
Zebrafish: A New Promise to Study the Impact of Metabolic Disorders on the Brain


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
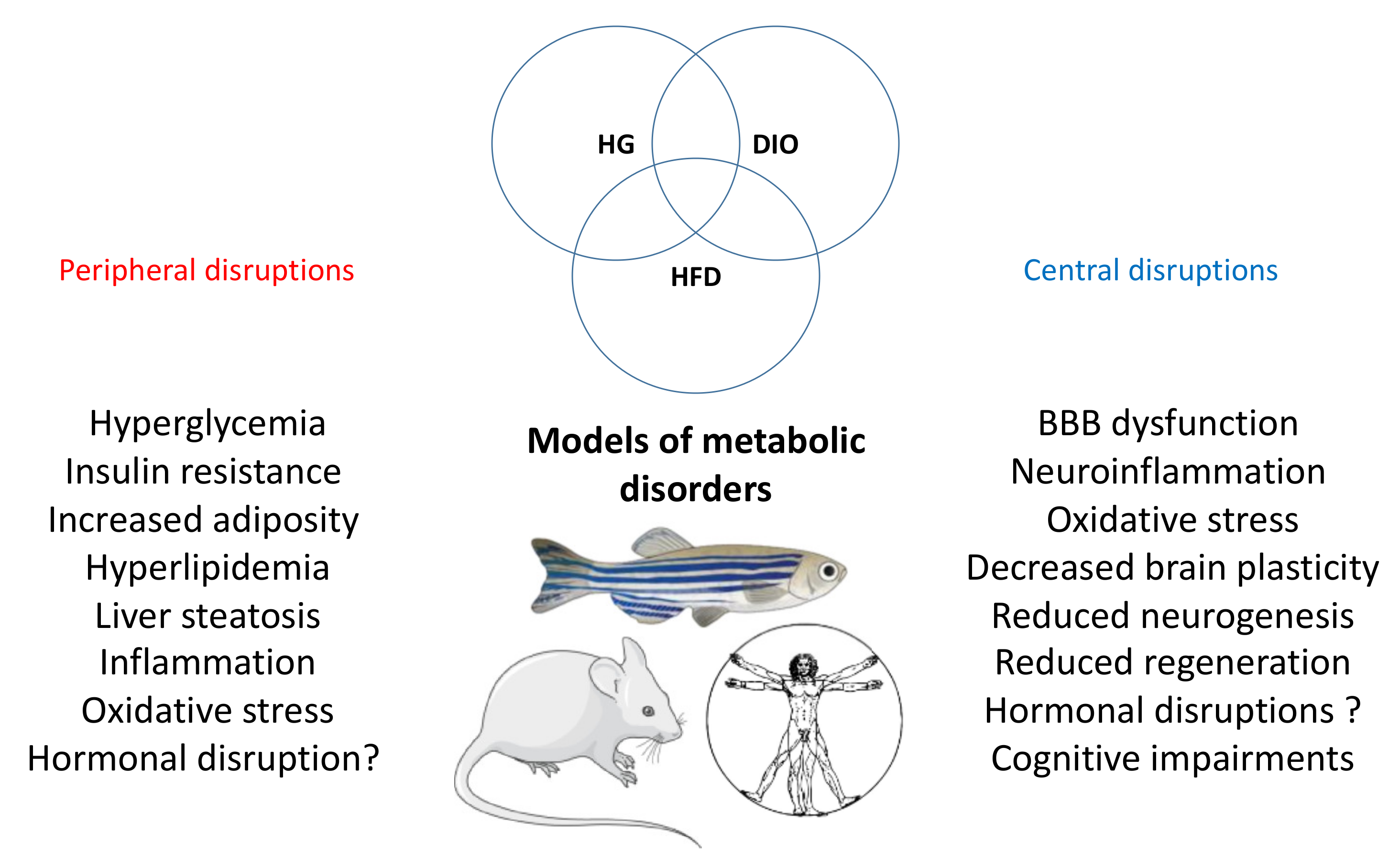
