
Structural and Dynamic Determinants of Molecular Recognition in Bile Acid-Binding Proteins


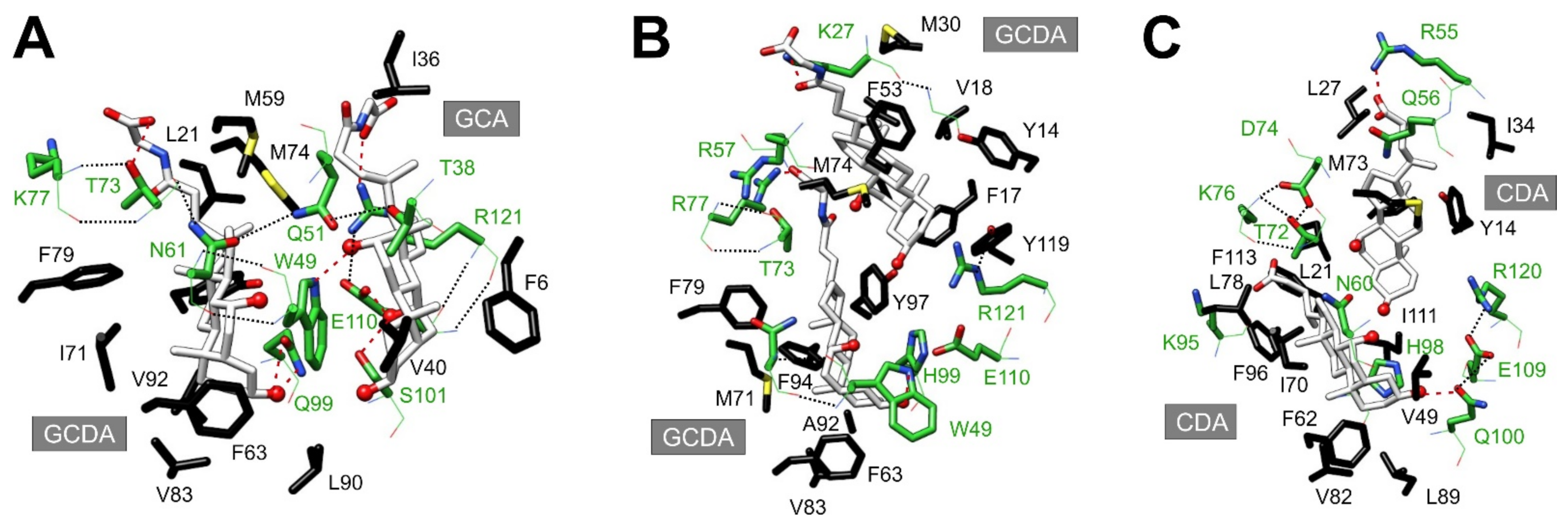
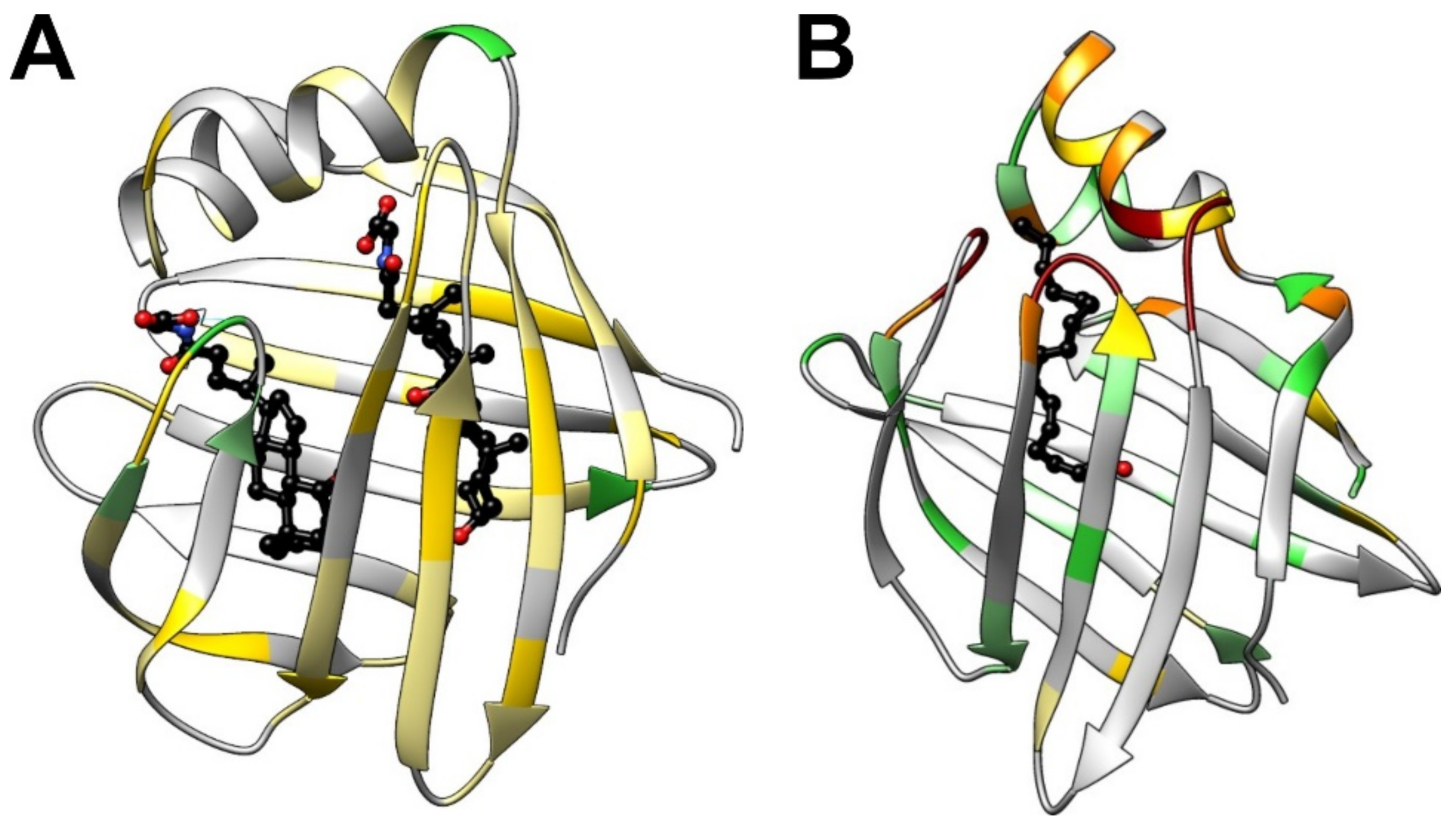
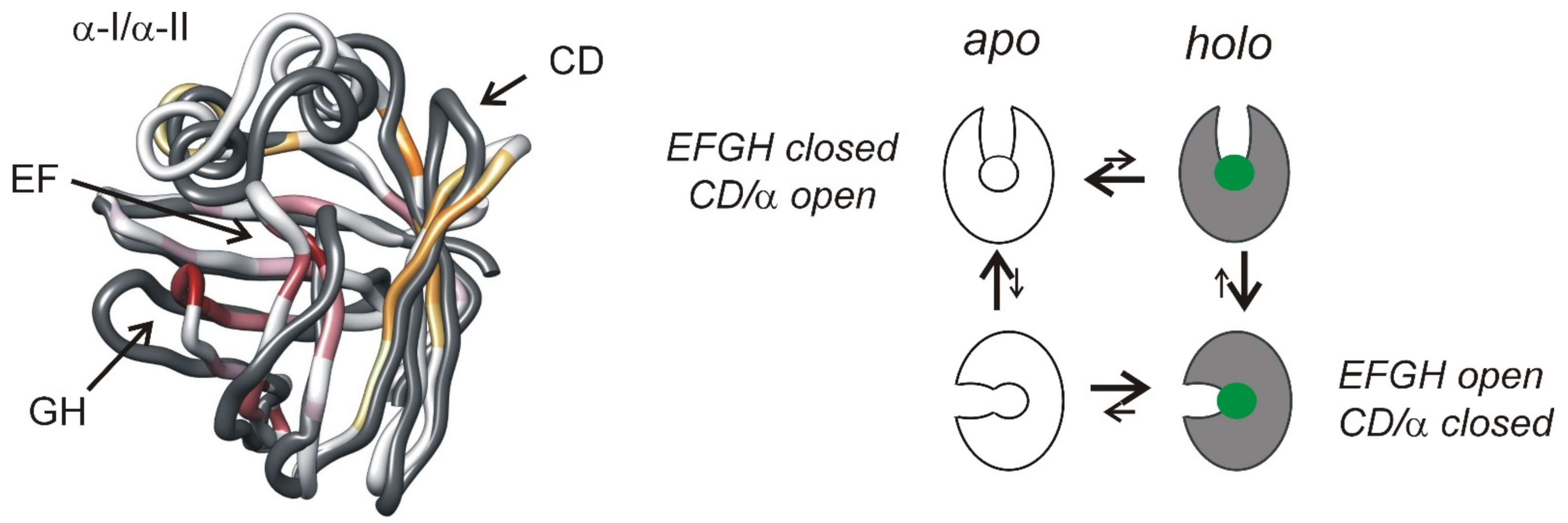
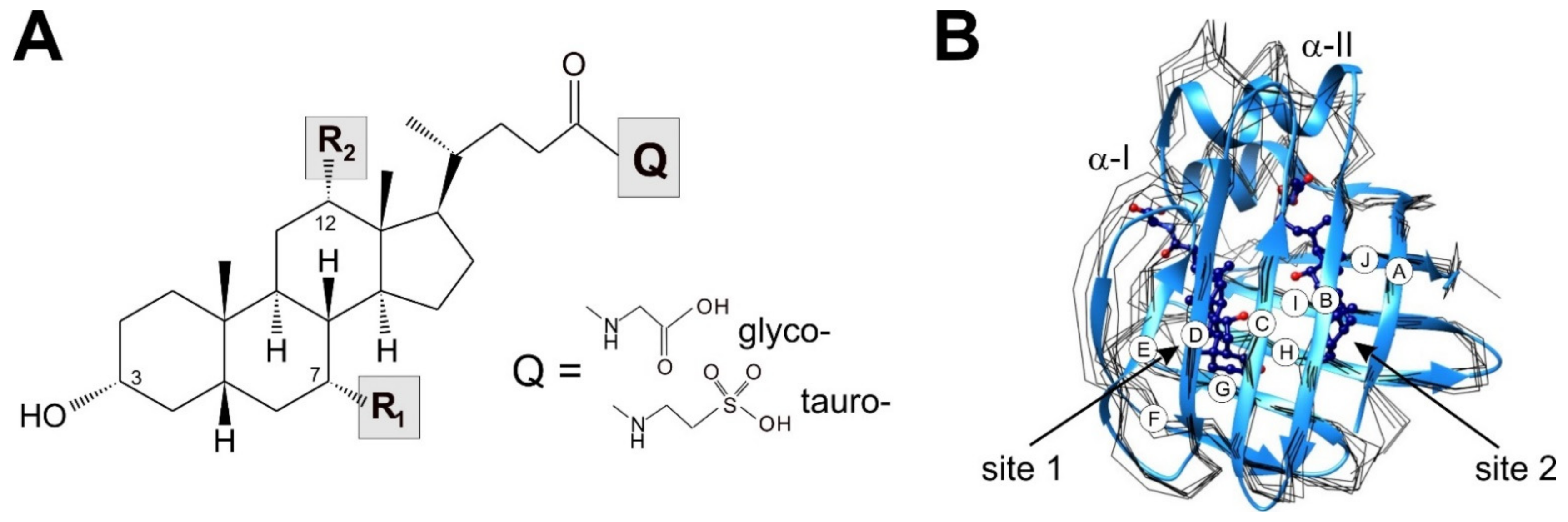{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Positive Binding Cooperativity
3. Site Preference of Bile Salts
4. Conformational Changes of the Protein Backbone upon Bile Salt Binding

5. Binding Cavity in BABP Complexes
6. Backbone Mobility on the Fast Time Scale
7. Conformational Fluctuations on the Millisecond Timescale
8. Ligand Entry and Release Mechanisms in BABPs and Other iLBPs
9. Ligand Transfer between BABPs and the Cell Membrane
10. Concluding Remarks
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BABP | Bile acid binding protein |
| CDA | Chenodeoxycholic acid |
| cI-BABP | Chicken ileal bile acid binding protein |
| cL-BABP | Chicken liver bile acid binding protein |
| CPMG | Carr–Purcell–Meiboom–Gill |
| CR(A)BP | Cellular retinol (retinoic acid) binding protein |
| FABP | Fatty acid binding protein |
| FXR | Farnesoid X receptor |
| FTIR | Fourier-transform infrared |
| GCA | Glycocholic acid |
| GCDA | Glycochenodeoxycholic acid |
| hI-BABP | Human ileal bile acid binding protein |
| IBAT | Ileal bile acid transporter |
| iLBP | Intracellular lipid binding protein |
| NMR | Nuclear magnetic resonance |
| PPAR | Peroxisomal proliferator-activated receptor |
| RAR | Retinoic acid receptor |
References
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borgström, B.; Barrowman, J.A.; Lindström, M. Roles of Bile Acids in Intestinal Lipid Digestion and Absorption. In Sterols and Bile Acids; Danielsson, H., Sjövall, J., Eds.; Elsevier: Amsterdam, The Netherlands, 1985; Volume 12, pp. 405–425. [Google Scholar]
- Houten, S.M.; Watanabe, M.; Auwerx, J. Endocrine functions of bile acids. EMBO J. 2006, 25, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef]
- Chawla, A.; Saez, E.; Evans, R.M. Don’t know much bile-ology. Cell 2000, 103, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Comm. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Stravitz, R.T.; Dent, P.; Hylemon, P.B. Down-regulation of cholesterol 7alpha-hydroxylase (CYP7A1) gene expression by bile acids in primary rat hepatocytes is mediated by the c-Jun N-terminal kinase pathway. J. Biol. Chem. 2001, 276, 15816–15822. [Google Scholar] [CrossRef] [Green Version]
- Qiao, L.; Han, S.I.; Fang, Y.; Park, J.S.; Gupta, S.; Gilfor, D.; Amorino, G.; Valerie, K.; Sealy, L.; Engelhardt, J.F.; et al. Bile acid regulation of C/EBPbeta, CREB, and c-Jun function, via the extracellular signal-regulated kinase and c-Jun NH2-terminal kinase pathways, modulates the apoptotic response of hepatocytes. Mol. Cell. Biol. 2003, 23, 3052–3066. [Google Scholar] [CrossRef] [Green Version]
- Potter, G.D. Bile acid diarrhea. Dig. Dis. 1998, 16, 118–124. [Google Scholar] [CrossRef]
- Alrefai, W.A.; Saksena, S.; Tyagi, S.; Gill, R.K.; Ramaswamy, K.; Dudeja, P.K. Taurodeoxycholate modulates apical Cl−/OH− exchange activity in Caco2 cells. Dig. Dis. Sci. 2007, 52, 1270–1278. [Google Scholar] [CrossRef]
- Kullak-Ublick, G.A.; Stieger, B.; Meier, P.J. Enterohepatic bile salt transporters in normal physiology and liver disease. Gastroenterology 2004, 126, 322–342. [Google Scholar] [CrossRef]
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorak, K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J. Gastroenterol. WJG 2009, 15, 3329–3340. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar]
