
Age-Related DNA Methylation in Normal Kidney Tissue Identifies Epigenetic Cancer Risk Susceptibility Loci in the ANKRD34B and ZIC1 Genes

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Biometrical Analysis
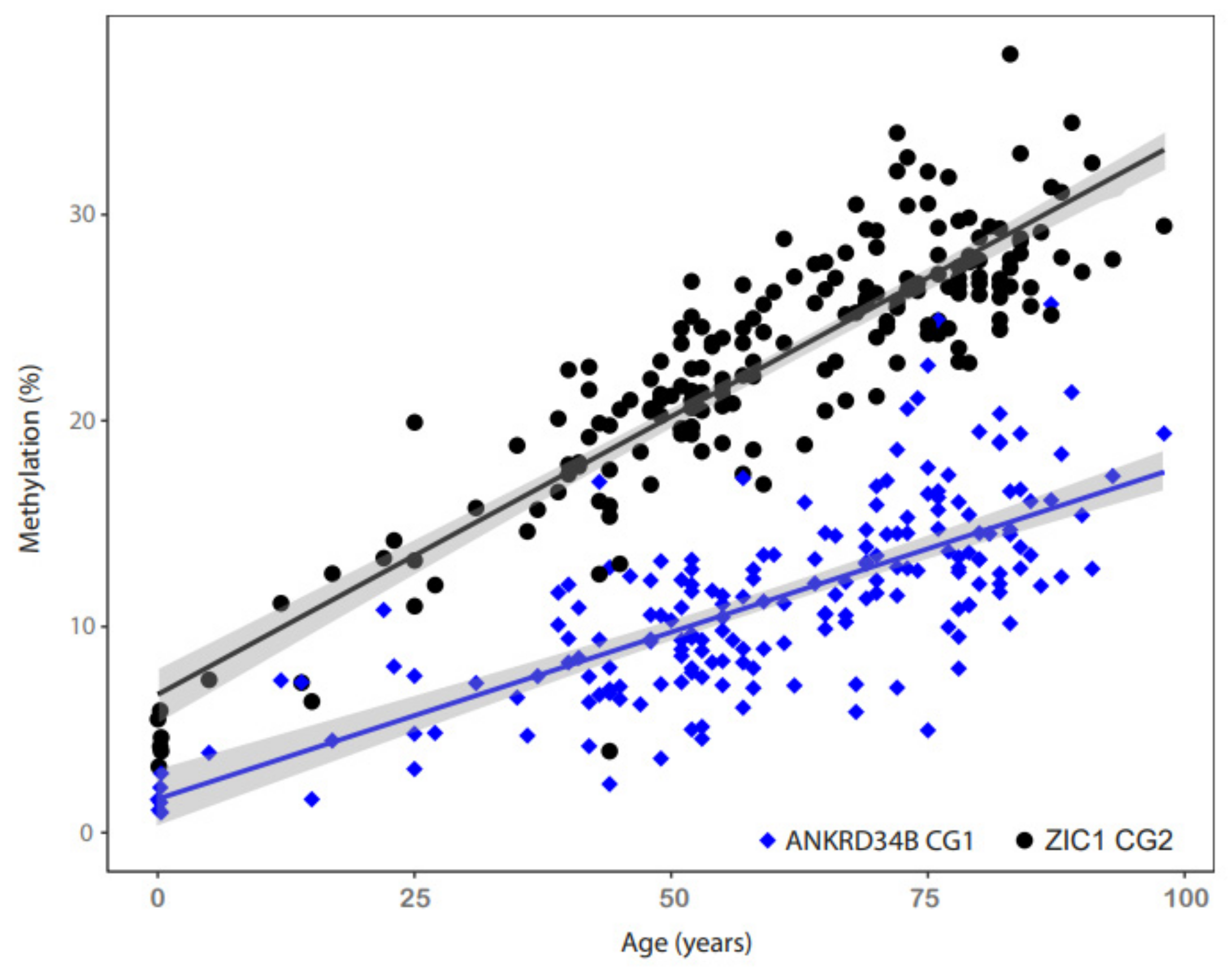
2.2. ANKRD34B and ZIC1 Loci Show a Strong Correlation with Age in Normal Kidney Tissues
2.3. Chronological Age Prediction of Normal Kidney Tissues
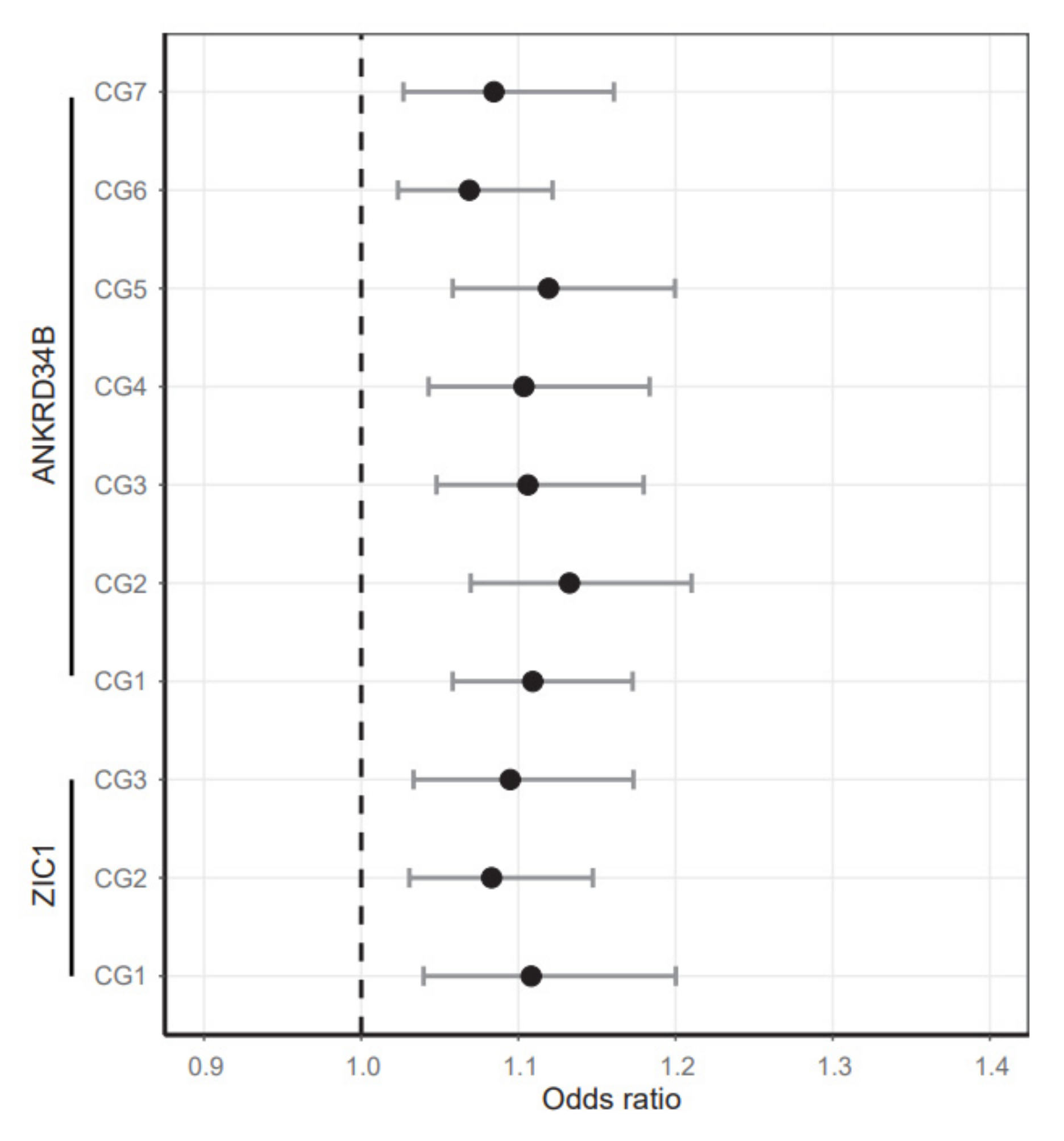
2.4. Age-Independent Increase of ANKRD34B and ZIC1 Methylation in Normal High-Risk Tissues
2.5. Analysis of CpG-Specific Methylation in Paired Tumor-Adjacent and Tumor Tissue Samples Shows Heterogeneous Alterations
2.6. Analysis of Human Cancer Cell Lines
3. Discussion
4. Methods
4.1. In Silico Analysis for Candidate Identification
4.2. Primary Cells and Tumor Cell Lines
4.3. Study Design
4.4. Analysis of DNA Methylation
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Jones, A.; Evans, I.; Reisel, D.; Dillner, J.; Sundstrom, K.; Steyerberg, E.W.; Vergouwe, Y.; Wegwarth, O.; Rebitschek, F.G.; et al. Epigenome-based cancer risk prediction: Rationale, opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Kotecha, R.R.; Motzer, R.J.; Voss, M.H. Towards individualized therapy for metastatic renal cell carcinoma. Nat. Rev. Clin. Oncol. 2019, 16, 621–633. [Google Scholar] [CrossRef]
- Pischon, T.; Lahmann, P.H.; Boeing, H.; Tjonneland, A.; Halkjaer, J.; Overvad, K.; Klipstein-Grobusch, K.; Linseisen, J.; Becker, N.; Trichopoulou, A.; et al. Body size and risk of renal cell carcinoma in the European Prospective Investigation into Cancer and Nutrition (EPIC). Int. J. Cancer 2006, 118, 728–738. [Google Scholar] [CrossRef] [Green Version]
- Hunt, J.D.; van der Hel, O.L.; McMillan, G.P.; Boffetta, P.; Brennan, P. Renal cell carcinoma in relation to cigarette smoking: Meta-analysis of 24 studies. Int. J. Cancer 2005, 114, 101–108. [Google Scholar] [CrossRef]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Moller, S.; Unger, R.H.; et al. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 2016, 315, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Joosten, S.C.; Smits, K.M.; Aarts, M.J.; Melotte, V.; Koch, A.; Tjan-Heijnen, V.C.; van Engeland, M. Epigenetics in renal cell cancer: Mechanisms and clinical applications. Nat. Rev. Urol. 2018, 15, 430–451. [Google Scholar] [CrossRef]
