G.S.; Whetstone, H.; Ng, A.; Wei, Q.; Poon, R.; Mylvaganam, S.; Grynpas, M.; Alman, B.A. Macrophages promote osteoblastic differentiation in-vivo: Implications in fracture repair and bone homeostasis. J. Bone Miner. Res. 2015, 30, 1090–1102. [Google Scholar] [CrossRef]
- Miron, R.J.; Bosshardt, D.D. OsteoMacs: Key players around bone biomaterials. Biomaterials 2016, 82, 1–19. [Google Scholar] [CrossRef]
- Kawao, N.; Tamura, Y.; Okumoto, K.; Yano, M.; Okada, K.; Matsuo, O.; Kaji, H. Plasminogen plays a crucial role in bone repair. J. Bone Miner. Res. 2013, 28, 1561–1574. [Google Scholar] [CrossRef]
- Kawao, N.; Tamura, Y.; Horiuchi, Y.; Okumoto, K.; Yano, M.; Okada, K.; Matsuo, O.; Kaji, H. The tissue fibrinolytic system contributes to the induction of macrophage function and CCL3 during bone repair in mice. PLoS ONE 2015, 10, e0123982. [Google Scholar] [CrossRef] [Green Version]
- Retzepi, M.; Donos, N. The effect of diabetes mellitus on osseous healing. Clin. Oral Implants Res. 2010, 21, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Koyama, H.; Morioka, T.; Tanaka, S.; Kizu, A.; Motoyama, K.; Mori, K.; Fukumoto, S.; Shioi, A.; Shimogaito, N.; et al. Receptor for advanced glycation end products is involved in impaired angiogenic response in diabetes. Diabetes 2006, 55, 2245–2255. [Google Scholar] [CrossRef] [Green Version]
- Ko, K.I.; Coimbra, L.S.; Tian, C.; Alblowi, J.; Kayal, R.A.; Einhorn, T.A.; Gerstenfeld, L.C.; Pignolo, R.J.; Graves, D.T. Diabetes reduces mesenchymal stem cells in fracture healing through a TNF-α-mediated mechanism. Diabetologia 2015, 58, 633–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, L.; Kawao, N.; Tamura, Y.; Okumoto, K.; Okada, K.; Yano, M.; Matsuo, O.; Kaji, H. Plasminogen activator inhibitor-1 is involved in impaired bone repair associated with diabetes in female mice. PLoS ONE 2014, 9, e92686. [Google Scholar]
- Tamura, Y.; Kawao, N.; Okada, K.; Yano, M.; Okumoto, K.; Matsuo, O.; Kaji, H. Plasminogen activator inhibitor-1 is involved in streptozotocin-induced bone loss in female mice. Diabetes 2013, 62, 3170–3179. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Tamura, Y.; Kawao, N.; Okada, K.; Yano, M.; Okumoto, K.; Kaji, H. Influence of diabetic state and vitamin D deficiency on bone repair in female mice. Bone 2014, 61, 102–108. [Google Scholar] [CrossRef]
- Okada, K.; Kawao, N.; Yano, M.; Tamura, Y.; Kurashimo, S.; Okumoto, K.; Kojima, K.; Kaji, H. Stromal cell-derived factor-1 mediates changes of bone marrow stem cells during the bone repair process. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E15–E23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrchen, J.M.; Roth, J.; Barczyk-Kahlert, K. Front more than suppression: Glucocorticoid action on monocytes and macrophages. Front. Immunol. 2019, 10, 2028. [Google Scholar] [CrossRef] [Green Version]
