
Role of Tyrosine Kinase Syk in Thrombus Stabilisation at High Shear

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Blood Withdrawal and Platelet Preparation
2.3. Platelet Aggregation
2.4. Western Blotting
2.5. Flow Adhesion
2.6. Epifluorescence Microscopy
2.7. Statistical Analysis
3. Results
3.1. Thrombin Stimulates Sustained Phosphorylation of Syk via Integrin αIIbβ3 during Platelet Aggregation
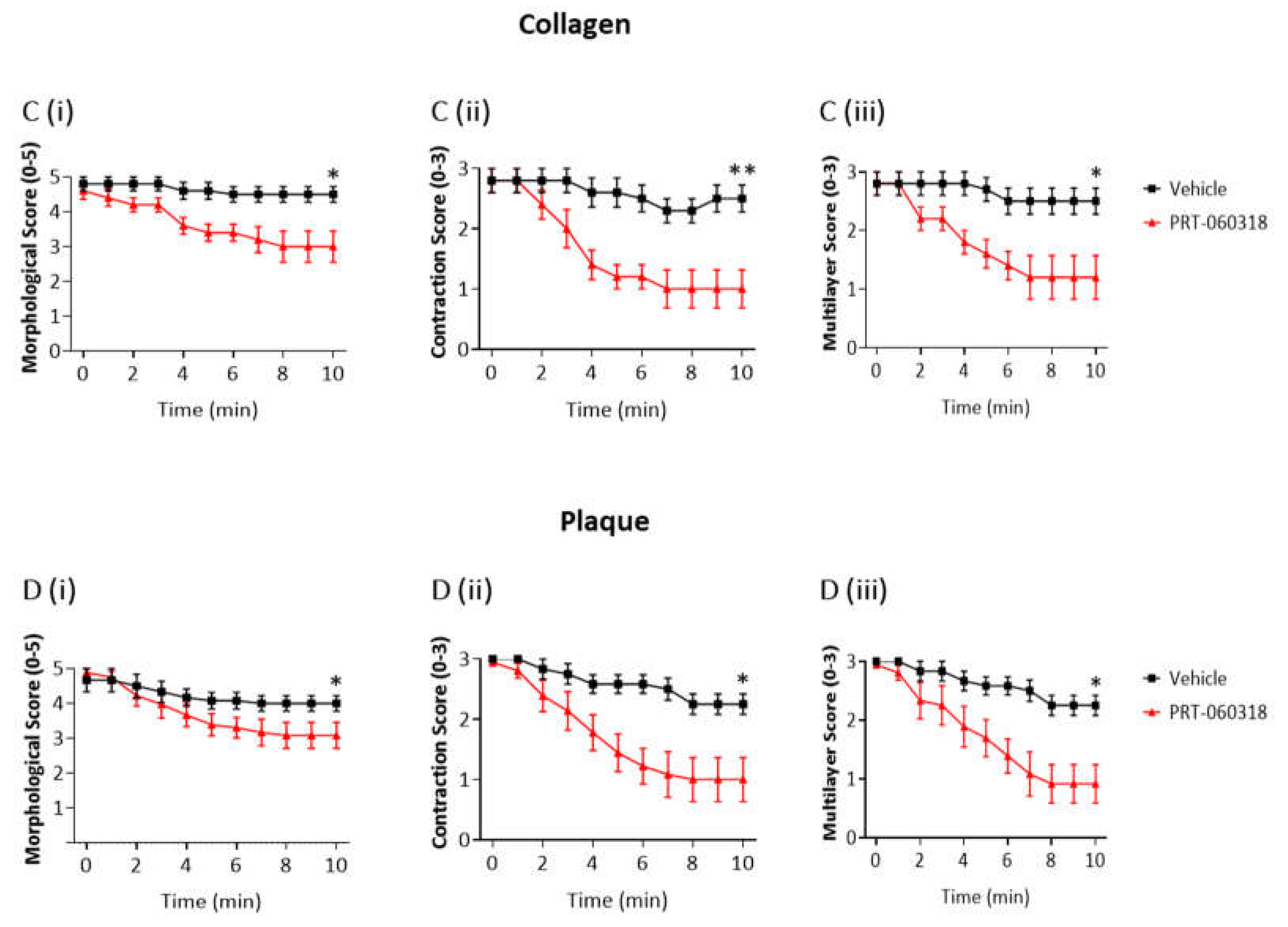
3.2. Inhibition of Syk on Collagen and Plaque Homogenate Causes Thrombus Shell Instability
3.3. Inhibition of Syk Promotes Thrombus Breakdown at 37 °C
3.4. Inhibition of Src and Secondary Agonists but Not Btk Promotes Thrombus Breakdown
3.5. Blocking of GPVI with Nanobody 21 Causes Only a Minor Thrombus Breakdown on Collagen
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tomaiuolo, M.; Brass, L.F.; Stalker, T.J. Regulation of Platelet Activation and Coagulation and Its Role in Vascular Injury and Arterial Thrombosis. Interv. Cardiol. Clin. 2017, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh, J.D.; Stalker, T.J.; Voronov, R.; Muthard, R.W.; Tomaiuolo, M.; Diamond, S.L.; Brass, L.F. A systems approach to hemostasis: 1. The interdependence of thrombus architecture and agonist movements in the gaps between platelets. Blood 2014, 124, 1808–1815. [Google Scholar] [CrossRef]
- Loyau, S.; Ho-Tin-Noe, B.; Bourrienne, M.C.; Boulaftali, Y.; Jandrot-Perrus, M. Microfluidic Modeling of Thrombolysis. Arter. Thromb. Vasc. Biol. 2018, 38, 2626–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.U.; Kaneva, V.; Loyau, S.; Nechipurenko, D.; Receveur, N.; Le Bris, M.; Janus-Bell, E.; Didelot, M.; Rauch, A.; Susen, S.; et al. Pharmacological Blockade of Glycoprotein VI Promotes Thrombus Disaggregation in the Absence of Thrombin. Arter. Thromb. Vasc. Biol. 2020, 40, 2127–2142. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Watson, S.P. Platelet-collagen interaction: Is GPVI the central receptor? Blood 2003, 102, 449–461. [Google Scholar] [CrossRef]
- Montague, S.J.; Patel, P.; Martin, E.M.; Slater, A.; Quintanilla, L.G.; Perrella, G.; Kardeby, C.; Nagy, M.; Mezzano, D.; Mendes, P.M.; et al. Platelet activation by charged ligands and nanoparticles: Platelet glycoprotein receptors as pattern recognition receptors. Platelets 2021, 32, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Perrella, G.; Huang, J.; Provenzale, I.; Swieringa, F.; Heubel-Moenen, F.; Farndale, R.W.; Roest, M.; van der Meijden, P.E.J.; Thomas, M.; Ariens, R.A.S.; et al. Nonredundant Roles of Platelet Glycoprotein VI and Integrin alphaIIbbeta3 in Fibrin-Mediated Microthrombus Formation. Arter. Thromb. Vasc. Biol. 2020, 41, e97–e111. [Google Scholar] [CrossRef]