- Waki, T.; Tamura, G.; Sato, M.; Motoyama, T. Age-related methylation of tumor suppressor and tumor-related genes: An analysis of autopsy samples. Oncogene 2003, 22, 4128–4133. [Google Scholar] [CrossRef] [Green Version]
- Atschekzei, F.; Hennenlotter, J.; Janisch, S.; Grosshennig, A.; Trankenschuh, W.; Waalkes, S.; Peters, I.; Dork, T.; Merseburger, A.S.; Stenzl, A.; et al. SFRP1 CpG island methylation locus is associated with renal cell cancer susceptibility and disease recurrence. Epigenetics 2012, 7, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Serth, J.; Peters, I.; Dubrowinskaja, N.; Reese, C.; Albrecht, K.; Klintschar, M.; Lafos, M.; Grote, A.; Becker, A.; Hennenlotter, J.; et al. Age-, tumor-, and metastatic tissue-associated DNA hypermethylation of a T-box brain 1 locus in human kidney tissue. Clin. Epigenetics 2020, 12, 33. [Google Scholar] [CrossRef] [Green Version]
- TCGA. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Al-Shaibi, N.; Ghosh, S.K. A novel phosphoprotein is induced during bone marrow commitment to dendritic cells. Biochem. Biophys. Res. Commun. 2004, 321, 26–30. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, B.; Wei, D.; Zhang, Z. Identification of Methylated Gene Biomarkers in Patients with Alzheimer’s Disease Based on Machine Learning. BioMed Res. Int. 2020, 2020, 8348147. [Google Scholar] [CrossRef] [Green Version]
- Nikitina, A.S.; Sharova, E.I.; Danilenko, S.A.; Butusova, T.B.; Vasiliev, A.O.; Govorov, A.V.; Prilepskaya, E.A.; Pushkar, D.Y.; Kostryukova, E.S. Novel RNA biomarkers of prostate cancer revealed by RNA-seq analysis of formalin-fixed samples obtained from Russian patients. Oncotarget 2017, 8, 32990–33001. [Google Scholar] [CrossRef]
- Ali, R.G.; Bellchambers, H.M.; Arkell, R.M. Zinc fingers of the cerebellum (Zic): Transcription factors and co-factors. Int. J. Biochem. Cell Biol. 2012, 44, 2065–2068. [Google Scholar] [CrossRef]
- Gan, L.; Chen, S.; Zhong, J.; Wang, X.; Lam, E.K.; Liu, X.; Zhang, J.; Zhou, T.; Yu, J.; Si, J.; et al. ZIC1 is downregulated through promoter hypermethylation, and functions as a tumor suppressor gene in colorectal cancer. PLoS ONE 2011, 6, e16916. [Google Scholar] [CrossRef]
- Wang, L.J.; Jin, H.C.; Wang, X.; Lam, E.K.; Zhang, J.B.; Liu, X.; Chan, F.K.; Si, J.M.; Sung, J.J. ZIC1 is downregulated through promoter hypermethylation in gastric cancer. Biochem. Biophys. Res. Commun. 2009, 379, 959–963. [Google Scholar] [CrossRef]