- Ficker, P.; Wagner, M. Biliary bile acids in hepatobiliary injury—what is the link? J. Hepatol. 2017, 67, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Perino, A.; Demagny, H.; Velazquez-Villegas, L.; Schoonjans, K. Molecular physiology of bile acid signaling in health, disease, and aging. Physiol. Rev. 2021, 101, 683–731. [Google Scholar] [CrossRef]
- Hendrick, A.G.; Muller, I.; Willems, H.; Leonard, P.M.; Irving, S.; Davenport, R.; Ito, T.; Reeves, J.; Wright, S.; Allen, V.; et al. Identification and investigation of novel binding fragments in the fatty acid binding protein (FABP6). J. Med. Chem. 2016, 59, 8094–8102. [Google Scholar] [CrossRef]
- Capaldi, S.; Saccomani, G.; Fessas, D.; Signorelli, M.; Perduca, M.; Monaco, H.L. The X-ray structure of zebrafish (Danio rerio) ileal bile acid-binding protein reveals the presence of binding sites on the surface of the protein molecule. J. Mol. Biol. 2009, 385, 99–116. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, A. Fatty acid induced remodelling within the Human liver fatty acid binding protein. J. Biol. Chem. 2011, 286, 31924–31928. [Google Scholar] [CrossRef] [Green Version]
- Nichesola, D.; Perduca, M.; Capaldi, S.; Carrizo, M.E.; Righetti, P.G.; Monaco, H.L. Crystal structure of chicken liver basic fatty acid-binding protein complexed with cholic acid. Biochemistry 2004, 43, 14072–14079. [Google Scholar] [CrossRef]
- Tassone, G.; Orlandini, M.; Olivucci, M.; Pozzi, C. Validation of recombinant chicken liver bile acid binding protein as a tool for cholic acid hosting. Biomolecules 2021, 11, 645. [Google Scholar] [CrossRef]
- Di Pietro, S.M.; Corsico, B.; Perduca, M.; Monaco, H.L.; Santome, J.A. Structural and biochemical characterization of toad liver basic fatty acid-binding protein. Biochemistry 2003, 42, 8192–8203. [Google Scholar] [CrossRef]
- Horváth, G.; Bencsura, Á.; Simon, Á.; Tochtrop, G.P.; DeKoster, G.T.; Covey, D.F.; Cistola, D.P.; Toke, O. Structural determinants of ligand binding in the ternary complex of human ileal bile acid binding protein with glycocholate and glycochenodeoxycholate obtained from solution NMR. FEBS J. 2016, 283, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Moschetta, A.; Xu, F.; Hagey, L.R.; Berge-Henegouwen, G.P.; van Erpecum, K.J.; Brouwers, J.F.; Cohen, J.C.; Bierman, M.; Hobbs, H.H.; Steinbach, J.H.; et al. A phylogenetic survey of biliary lipids in vertebrates. J. Lipid Res. 2005, 46, 2221–2232. [Google Scholar] [CrossRef] [Green Version]
- Bortolini, O.; Medici, A.; Poli, S. Biotransformations on steroid nucleus of bile acids. Steroids 1997, 62, 564–577. [Google Scholar] [CrossRef]
- Begley, M.; Gahan, C.G.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Dawson, P.A. Animal models to study bile acid metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 895–911. [Google Scholar] [CrossRef]
- Joyce, S.A.; Gahan, C.G. Disease-associated changes in bile acid profiles and links to altered gut microbiota. Dig. Dis. 2017, 35, 169–177. [Google Scholar] [CrossRef]
- Small, D.M.; Dowling, R.H.; Redinger, R.N. The enterohepatic circulation of bile salts. Arch. Intern. Med. 1972, 130, 552–573. [Google Scholar] [CrossRef]
- Alrefai, W.A.; Gill, R.K. Bile acid transporters: Structure, Function, Regulation, and Patophysiological implications. Pharmac. Res. 2007, 24, 1803–1823. [Google Scholar] [CrossRef] [PubMed]
- Banaszak, L.; Winter, N.; Xu, Z.; Bernlohr, D.A.; Cowan, S.; Jones, T.A. Lipid-binding proteins: A family of fatty acid and retinoid transport proteins. Adv. Protein Chem. 1994, 45, 89–151. [Google Scholar] [PubMed]