- Watson, S.P.; Auger, J.M.; McCarty, O.J.; Pearce, A.C. GPVI and integrin alphaIIb beta3 signaling in platelets. J. Thromb. Haemost 2005, 3, 1752–1762. [Google Scholar] [CrossRef]
- Durrant, T.N.; van den Bosch, M.T.; Hers, I. Integrin alphaIIbbeta3 outside-in signaling. Blood 2017, 130, 1607–1619. [Google Scholar] [CrossRef] [Green Version]
- Cooper, N.; Altomare, I.; Thomas, M.R.; Nicolson, P.L.R.; Watson, S.P.; Markovtsov, V.; Todd, L.K.; Masuda, E.; Bussel, J.B. Assessment of thrombotic risk during long-term treatment of immune thrombocytopenia with fostamatinib. Ther. Adv. Hematol. 2021, 12, 20406207211010875. [Google Scholar] [CrossRef]
- Slater, A.; Di, Y.; Clark, J.C.; Jooss, N.J.; Martin, E.M.; Alenazy, F.O.; Thomas, M.R.; Ariens, R.A.; Herr, A.B.; Poulter, N.S.; et al. Structural characterisation of a novel GPVI nanobody-complex reveals a biologically active domain-swapped GPVI dimer. Blood 2021, 137, 3443–3453. [Google Scholar] [CrossRef] [PubMed]
- Jamasbi, J.; Megens, R.T.; Bianchini, M.; Munch, G.; Ungerer, M.; Faussner, A.; Sherman, S.; Walker, A.; Goyal, P.; Jung, S.; et al. Differential Inhibition of Human Atherosclerotic Plaque-Induced Platelet Activation by Dimeric GPVI-Fc and Anti-GPVI Antibodies: Functional and Imaging Studies. J. Am. Coll. Cardiol. 2015, 65, 2404–2415. [Google Scholar] [CrossRef] [Green Version]
- Asazuma, N.; Wilde, J.I.; Berlanga, O.; Leduc, M.; Leo, A.; Schweighoffer, E.; Tybulewicz, V.; Bon, C.; Liu, S.K.; McGlade, C.J.; et al. Interaction of linker for activation of T cells with multiple adapter proteins in platelets activated by the glycoprotein VI-selective ligand, convulxin. J. Biol. Chem. 2000, 275, 33427–33434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onselaer, M.B.; Nagy, M.; Pallini, C.; Pike, J.A.; Perrella, G.; Quintanilla, L.G.; Eble, J.A.; Poulter, N.S.; Heemskerk, J.W.M.; Watson, S.P. Comparison of the GPVI inhibitors losartan and honokiol. Platelets 2020, 31, 187–197. [Google Scholar] [CrossRef]
- de Witt, S.M.; Swieringa, F.; Cavill, R.; Lamers, M.M.; van Kruchten, R.; Mastenbroek, T.; Baaten, C.; Coort, S.; Pugh, N.; Schulz, A.; et al. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat. Commun. 2014, 5, 4257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Geffen, J.P.; Brouns, S.L.N.; Batista, J.; McKinney, H.; Kempster, C.; Nagy, M.; Sivapalaratnam, S.; Baaten, C.; Bourry, N.; Frontini, M.; et al. High-throughput elucidation of thrombus formation reveals sources of platelet function variability. Haematologica 2019, 104, 1256–1267. [Google Scholar] [CrossRef]
- Sanchez-Cortes, J.; Mrksich, M. The platelet integrin alphaIIbbeta3 binds to the RGD and AGD motifs in fibrinogen. Chem. Biol. 2009, 16, 990–1000. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Sampietro, S.; Wu, J.; Tang, J.; Gupta, S.; Matzko, C.N.; Tang, C.; Yu, Y.; Brass, L.F.; Zhu, L.; et al. Coordination of platelet agonist signaling during the hemostatic response in vivo. Blood Adv. 2017, 1, 2767–2775. [Google Scholar] [CrossRef] [Green Version]