- Hou, Y.; Chen, K.; Liao, R.; Li, Y.; Yang, H.; Gong, J. LINC01419-mediated epigenetic silencing of ZIC1 promotes metastasis in hepatocellular carcinoma through the PI3K/Akt signaling pathway. Lab. Invest. 2021, 101, 570–587. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Jiang, J.X.; Ma, H.; Han, J.; Sun, Z.Y.; Liu, Z.M.; Xu, Z.G. Role of ZIC1 methylation in hepatocellular carcinoma and its clinical significance. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2014, 35, 7429–7433. [Google Scholar] [CrossRef]
- Qiang, W.; Zhao, Y.; Yang, Q.; Liu, W.; Guan, H.; Lv, S.; Ji, M.; Shi, B.; Hou, P. ZIC1 is a putative tumor suppressor in thyroid cancer by modulating major signaling pathways and transcription factor FOXO3a. J. Clin. Endocrinol. Metab. 2014, 99, E1163–E1172. [Google Scholar] [CrossRef] [Green Version]
- Pourebrahim, R.; Van Dam, K.; Bauters, M.; De Wever, I.; Sciot, R.; Cassiman, J.J.; Tejpar, S. ZIC1 gene expression is controlled by DNA and histone methylation in mesenchymal proliferations. FEBS Lett. 2007, 581, 5122–5126. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.Y.; Kirschner, M.B.; Cheng, N.C.; Gattani, S.; Klebe, S.; Edelman, J.J.; Vallely, M.P.; McCaughan, B.C.; Jin, H.C.; van Zandwijk, N.; et al. ZIC1 is silenced and has tumor suppressor function in malignant pleural mesothelioma. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2013, 8, 1317–1328. [Google Scholar] [CrossRef] [Green Version]
- Baribault, C.; Ehrlich, K.C.; Ponnaluri, V.K.C.; Pradhan, S.; Lacey, M.; Ehrlich, M. Developmentally linked human DNA hypermethylation is associated with down-modulation, repression, and upregulation of transcription. Epigenetics 2018, 13, 275–289. [Google Scholar] [CrossRef]
- Dick, S.; Verhoef, L.; De Strooper, L.M.; Ciocanea-Teodorescu, I.; Wisman, G.B.A.; Meijer, C.J.; Bleeker, M.C.; Steenbergen, R.D.; Heideman, D.A. Evaluation of six methylation markers derived from genome-wide screens for detection of cervical precancer and cancer. Epigenomics 2020, 12, 1569–1578. [Google Scholar] [CrossRef]