- Veerkamp, J.H.; Maatman, R.G. Cytoplasmic fatty acid-binding proteins: Their structure and genes. Prog. Lipid Res. 1995, 34, 17–52. [Google Scholar] [CrossRef]
- Nakahara, M.; Furuya, N.; Takagaki, K.; Sugaya, T.; Hirota, K.; Fukamizu, A.; Kanda, T.; Fujii, H.; Sato, R. Ileal bile acid-binding protein, functionally associated with the farnesoid X receptor or the ileal bile acid transporter, regulates bile acid activity in the small intestine. J. Biol. Chem. 2005, 280, 42283–42289. [Google Scholar] [CrossRef]
- Haunerland, N.H.; Spener, F. Fatty acid-binding proteins—Insights from genetic manipulations. Prog. Lipid Res. 2004, 43, 328–349. [Google Scholar] [CrossRef]
- Xu, H.; Diolintzi, A.; Storch, J. Fatty acid-binding proteins: Functional understanding and diagnostic implications. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 407–412. [Google Scholar] [CrossRef]
- Napoli, J.L. Cellular retinoid binding-proteins, CRBP, CRABP, FABP5: Effects on retinoid metabolism, function and related diseases. Pharmacol. Ther. 2017, 173, 19–39. [Google Scholar] [CrossRef] [Green Version]
- Kurz, M.; Brachvogel, V.; Matter, H.; Stengelin, S.; Thüring, H.; Kramer, W. Insights into the bile acid transportation system: The human ileal lipid-binding protein-cholyltaurine complex and its comparison with homologous structures. Proteins 2003, 50, 312–328. [Google Scholar] [CrossRef]
- Eliseo, T.; Ragona, L.; Catalano, M.; Assfalg, M.; Paci, M.; Zetta, L.; Molinari, H.; Cicero, D.O. Structural and dynamic determinants of ligand binding in the ternary complex of chicken liver bile acid binding protein with two bile salts revealed by NMR. Biochemistry 2007, 46, 12557–12567. [Google Scholar] [CrossRef]
- Tochtrop, G.P.; Richter, K.; Tang, C.; Toner, J.J.; Covey, D.F.; Cistola, D.P. Energetics by NMR: Site-specific binding in a positively cooperative system. Proc. Natl. Acad. Sci. USA 2002, 99, 1847–1852. [Google Scholar] [CrossRef] [Green Version]
- Tochtrop, G.P.; DeKoster, G.T.; Covey, D.F.; Cistola, D.P. A single hydroxyl group governs ligand site selectivity in human ileal bile acid binding protein. J. Am. Chem. Soc. 2004, 126, 11024–11029. [Google Scholar] [CrossRef]
- Toke, O.; Monsey, J.D.; DeKoster, G.T.; Tochtrop, G.P.; Tang, C.; Cistola, D.P. Determinants of cooperativity and site-selectivity in human ileal bile acid-binding protein. Biochemistry 2006, 45, 727–737. [Google Scholar] [CrossRef]
- Toke, O.; Monsey, J.D.; Cistola, D.P. Kinetic mechanism of ligand binding in human ileal bile acid binding protein as determined by stopped-flow fluorescence analysis. Biochemistry 2007, 46, 5427–5436. [Google Scholar] [CrossRef]
- Guariento, M.; Assfalg, M.; Zanzoni, S.; Fessas, D.; Longhi, R.; Molinari, H. Chicken ileal bile-acid-binding protein: A promising target of investigation to understand binding co-operativity across the protein family. Biochem. J. 2010, 425, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Cogliati, C.; Ragona, L.; D’Onofrio, M.; Günther, U.; Whittaker, S.; Ludwig, C.; Tomaselli, S.; Assfalg, M.; Molinari, H. Site-specific investigation of the steady-state kinetics and dynamics of the multistep binding of bile acid molecules to a lipid carrier protein. Chem. Eur. J. 2010, 16, 11300–11310. [Google Scholar] [CrossRef]
- Turpin, E.R.; Fang, H.-J.; Thomas, N.R.; Hirst, J.D. Cooperativity and site selectivity in the ileal lipid binding protein. Biochemistry 2013, 52, 4723–4733. [Google Scholar] [CrossRef]