- Ai, F.; Zhao, G.; Lv, W.; Liu, B.; Lin, J. Dexamethasone induces aberrant macrophage immune function and apoptosis. Oncol. Rep. 2020, 43, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Tolmeijer, S.; Oskam, J.M.; Tonkens, T.; Meijer, A.H.; Schaaf, M.J.M. Glucocorticoids inhibit macrophage differentiation towards a pro-inflammatory phenotype upon wounding without affecting their migration. Dis. Models Mech. 2019, 12, dmm037887. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Deng, R.; Chai, Y.; Chen, H.; Hu, B.; Wang, X.; Zhu, S.; Cao, Y.; Ni, S.; Wan, M.; et al. Macrophage-lineage TRAP+ cells recruit periosteum-derived cells for periosteal osteogenesis and regeneration. J. Clin. Investig. 2019, 129, 2578–2594. [Google Scholar] [CrossRef] [Green Version]
- Vi, L.; Baht, G.S.; Soderblom, E.J.; Whetstone, H.; Wei, Q.; Furman, B.; Puviindran, V.; Nadesan, P.; Foster, M.; Poon, R.; et al. Macrophage cells secrete factors including LRP1 that orchestrate the rejuvenation of bone repair in mice. Nat. Commun. 2018, 9, 5191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.J.; Liu, H.; Chen, J.; Qian, D.F.; Kong, F.Q.; Jie, J.; Yin, G.Y.; Li, Q.Q.; Fan, J.J. Macrophage GIT1 contributes to bone regeneration by regulating inflammatory responses in an ERK/NRF2-dependent way. Bone Miner. Res. 2020, 35, 2015–2031. [Google Scholar] [CrossRef]
- Qiao, W.; Xie, H.; Fang, J.; Shen, J.; Li, W.; Shen, D.; Wu, J.; Wu, S.; Liu, X.; Zheng, Y.; et al. Sequential activation of heterogeneous macrophage phenotypes is essential for biomaterials-induced bone regeneration. Biomaterials 2021, 276, 121038. [Google Scholar] [CrossRef]
- Shimoide, T.; Kawao, N.; Tamura, Y.; Okada, K.; Horiuchi, Y.; Okumoto, K.; Kurashimo, S.; Ishida, M.; Tatsumi, K.; Matsuo, O.; et al. Role of macrophages and plasminogen activator inhibitor-1 in delayed bone repair in diabetic female mice. Endocrinology 2018, 159, 1875–1885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albiero, M.; Poncina, N.; Ciciliot, S.; Cappellari, R.; Menegazzo, L.; Ferraro, F.; Bolego, C.; Cignarella, A.; Avogaro, A.; Fadini, G.P. Bone marrow macrophages contribute to diabetic stem cell mobilopathy by producing oncostatin M. Diabetes 2015, 64, 2957–2968. [Google Scholar] [CrossRef] [Green Version]
- Hazra, S.; Jarajapu, Y.P.; Stepps, V.; Caballero, S.; Thinschmidt, J.S.; Sautina, L.; Bengtsson, N.; Licalzi, S.; Dominguez, J.; Kern, T.S.; et al. Long-term type 1 diabetes influences haematopoietic stem cells by reducing vascular repair potential and increasing inflammatory monocyte generation in a murine model. Diabetologia 2013, 56, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, A.; Siragusa, M.; Quaini, F.; Mangialardi, G.; Katare, R.G.; Caporali, A.; van Buul, J.D.; van Alphen, F.P.; Graiani, G.; Spinetti, G.; et al. Diabetes mellitus induces bone marrow microangiopathy. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 498–508. [Google Scholar] [CrossRef]
- Fadini, G.P.; Albiero, M.; de Kreutzenberg, S.V.; Boscaro, E.; Cappellari, R.; Marescotti, M.; Poncina, N.; Agostini, C.; Avogaro, A. Diabetes impairs stem cell and proangiogenic cell mobilization in humans. Diabetes Care 2013, 36, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Kaji, H. Adipose tissue-derived plasminogen activator inhibitor-I function and regulation. Comp. Physiol. 2016, 6, 1873–1896. [Google Scholar]