- Verhoef, L.; Bleeker, M.C.G.; Polman, N.; Steenbergen, R.D.M.; Meijer, C.; Melchers, W.J.G.; Bekkers, R.L.; Molijn, A.C.; Quint, W.G.; van Kemenade, F.J.; et al. Performance of DNA methylation analysis of ASCL1, LHX8, ST6GALNAC5, GHSR, ZIC1 and SST for the triage of HPV-positive women: Results from a Dutch primary HPV-based screening cohort. Int. J. Cancer 2022, 150, 440–449. [Google Scholar] [CrossRef]
- Verlaat, W.; Snijders, P.J.F.; Novianti, P.W.; Wilting, S.M.; De Strooper, L.M.A.; Trooskens, G.; Vandersmissen, J.; Van Criekinge, W.; Wisman, G.B.A.; Meijer, C.; et al. Genome-wide DNA Methylation Profiling Reveals Methylation Markers Associated with 3q Gain for Detection of Cervical Precancer and Cancer. Clin. Cancer Res. 2017, 23, 3813–3822. [Google Scholar] [CrossRef] [Green Version]
- van den Helder, R.; Wever, B.M.M.; van Trommel, N.E.; van Splunter, A.P.; Mom, C.H.; Kasius, J.C.; Bleeker, M.C.G.; Steenbergen, R.D.M. Non-invasive detection of endometrial cancer by DNA methylation analysis in urine. Clin. Epigenetics 2020, 12, 165. [Google Scholar] [CrossRef]
- Lin, Z.; Luo, M.; Chen, X.; He, X.; Qian, Y.; Lai, S.; Si, J.; Chen, S. Combined Detection of Plasma ZIC1, HOXD10 and RUNX3 Methylation is a Promising Strategy for Early Detection of Gastric Cancer and Precancerous Lesions. J. Cancer 2017, 8, 1038–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Lin, Z.; Xue, M.; Si, J.; Chen, S. Zic1 Promoter Hypermethylation in Plasma DNA Is a Potential Biomarker for Gastric Cancer and Intraepithelial Neoplasia. PLoS ONE 2015, 10, e0133906. [Google Scholar] [CrossRef] [PubMed]
- van den Helder, R.; van Trommel, N.E.; van Splunter, A.P.; Lissenberg-Witte, B.I.; Bleeker, M.C.G.; Steenbergen, R.D.M. Methylation analysis in urine fractions for optimal CIN3 and cervical cancer detection. Papillomavirus Res. 2020, 9, 100193. [Google Scholar] [CrossRef] [PubMed]
- van der Zee, R.P.; van Noesel, C.J.M.; Martin, I.; Ter Braak, T.J.; Heideman, D.A.M.; de Vries, H.J.C.; Prins, J.M.; Steenbergen, R.D.M. DNA methylation markers have universal prognostic value for anal cancer risk in HIV-negative and HIV-positive individuals. Mol. Oncol. 2021, 15, 3024–3036. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rodero, S.; Fernandez, A.F.; Fernandez-Morera, J.L.; Castro-Santos, P.; Bayon, G.F.; Ferrero, C.; Urdinguio, R.G.; Gonzalez-Marquez, R.; Suarez, C.; Fernandez-Vega, I.; et al. DNA methylation signatures identify biologically distinct thyroid cancer subtypes. J. Clin. Endocrinol. Metab. 2013, 98, 2811–2821. [Google Scholar] [CrossRef]