- Kouvatsos, N.; Thurston, V.; Ball, K.; Oldham, N.J.; Thomas, N.R.; Searle, M.S. Bile acid interactions with rabbit ileal lipid binding protein and an engineered helixless variant reveal novel ligand binding properties of a versatile beta-clam shell protein scaffold. J. Mol. Biol. 2007, 371, 1365–1377. [Google Scholar] [CrossRef]
- Capaldi, S.; Guariento, M.; Saccomani, G.; Fessas, D.; Perduca, M.; Monaco, H.L. A single amino acid mutation in zebrafish (Danio rerio) liver bile acid-binding protein can change the stoichiometry of ligand binding. J. Biol. Chem. 2007, 282, 31008–31018. [Google Scholar] [CrossRef] [Green Version]
- Tochtrop, G.P.; Bruns, J.M.; Tang, C.; Covey, D.F.; Cistola, D.P. Steroid ring hydroxylation patterns govern cooperativity in human bile acid binding protein. Biochemistry 2003, 42, 11561–11567. [Google Scholar] [CrossRef]
- Zanzoni, S.; Assfalg, M.; Giorgetti, A.; D’Onofrio, M.; Molinari, H. Structural requirements for cooperativity in ileal bile acid-binding proteins. J. Biol. Chem. 2011, 286, 39307–39317. [Google Scholar] [CrossRef] [Green Version]
- Pedò, M.; D’Onofrio, M.; Ferranti, P.; Molinari, H.; Assfalg, M. Towards the elucidation of molecular determinants of cooperativity in the liver bile acid binding protein. Proteins 2009, 77, 718–731. [Google Scholar] [CrossRef]
- Tomaselli, S.; Assfalg, M.; Pagano, K.; Cogliati, C.; Zanzoni, S.; Molinari, H.; Ragona, L. A disulfide bridge allows for site-selective binding in liver bile acid binding protein thereby stabilising the orientation of key amino acid side chains. Chem. Eur. J. 2012, 18, 2857–2866. [Google Scholar] [CrossRef]
- Horváth, G.; Micsonai, A.; Kardos, J.; Toke, O. Multiple timescale dynamic analysis of functionally impairing mutations in human ileal bile acid-binding protein. FEBS J. 2017, 284, 3637–3661. [Google Scholar]
- Cogliati, C.; Tomaselli, S.; Assfalg, M.; Pedò, M.; Ferranti, P.; Zetta, L.; Molinari, H.; Ragona, L. Disulfide bridge regulates ligand-binding site selectivity in liver bile acid-binding proteins. FEBS J. 2009, 276, 6011–6023. [Google Scholar] [CrossRef]
- Ragona, L.; Catalano, M.; Luppi, M.; Cicero, D.; Eliseo, T.; Foote, J.; Fogolari, F.; Zetta, L.; Molinari, H. NMR dynamic studies suggest that allosteric activation regulates ligand binding in chicken liver bile acid-binding protein. J. Biol. Chem. 2006, 281, 9697–9709. [Google Scholar] [CrossRef] [Green Version]
- Sacchettini, J.C.; Gordon, J.I.; Banaszak, L.J. Crystal Structure of rat intestinal fatty-acid-binding protein. Refinement and analysis of the Escherichia coli-derived protein with bound palmitate. J. Mol. Biol. 1989, 208, 327–339. [Google Scholar] [CrossRef]
- Sacchettini, J.C.; Scapin, G.; Gopaul, D.; Gordon, J.I. Refinement of the structure of Excherichia coli-derived rat intestinal fatty acid binding protein with bound oleate to 1.75-Å resolution. Correlation with the structure of the apoprotein and the protein with bound palmitate. J. Biol. Chem. 1992, 267, 23534–23545. [Google Scholar] [CrossRef]
- Hodsdon, M.E.; Ponder, J.W.; Cistola, D.P. The NMR solution structure of intestinal fatty acid-binding protein complexed with palmitate: Application of a novel distance geometry algorithm. J. Mol. Biol. 1996, 264, 585–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodsdon, M.E.; Cistola, D.P. Discrete backbone disorder in the nuclear magnetic resonance structure of apo intestinal fatty acid-binding protein: Implications for the mechanism of ligand entry. Biochemistry 1997, 36, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Franzoni, L.; Lücke, C.; Pérez, C.; Cavazzini, D.; Rademacher, M.; Ludwig, C.; Spisni, A.; Rossi, G.L.; Rüterjans, H. Structure and backbone dynamics of apo- and holo-cellular retinol-binding protein in solution. J. Biol. Chem. 2002, 277, 21983–21997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horváth, G.; Király, P.; Tárkányi, G.; Toke, O. Internal motions and exchange processes in human ileal bile acid binding protein as studied by backbone 15N nuclear magnetic resonance spectroscopy. Biochemistry 2012, 51, 1848–1861, Erratum in Biochemistry, 51, 10119. [Google Scholar] [CrossRef]