- Furie, B.; Furie, B.C. Mechanisms of thrombus formation. N. Engl. J. Med. 2008, 359, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Tomaiuolo, M.; Stalker, T.J.; Welsh, J.D.; Diamond, S.L.; Sinno, T.; Brass, L.F. A systems approach to hemostasis: 2. Computational analysis of molecular transport in the thrombus microenvironment. Blood 2014, 124, 1816–1823. [Google Scholar] [CrossRef] [Green Version]
- Mattheij, N.J.; Gilio, K.; van Kruchten, R.; Jobe, S.M.; Wieschhaus, A.J.; Chishti, A.H.; Collins, P.; Heemskerk, J.W.; Cosemans, J.M. Dual mechanism of integrin alphaIIbbeta3 closure in procoagulant platelets. J. Biol. Chem. 2013, 288, 13325–13336. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, D.I.; Kuijpers, M.J.E.; Heemskerk, J.W.M. Platelet calcium signaling by G-protein coupled and ITAM-linked receptors regulating anoctamin-6 and procoagulant activity. Platelets 2021, 32, 863–871. [Google Scholar] [CrossRef]
- Jooss, N.J.; De Simone, I.; Provenzale, I.; Fernandez, D.I.; Brouns, S.L.N.; Farndale, R.W.; Henskens, Y.M.C.; Kuijpers, M.J.E.; Ten Cate, H.; van der Meijden, P.E.J.; et al. Role of Platelet Glycoprotein VI and Tyrosine Kinase Syk in Thrombus Formation on Collagen-Like Surfaces. Int. J. Mol. Sci. 2019, 20, 2788. [Google Scholar] [CrossRef] [Green Version]
- Reilly, M.P.; Sinha, U.; Andre, P.; Taylor, S.M.; Pak, Y.; Deguzman, F.R.; Nanda, N.; Pandey, A.; Stolla, M.; Bergmeier, W.; et al. PRT-060318, a novel Syk inhibitor, prevents heparin-induced thrombocytopenia and thrombosis in a transgenic mouse model. Blood 2011, 117, 2241–2246. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.C.; Damaskinaki, F.N.; Cheung, Y.F.H.; Slater, A.; Watson, S.P. Structure-function relationship of the platelet glycoprotein VI (GPVI) receptor: Does it matter if it is a dimer or monomer? Platelets 2021, 32, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, P.L.R.; Nock, S.H.; Hinds, J.; Garcia-Quintanilla, L.; Smith, C.W.; Campos, J.; Brill, A.; Pike, J.A.; Khan, A.O.; Poulter, N.S.; et al. Low-dose Btk inhibitors selectively block platelet activation by CLEC-2. Haematologica 2021, 106, 208–219. [Google Scholar] [CrossRef] [Green Version]
- Cosemans, J.M.; Munnix, I.C.; Wetzker, R.; Heller, R.; Jackson, S.P.; Heemskerk, J.W. Continuous signaling via PI3K isoforms beta and gamma is required for platelet ADP receptor function in dynamic thrombus stabilization. Blood 2006, 108, 3045–3052. [Google Scholar] [CrossRef] [PubMed]