- Paluszczak, J.; Wisniewska, D.; Kostrzewska-Poczekaj, M.; Kiwerska, K.; Grenman, R.; Mielcarek-Kuchta, D.; Jarmuz-Szymczak, M. Prognostic significance of the methylation of Wnt pathway antagonists-CXXC4, DACT2, and the inhibitors of sonic hedgehog signaling-ZIC1, ZIC4, and HHIP in head and neck squamous cell carcinomas. Clin. Oral Investig. 2017, 21, 1777–1788. [Google Scholar] [CrossRef] [Green Version]
- Kitchen, M.O.; Bryan, R.T.; Haworth, K.E.; Emes, R.D.; Luscombe, C.; Gommersall, L.; Cheng, K.K.; Zeegers, M.P.; James, N.D.; Devall, A.J.; et al. Methylation of HOXA9 and ISL1 Predicts Patient Outcome in High-Grade Non-Invasive Bladder Cancer. PLoS ONE 2015, 10, e0137003. [Google Scholar] [CrossRef] [Green Version]
- Schneider, B.G.; Mera, R.; Piazuelo, M.B.; Bravo, J.C.; Zabaleta, J.; Delgado, A.G.; Bravo, L.E.; Wilson, K.T.; El-Rifai, W.; Peek, R.M., Jr.; et al. DNA Methylation Predicts Progression of Human Gastric Lesions. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1607–1613. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.L.; Gu, F.; Kirma, N.B.; Ruan, J.; Chen, C.L.; Wang, H.C.; Liao, Y.P.; Chang, C.C.; Yu, M.H.; Pilrose, J.M.; et al. Comprehensive methylome analysis of ovarian tumors reveals hedgehog signaling pathway regulators as prognostic DNA methylation biomarkers. Epigenetics 2013, 8, 624–634. [Google Scholar] [CrossRef] [Green Version]
- Joehanes, R.; Just, A.C.; Marioni, R.E.; Pilling, L.C.; Reynolds, L.M.; Mandaviya, P.R.; Guan, W.; Xu, T.; Elks, C.E.; Aslibekyan, S.; et al. Epigenetic Signatures of Cigarette Smoking. Circulation. Cardiovasc. Genet. 2016, 9, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Markunas, C.A.; Semick, S.A.; Quach, B.C.; Tao, R.; Deep-Soboslay, A.; Carnes, M.U.; Bierut, L.J.; Hyde, T.M.; Kleinman, J.E.; Johnson, E.O.; et al. Genome-wide DNA methylation differences in nucleus accumbens of smokers vs. nonsmokers. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2021, 46, 554–560. [Google Scholar] [CrossRef]
- Zheng, Y.; Huang, G.; Silva, T.C.; Yang, Q.; Jiang, Y.Y.; Koeffler, H.P.; Lin, D.C.; Berman, B.P. A pan-cancer analysis of CpG Island gene regulation reveals extensive plasticity within Polycomb target genes. Nat. Commun. 2021, 12, 2485. [Google Scholar] [CrossRef]