- Hodsdon, M.E.; Cistola, D.P. Ligand binding alters the backbone mobility of intestinal fatty acid-binding protein as monitored by 15N NMR relaxation and 1H exchange. Biochemistry 1997, 36, 2278–2290. [Google Scholar] [CrossRef]
- Lu, J.; Cistola, D.P.; Li, E. Two homologous rat cellular retinol-binding proteins differ in local conformational flexibility. J. Mol. Biol. 2003, 330, 799–812. [Google Scholar] [CrossRef]
- Horváth, G.; Egyed, O.; Tang, C.; Kovács, M.; Micsonai, A.; Kardos, J.; Toke, O. Ligand entry in human ileal bile acid-binding protein is mediated by histidine protonation. Sci. Rep. 2019, 9, 4825. [Google Scholar] [CrossRef]
- Cowan, S.W.; Newcomer, M.E.; Jones, T.A. Crystallographic studies on a family of cellular lipophilic transport proteins. Refinement of P2 myelin protein and the structure determination and refinement of cellular retinol-binding protein in complex with all-trans-retinol. J. Mol. Biol. 1993, 230, 1225–1246. [Google Scholar] [CrossRef]
- Winter, N.S.; Bratt, J.M.; Banaszak, L.J. Crystal structure of holo- and apo-cellular retinol-binding protein II. J. Mol. Biol. 1993, 230, 1247–1259. [Google Scholar] [CrossRef]
- Kleywegt, G.J.; Bergfors, T.; Senn, H.; Le Motte, P.; Gsell, B.; Shudo, K.; Jones, T.A. Crystal structures of cellular retinoic acid binding proteins I and II in complex with all-trans retinoic acid and a synthetic retinoid. Structure 1994, 2, 1241–1258. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Lin, C.L.; Tang, C.; Ponder, J.W.; Kao, J.L.F.; Cistola, D.P.; Li, E. The structure and dynamics of rat apo-cellular retinol-binding protein II in solution: Comparison with the X-ray structure. J. Mol. Biol. 1999, 286, 1179–1195. [Google Scholar] [CrossRef] [Green Version]
- Scapin, G.; Gordon, J.I.; Sacchettini, J.C. Refinement of the structure of recombinant rat intestinal fatty acid-binding apoprotein at 1.2-Å resolution. J. Biol. Chem. 1992, 267, 4253–4269. [Google Scholar] [CrossRef]
- Xu, Z.; Bernlohr, D.A.; Banaszak, L.J. Crystal structure of recombinant murine adipocyte lipid-binding protein. Biochemistry 1992, 31, 3484–3492. [Google Scholar] [CrossRef]
- Young, A.C.M.; Scapin, G.; Kromminga, A.; Patel, S.B.; Veerkamp, J.H.; Sacchettini, J.C. Structural studies on human muscle fatty acid binding protein at 1.4 Å resolution: Binding interactions with three C18 fatty acids. Structure 1994, 2, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Lücke, C.; Huang, S.; Rademacher, M.; Rüterjans, H. New insight into intracellular lipid binding proteins: The role of buried water. Prot. Sci. 2002, 11, 2382–2392. [Google Scholar] [CrossRef]
- Palmer, A.G. NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 2004, 104, 3623–3640. [Google Scholar] [CrossRef]
- Constantine, K.L.; Friedrichs, M.S.; Wittekind, M.; Jamil, H.; Chu, C.H.; Parker, R.A.; Goldfarb, V.; Mueller, L.; Farmer II, B.T. Backbone and side chain dynamics of uncomplexed human adipocyte and muscle fatty acid-binding proteins. Biochemistry 1998, 37, 7965–7980. [Google Scholar] [CrossRef]