- Mangin, P.H.; Onselaer, M.B.; Receveur, N.; Le Lay, N.; Hardy, A.T.; Wilson, C.; Sanchez, X.; Loyau, S.; Dupuis, A.; Babar, A.K.; et al. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica 2018, 103, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Andre, P.; Morooka, T.; Sim, D.; Abe, K.; Lowell, C.; Nanda, N.; Delaney, S.; Siu, G.; Yan, Y.; Hollenbach, S.; et al. Critical role for Syk in responses to vascular injury. Blood 2011, 118, 5000–5010. [Google Scholar] [CrossRef] [Green Version]
- van Eeuwijk, J.M.; Stegner, D.; Lamb, D.J.; Kraft, P.; Beck, S.; Thielmann, I.; Kiefer, F.; Walzog, B.; Stoll, G.; Nieswandt, B. The novel oral Syk inhibitor, Bl1002494, protects mice from arterial thrombosis and thromboinflammatory brain infarction. Arter. Thromb. Vasc. Biol. 2016, 36, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Borst, O.; Gawaz, M. Glycoprotein VI—novel target in antiplatelet medication. Pharmacol. Ther. 2021, 217, 107630. [Google Scholar] [CrossRef]
- Voors-Pette, C.; Lebozec, K.; Dogterom, P.; Jullien, L.; Billiald, P.; Ferlan, P.; Renaud, L.; Favre-Bulle, O.; Avenard, G.; Machacek, M.; et al. Safety and Tolerability, Pharmacokinetics, and Pharmacodynamics of ACT017, an Antiplatelet GPVI (Glycoprotein VI) Fab. Arter. Thromb. Vasc. Biol. 2019, 39, 956–964. [Google Scholar] [CrossRef]
- Mahtta, D.; Bavry, A.A. 52-αIIbβ3 (GPIIb-IIIa) Antagonists. In Platelets, 4th ed.; Michelson, A.D., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 957–971. [Google Scholar]
- Harbi, M.H.; Smith, C.W.; Nicolson, P.L.R.; Watson, S.P.; Thomas, M.R. Novel antiplatelet strategies targeting GPVI, CLEC-2 and tyrosine kinases. Platelets 2021, 32, 29–41. [Google Scholar] [CrossRef]
- Coffey, G.P.; Feng, J.; Betz, A.; Pandey, A.; Birrell, M.; Leeds, J.M.; Der, K.; Kadri, S.; Lu, P.; Segal, J.; et al. Cerdulatinib Pharmacodynamics and Relationships to Tumor Response Following Oral Dosing in Patients with Relapsed/Refractory B-cell Malignancies. Clin. Cancer Res. 2019, 25, 1174–1184. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrella, G.; Montague, S.J.; Brown, H.C.; Garcia Quintanilla, L.; Slater, A.; Stegner, D.; Thomas, M.; Heemskerk, J.W.M.; Watson, S.P. Role of Tyrosine Kinase Syk in Thrombus Stabilisation at High Shear. Int. J. Mol. Sci. 2022, 23, 493. https://doi.org/10.3390/ijms23010493
Perrella G, Montague SJ, Brown HC, Garcia Quintanilla L, Slater A, Stegner D, Thomas M, Heemskerk JWM, Watson SP. Role of Tyrosine Kinase Syk in Thrombus Stabilisation at High Shear. International Journal of Molecular Sciences. 2022; 23(1):493. https://doi.org/10.3390/ijms23010493
Chicago/Turabian StylePerrella, Gina, Samantha J. Montague, Helena C. Brown, Lourdes Garcia Quintanilla, Alexandre Slater, David Stegner, Mark Thomas, Johan W. M. Heemskerk, and Steve P. Watson. 2022. "Role of Tyrosine Kinase Syk in Thrombus Stabilisation at High Shear" International Journal of Molecular Sciences 23, no. 1: 493. https://doi.org/10.3390/ijms23010493