- Hong, S.R.; Jung, S.E.; Lee, E.H.; Shin, K.J.; Yang, W.I.; Lee, H.Y. DNA methylation-based age prediction from saliva: High age predictability by combination of 7 CpG markers. Forensic Sci. Int. Genet. 2017, 29, 118–125. [Google Scholar] [CrossRef]
- Peters, I.; Rehmet, K.; Wilke, N.; Kuczyk, M.; Hennenlotter, J.; Eilers, T.; Machtens, S.; Jonas, U.; Serth, J. RASSF1A promoter methylation and expression analysis in normal and neoplastic kidney indicates a role in early tumorigenesis. Mol. Cancer 2007, 6, 49. [Google Scholar] [CrossRef] [Green Version]
- Bar, W.; Kratzer, A.; Machler, M.; Schmid, W. Postmortem stability of DNA. Forensic. Sci. Int. 1988, 39, 59–70. [Google Scholar] [CrossRef]
- Lokk, K.; Modhukur, V.; Rajashekar, B.; Martens, K.; Magi, R.; Kolde, R.; Koltsina, M.; Nilsson, T.K.; Vilo, J.; Salumets, A.; et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014, 15, r54. [Google Scholar] [CrossRef] [Green Version]
- Slieker, R.C.; Relton, C.L.; Gaunt, T.R.; Slagboom, P.E.; Heijmans, B.T. Age-related DNA methylation changes are tissue-specific with ELOVL2 promoter methylation as exception. Epigenetics Chromatin 2018, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Teschendorff, A.E.; Jones, A.; Fiegl, H.; Sargent, A.; Zhuang, J.J.; Kitchener, H.C.; Widschwendter, M. Epigenetic variability in cells of normal cytology is associated with the risk of future morphological transformation. Genome Med. 2012, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- Marumo, T.; Hoshino, J.; Kawarazaki, W.; Nishimoto, M.; Ayuzawa, N.; Hirohama, D.; Yamanouchi, M.; Ubara, Y.; Okaneya, T.; Fujii, T.; et al. Methylation pattern of urinary DNA as a marker of kidney function decline in diabetes. BMJ Open Diabetes Res. Care 2020, 8, e001501. [Google Scholar] [CrossRef]
- Gebauer, K.; Peters, I.; Dubrowinskaja, N.; Hennenlotter, J.; Abbas, M.; Scherer, R.; Tezval, H.; Merseburger, A.S.; Stenzl, A.; Kuczyk, M.A.; et al. Hsa-mir-124-3 CpG island methylation is associated with advanced tumours and disease recurrence of patients with clear cell renal cell carcinoma. Br. J. Cancer 2013, 108, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Tezval, H.; Dubrowinskaja, N.; Peters, I.; Reese, C.; Serth, K.; Atschekzei, F.; Hennenlotter, J.; Stenzl, A.; Kuczyk, M.A.; Serth, J. Tumor Specific Epigenetic Silencing of Corticotropin Releasing Hormone -Binding Protein in Renal Cell Carcinoma: Association of Hypermethylation and Metastasis. PLoS ONE 2016, 11, e0163873. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 21 March 2022).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene. | Chrom. | Pos. | HM450K | R | R2 | p.adj* | N | Abbr |