- Cai, J.; Lucke, C.; Chen, Z.; Qiao, Y.; Klimtchuk, E.; Hamilton, J.A. Solution structure and backbone dynamics of human liver fatty acid binding protein: Fatty acid binding revisited. Biophys. J. 2012, 102, 2585–2594. [Google Scholar] [CrossRef] [Green Version]
- Horváth, G.; Egyed, O.; Toke, O. Temperature dependence of backbone dynamics in human ileal bile acid-binding protein: Implications for the mechanism of ligand binding. Biochemistry 2014, 53, 5186–5198. [Google Scholar] [CrossRef] [Green Version]
- Eberini, I.; Rocco, A.G.; Ientile, A.R.; Baptista, A.M.; Gianazza, E.; Tomaselli, S.; Molinari, H.; Ragona, L. Conformational and dynamics changes induced by bile acids binding to chicken liver bile acid binding protein. Proteins 2008, 71, 1889–1898. [Google Scholar] [CrossRef]
- Kim, K.; Cistola, D.P.; Frieden, C. Intestinal fatty acid-binding protein: The structure and stability of a helix-less variant. Biochemistry 1996, 35, 7553–7558. [Google Scholar] [CrossRef]
- Baldwin, A.J.; Kay, L.E. NMR spectroscopy brings invisible states into focus. Nature Chem. Biol. 2009, 5, 808–814. [Google Scholar] [CrossRef]
- Lücke, C.; Fushman, D.; Ludwig, C.; Hamilton, J.A.; Sacchettini, J.C.; Rüterjans, H. A comparative study of the backbone dynamics of two closely related lipid binding proteins: Bovine heart fatty acid binding protein and porcine ileal lipid binding protein. Mol. Cell. Biochem. 1999, 192, 109–121. [Google Scholar] [CrossRef]
- Cistola, D.P.; Kim, K.; Rogl, H.; Frieden, C. Fatty acid interactions with a helix-less variant of intestinal fatty acid-binding protein. Biochemistry 1996, 35, 7559–7565. [Google Scholar] [CrossRef] [PubMed]
- Horváth, G.; Biczók, L.; Majer, Z.; Kovács, M.; Micsonai, A.; Kardos, J.; Toke, O. Structural insight into a partially unfolded state preceding aggregation in an intracellular lipid-binding protein. FEBS J. 2017, 284, 3637–3661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Onofrio, M.; Ragona, L.; Fessas, D.; Signorelli, M.; Ugolini, R.; Pedò, M.; Assfalg, M.; Molinari, H. NMR unfolding studies on a liver bile acid binding protein reveal a global two-state unfolding and localized singular behaviors. Arch. Biochem. Biophys. 2009, 481, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Ferrolino, M.C.; Zhuravleva, A.; Budyak, I.L.; Krishnan, B.; Gierasch, L.M. A delicate balance between functionally required flexibility and aggregation risk in a β-rich protein. Biochemistry 2013, 52, 8843–8854. [Google Scholar] [CrossRef] [Green Version]
- Budyak, I.L.; Krishnan, B.; Marcelino-Cruz, A.M.; Ferrolino, M.C.; Zhuravleva, A.; Gierasch, L.M. Early folding events protect aggregation-prone regions of a β-rich protein. Structure 2013, 21, 476–485. [Google Scholar] [CrossRef] [Green Version]
- Gershenson, A.; Gierasch, L.M.; Pastore, A.; Radford, S.E. Energy landscape of functional proteins are inherently risky. Nat. Chem. Biol. 2014, 10, 884–891. [Google Scholar] [CrossRef] [Green Version]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef]
- Krishnan, V.V.; Sukumar, M.; Gierasch, L.M.; Cosman, M. Dynamics of cellular retinoic acid binding protein I on multiple time scales with implications for ligand binding. Biochemistry 2000, 39, 9119–9129. [Google Scholar] [CrossRef]
- Ragona, L.; Pagano, K.; Tomaselli, S.; Favretto, F.; Ceccon, A.; Zanzoni, S.; D’Onofrio, M.; Assfalg, M.; Molinari, H. The role of dynamics in modulating ligand exchange in intracellular lipid binding proteins. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 1268–1278. [Google Scholar] [CrossRef]