- Tamura, Y.; Kawao, N.; Yano, M.; Okada, K.; Okumoto, K.; Chiba, Y.; Matsuo, O.; Kaji., H. Role of plasminogen activator inhibitor-1 in glucocorticoid-induced diabetes and osteopenia in mice. Diabetes 2015, 64, 2194–2206. [Google Scholar] [CrossRef] [Green Version]
- Tamura, Y.; Kawao, N.; Shimoide, T.; Okada, K.; Matsuo, O.; Kaji, H. Role of plasminogen activator inhibitor-1 in glucocorticoid-induced muscle change in mice. J. Bone Miner. Metab. 2018, 36, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Takayanagi, H. Osteoimmunology in bone fracture healing. Curr. Osteoporos. Rep. 2017, 15, 367–375. [Google Scholar] [CrossRef]
- McCauley, J.; Bitsaktsis, C.; Cottrell, J. Macrophage subtype and cytokine expression characterization during the acute inflammatory phase of mouse bone fracture repair. J. Orthop. Res. 2020, 38, 1693–1702. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name and Symbol | Forward Primer | Reverse Primer |
|---|---|---|
| Macrophage colony-stimulating factor (MCSF) | GACTTCATGCCAGATTGCC | GGTGGCTTTAGGGTACAGG |
| Monocyte chemoattractant protein-1 (MCP-1) | CCACTCACCTGCTGCTACTCA | TGGTGATCCTCTTGTAGCTCTCC |
| Macrophage inflammatory protein-1α (MIP-1α) | CCTCTGTCACCTGCTCAACA | GATGAATTGGCGTGGAATCT |
| Interleukin-4 (IL-4) | ACAGGAGAAGGGACGCCAT | GAAGCCCTACAGACGAGCTCA |
| Interleukin-6 (IL-6) | GTTCTCTGGGAAATCGTGGA | GGAAATTCGGGGTAGGAAGGA |
| Tumor necrosis factor-α (TNF-α) | CCCAGACCCTCACACTCAGATC | GCCACTCCAGCTGCTCCTC |
| Interleukin-1β (IL-1β) | GGTCAAAGGTTTGGAAGCAG | TGTGAAATGCCACCTTTTGA |
| Inducible nitric oxide synthase (iNOS) | TTTGCTTCCATGCTAATGCGAAAG | GCTCTGTTGAGGTCTAAAGGCTCCG |
| Interleukin-10 (IL-10) | GCTCTTACTGACTGGCATGAG | CGCAGCTCTAGGAGCATGTG |
| CD206 | TTTGGAATCAAGGGCACAGAG | TGCTCCACAATCCCGAACC |
| Arginase1 | CTCCAAGCCAAAGTCCTTAGAG | AGGAGCTGTCATTAGGGACATC |
| Transforming growth factor-β1 (TGF-β1) | GCAACAATTCCTGGCGTTACC | CGCTGAATCGAAAGCCCTGTA |
| Stromal-derived factor-1 (SDF-1) | CTGTGCCCTTACGATTGTTG | TCAGCCTTCCTCGGGGGTCT |
| β-actin | TACCACAGGCATTGTGATGG | TTTGATGTCACGCACGATTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okada, K.; Kawao, N.; Nakai, D.; Wakabayashi, R.; Horiuchi, Y.; Okumoto, K.; Kurashimo, S.; Takafuji, Y.; Matsuo, O.; Kaji, H. Role of Macrophages and Plasminogen Activator Inhibitor-1 in Delayed Bone Repair Induced by Glucocorticoids in Mice. Int. J. Mol. Sci. 2022, 23, 478. https://doi.org/10.3390/ijms23010478
Okada K, Kawao N, Nakai D, Wakabayashi R, Horiuchi Y, Okumoto K, Kurashimo S, Takafuji Y, Matsuo O, Kaji H. Role of Macrophages and Plasminogen Activator Inhibitor-1 in Delayed Bone Repair Induced by Glucocorticoids in Mice. International Journal of Molecular Sciences. 2022; 23(1):478. https://doi.org/10.3390/ijms23010478
Chicago/Turabian StyleOkada, Kiyotaka, Naoyuki Kawao, Daisho Nakai, Rei Wakabayashi, Yoshitaka Horiuchi, Katsumi Okumoto, Shinji Kurashimo, Yoshimasa Takafuji, Osamu Matsuo, and Hiroshi Kaji. 2022. "Role of Macrophages and Plasminogen Activator Inhibitor-1 in Delayed Bone Repair Induced by Glucocorticoids in Mice" International Journal of Molecular Sciences 23, no. 1: 478. https://doi.org/10.3390/ijms23010478