|---|---|---|---|---|---|---|---|---|
| ANKRD34B | chr5 | 79,866,347 | 0.75 | 0.56 | 0 | 214 | CG1 | |
| chr5 | 79,866,354 | 0.72 | 0.52 | 0 | 214 | CG2 | ||
| chr5 | 79,866,359 | 0.74 | 0.55 | 0 | 214 | CG3 | ||
| chr5 | 79,866,366 | 0.72 | 0.51 | 0 | 214 | CG4 | ||
| chr5 | 79,866,368 | cg21800232 | 0.70 | 0.49 | 0 | 214 | CG5 | |
| chr5 | 79,866,373 | 0.73 | 0.53 | 0 | 214 | CG6 | ||
| chr5 | 79,866,379 | cg25316339 | 0.61 | 0.37 | 0 | 214 | CG7 | |
| ZIC1 | chr3 | 147,126,196 | 0.70 | 0.49 | 0 | 214 | CG1 | |
| chr3 | 147,126,206 | cg16181396 | 0.88 | 0.77 | 0 | 214 | CG2 | |
| chr3 | 147,126,218 | 0.74 | 0.55 | 0 | 214 | CG3 |
| CpG Site | Mean N | Mean adN | Diff. Mean | OR | CI Low | CI High | p-Val | p-Val Bonf. | p-Val Age | p-Val Sex | n N | n adN |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ANKRD34B | ||||||||||||
| CG1 | 11.4 | 15.0 | 3.6 | 1.11 | 1.06 | 1.17 | 0.000 | 0.001 | 0.425 | 0.549 | 214 | 157 |
| CG2 | 9.9 | 13.1 | 3.2 | 1.13 | 1.07 | 1.21 | 0.000 | 0.001 | 0.492 | 0.576 | 214 | 157 |
| CG3 | 9.2 | 11.9 | 2.6 | 1.11 | 1.05 | 1.18 | 0.001 | 0.006 | 0.776 | 0.712 | 214 | 157 |
| CG4 | 6.8 | 9.1 | 2.3 | 1.10 | 1.04 | 1.18 | 0.002 | 0.016 | 0.932 | 0.690 | 214 | 157 |
| CG5 | 6.7 | 9.6 | 2.9 | 1.12 | 1.06 | 1.20 | 0.000 | 0.003 | 0.914 | 0.675 | 214 | 157 |
| CG6 | 11.6 | 13.8 | 2.2 | 1.07 | 1.02 | 1.12 | 0.004 | 0.029 | 0.814 | 0.693 | 214 | 157 |
| CG7 | 7.3 | 9.2 | 1.9 | 1.08 | 1.03 | 1.16 | 0.010 | 0.071 | 0.446 | 0.815 | 214 | 157 |
| ZIC1 | ||||||||||||
| CG1 | 14.7 | 16.8 | 2.1 | 1.11 | 1.04 | 1.20 | 0.006 | 0.017 | 0.981 | 0.822 | 214 | 145 |
| CG2 | 22.8 | 25.5 | 2.7 | 1.08 | 1.03 | 1.15 | 0.004 | 0.011 | 0.257 | 0.801 | 214 | 145 |
| CG3 | 18.8 | 20.7 | 2.0 | 1.09 | 1.03 | 1.17 | 0.005 | 0.015 | 0.871 | 0.968 | 214 | 145 |
| Region. | CpG | Diff. Mean | CI Low | CI High | p-Value | p-Value (Adj.) |
|---|---|---|---|---|---|---|
| ANKRD34B | CG1 | 11.4 | 8.3 | 14.6 | 4.80 × 10−11 | 1.44 × 10−10 |
| CG2 | 11.9 | 8.8 | 15.0 | 2.90 × 10−12 | 1.74 × 10−11 | |