- Favretto, F.; Assfalg, M.; Gallo, M.; Cicero, D.O.; D’Onofrio, M.; Molinari, H. Ligand binding promiscuity of human liver fatty acid binding protein: Structural and dynamic insights from an interaction study with glycocholate and oleate. Chembiochem 2013, 14, 1807–1819. [Google Scholar] [CrossRef]
- Zhang, X.; Sui, X.; Yang, D. Probing methyl dynamics from 13C autocorrelated and cross-correlated relaxation. J. Am. Chem. Soc. 2006, 128, 5073–5081. [Google Scholar] [CrossRef]
- Jenkins, A.E.; Hockenberry, J.A.; Nguyen, T.; Bernlohr, D.A. Testing of the portal hypothesis: Analysis of a V32G, F57G, K85G mutant of the fatty acid binding protein of the murine adipocyte. Biochemistry 2002, 41, 2022–2027. [Google Scholar] [CrossRef]
- Menozzi, I.; Polverini, E.; Berni, R. Deciphering protein dynamics changes along the pathway of retinol by cellular retinol-binding proteins 1 and 2. Arch. Biochem. Biphys. 2018, 645, 107–116. [Google Scholar] [CrossRef]
- Sjoelund, V.; Kaltashov, I.A. Transporter-to-trap conversion: A disulphide bond formation in cellular retinoic acid binding protein I mutant triggered by retinoic acid binding irreversibly locks the ligand inside the protein. Biochemistry 2007, 46, 13382–13390. [Google Scholar] [CrossRef] [Green Version]
- Sun, A.Q.; Balasubramaniyan, N.; Liu, C.J.; Shahid, M.; Suchy, F.J. Association of the 16-kDa subunit c of vacuolar proton pump with the ileal Na+-dependent bile acid transporter. J. Biol. Chem. 2004, 279, 16295–16300. [Google Scholar] [CrossRef] [Green Version]
- Ceccon, A.; Busato, M.; Santero, S.P.; D’Onofrio, M.; Musiani, M.; Giorgetti, A.; Assfalg, M. Transient interactions of a cytosolic protein with macromolecular and vesicular cosolutes: Unspecific and specific effects. ChemBioChem 2015, 16, 2633–2645. [Google Scholar] [CrossRef]
- Nolan, V.; Perduca, M.; Monaco, H.L.; Maggio, B.; Montich, G.G. Interactions of chicken liver basic fatty acid-binding protein with lipid membranes. Biochim. Biophys. Acta 2003, 1611, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Pedò, M.; Löhr, F.; D’Onofrio, M.; Assfalg, M.; Dötsch, V.; Molinari, H. NMR studies reveal the role of biomembranes in modulating ligand binding and release by intracellular bile acid binding proteins. J. Mol. Biol. 2009, 394, 852–863. [Google Scholar] [CrossRef]
- Ceccon, A.; D’Onofrio, M.; Zanzoni, S.; Longo, D.L.; Aime, S.; Molinari, H.; Assfalg, M. NMR investigation of the equilibrium partitioning of a water-soluble bile salt protein carrier to phospholipid vesicles. Proteins 2013, 81, 1776–1791. [Google Scholar] [CrossRef]
- Nolan, V.; Perduca, M.; Monaco, H.L.; Montich, G.G. Chicken liver bile acid-binding protein is a compact partly folded state at acidic pH. Its relevance to the interaction with lipid membranes. Biochemistry 2005, 44, 8486–8493. [Google Scholar] [CrossRef]
- Decca, M.B.; Perduca, M.; Monaco, H.L.; Montich, G.G. Conformational changes of chicken liver bile acid-binding protein bound to anionic lipid membrane are coupled to the lipid phase transitions. Biochim. Biophys. Acta 2007, 1768, 1583–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galassi, V.; Nolan, V.; Villarreal, M.A.; Perduca, M.; Monaco, H.L.; Montich, G.G. Kinetics of lipid-membrane binding and conformational change of L-BABP. Biochim. Biopys. Res. Comm. 2009, 382, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, M.A.; Perduca, M.; Monaco, H.L.; Montich, G.G. Binding and interactions if L-BABP to lipid membranes studied by molecular dynamics simulations. Biochim. Biophys. Acta 2008, 1778, 1390–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-K.; Storch, J. Mechanism of free fatty acid transfer from rat heart fatty acid-binding protein to phospholipid membranes (Evidence for a collisional process). J. Biol. Chem. 1992, 267, 20051–20056. [Google Scholar] [CrossRef]
- Storch, J.; Veerkamp, J.H.; Hsu, K.-T. Similar mechanism of fatty acid transfer from human anal rodent fatty acid-binding proteins to membranes: Liver, intestine, heart muscle and adipose tissue FABPs. Mol. Cell. Biochem. 2002, 239, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Jakoby, M.G.; Miller, K.R.; Toner, J.J.; Bauman, A.; Cheng, L.; Cistola, D.P. Ligand-protein electrostatic interactions govern the specificity of retinol- and fatty acid-binding proteins. Biochemistry 1993, 32, 872–878. [Google Scholar] [CrossRef]
- Herr, F.M.; Aronson, J.; Storch, J. Role of portal region lysine residues in electrostatic interactions between heart fatty acid binding protein and phospholipid membranes. Biochemistry 1996, 35, 1296–1303. [Google Scholar] [CrossRef]
- Davies, J.K.; Hagan, R.M.; Wilton, D.C. Effect of charge reversal mutations on the ligand- and membrane-binding properties of liver fatty acid-binding protein. J. Biol. Chem. 2002, 277, 48395–48402. [Google Scholar] [CrossRef] [Green Version]
- Franchini, G.R.; Storch, J.; Corsico, B. The integrity of the α-helical domain of intestinal fatty acid binding protein is essential for the collision-mediated transfer of fatty acids to phospholipid membranes. Biochim. Biopys. Acta 2008, 1781, 192–199. [Google Scholar] [CrossRef] [Green Version]
- D’Onofrio, M.; Zanzoni, S.; Munari, F.; Monaco, H.L.; Assfalg, M.; Capaldi, S. The long variant of human ileal bile acid-binding protein associated with colorectal cancer exhibits sub-cellular localization and lipid binding behaviour distinct from those of the common isoform. BBA Gen. Subj. 2017, 1861, 2315–2324. [Google Scholar] [CrossRef]
- Sessler, R.J.; Noy, N. A ligand-activated nuclear localization signal in cellular retinoic acid binding protein-II. Mol. Cell 2005, 18, 343–353. [Google Scholar] [CrossRef]
- Ayers, S.D.; Nedrow, K.L.; Gillilan, R.E.; Noy, N. Continuous nucleocytoplasmic shuttling underlies transcriptional activation of PPARγ by FABP4. Biochemistry 2007, 46, 6744–6752. [Google Scholar] [CrossRef]
- Dong, D.; Ruuska, S.E.; Levinthal, D.J.; Noy, N. Distinct roles for cellular retinoic acid-binding proteins I and II in regulating signaling by retinoic acid. J. Biol. Chem. 1999, 274, 23695–23698. [Google Scholar] [CrossRef] [Green Version]
- Storch, J.; Thumser, A.E.A. The fatty acid transport function of fatty acid-binding proteins. Biochim. Biophys. Acta 2000, 1486, 28–44. [Google Scholar] [CrossRef] [Green Version]
- Dayhoff, M.O.; Schwartz, R.M.; Orcutt, B.C. A model of evolutionary change in proteins. In Atlas of Protein Sequence and Structure; Dayhoff, M.O., Ed.; National Biomedical Science Foundation: Washington, DC, USA, 1978; Volume 5, pp. 345–352. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toke, O. Structural and Dynamic Determinants of Molecular Recognition in Bile Acid-Binding Proteins. Int. J. Mol. Sci. 2022, 23, 505. https://doi.org/10.3390/ijms23010505
Toke O. Structural and Dynamic Determinants of Molecular Recognition in Bile Acid-Binding Proteins. International Journal of Molecular Sciences. 2022; 23(1):505. https://doi.org/10.3390/ijms23010505
Chicago/Turabian StyleToke, Orsolya. 2022. "Structural and Dynamic Determinants of Molecular Recognition in Bile Acid-Binding Proteins" International Journal of Molecular Sciences 23, no. 1: 505. https://doi.org/10.3390/ijms23010505