| CG3 | 10.4 | 7.5 | 13.3 | 5.41 × 10−11 | 1.44 × 10−10 | |
| CG4 | 11.1 | 8.3 | 13.9 | 2.09 × 10−12 | 1.46 × 10−11 | |
| CG5 | 11.1 | 8.1 | 14.0 | 7.10 × 10−12 | 3.55 × 10−11 | |
| CG6 | 10.5 | 7.7 | 13.4 | 1.89 × 10−11 | 7.58 × 10−11 | |
| CG7 | 7.7 | 5.1 | 10.3 | 4.41 × 10−08 | 4.41 × 10−08 | |
| ZIC1 | CG1 | 6.2 | 3.3 | 9.0 | 3.38 × 10−05 | 3.38 × 10−05 |
| CG2 | 8.4 | 5.8 | 11.1 | 3.93 × 10−09 | 1.18 × 10−08 | |
| CG3 | 7.6 | 4.9 | 10.3 | 1.17 × 10−07 | 2.35 × 10−07 |
| Tissue | N | adN | adN*/T | ||||
|---|---|---|---|---|---|---|---|
| Gene | ANKRD34B | ZIC1 | ANKRD34B | ZIC1 | ANKRD34B | ZIC1 | |
| Total (n) | 214 | 214 | 157 | 145 | 143 | 125 | |
| Age (y) | Median (min–max) | 61 (0–98) | 60 (0–99) | 66 (35–91) | 65 (35–91) | 66 (35–91) | 66 (35–91) |
| Sex (n) | female (%) | 77 (36.0) | 72 (33.6) | 58 (36.9) | 53 (36.6) | 53 (37.1) | 46 (36.8) |
| male (%) | 137 (64.0) | 142 (66.4) | 99 (63.1) | 92 (63.4) | 90 (62.9) | 79 (63.2) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serth, J.; Peters, I.; Hill, B.; Hübscher, T.; Hennenlotter, J.; Klintschar, M.; Kuczyk, M.A. Age-Related DNA Methylation in Normal Kidney Tissue Identifies Epigenetic Cancer Risk Susceptibility Loci in the ANKRD34B and ZIC1 Genes. Int. J. Mol. Sci. 2022, 23, 5327. https://doi.org/10.3390/ijms23105327
Serth J, Peters I, Hill B, Hübscher T, Hennenlotter J, Klintschar M, Kuczyk MA. Age-Related DNA Methylation in Normal Kidney Tissue Identifies Epigenetic Cancer Risk Susceptibility Loci in the ANKRD34B and ZIC1 Genes. International Journal of Molecular Sciences. 2022; 23(10):5327. https://doi.org/10.3390/ijms23105327
Chicago/Turabian StyleSerth, Jürgen, Inga Peters, Bastian Hill, Tatjana Hübscher, Jörg Hennenlotter, Michael Klintschar, and Markus Antonius Kuczyk. 2022. "Age-Related DNA Methylation in Normal Kidney Tissue Identifies Epigenetic Cancer Risk Susceptibility Loci in the ANKRD34B and ZIC1 Genes" International Journal of Molecular Sciences 23, no. 10: 5327. https://doi.org/10.3390/ijms23105327
APA StyleSerth, J., Peters, I., Hill, B., Hübscher, T., Hennenlotter, J., Klintschar, M., & Kuczyk, M. A. (2022). Age-Related DNA Methylation in Normal Kidney Tissue Identifies Epigenetic Cancer Risk Susceptibility Loci in the ANKRD34B and ZIC1 Genes. International Journal of Molecular Sciences, 23(10), 5327. https://doi.org/10.3390/ijms23105327