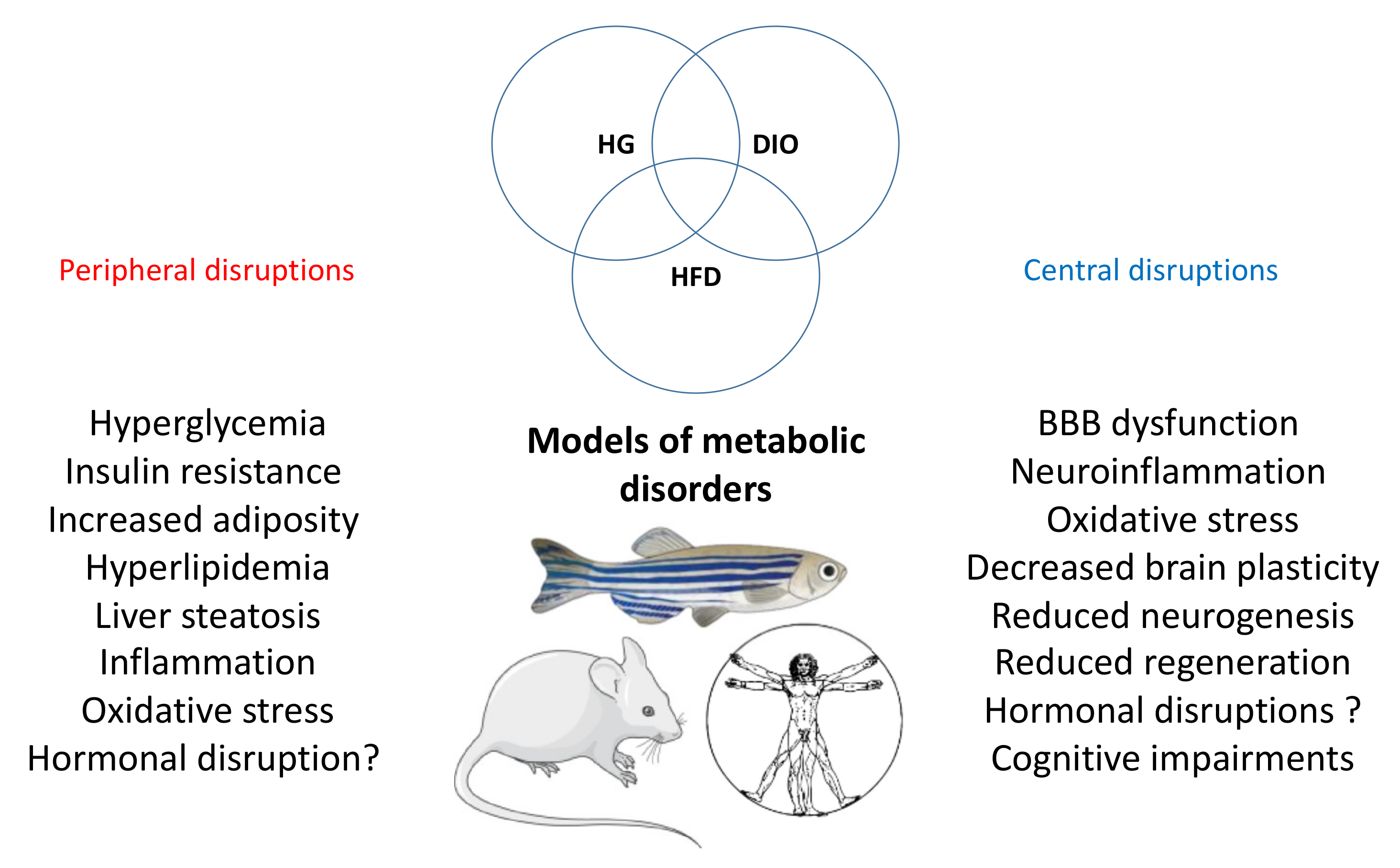
2. Different Models of Metabolic Disorders in Adult Zebrafish
2.1. Models of Acute and Chronic Hyperglycemia
2.2. Models of Overweight and Obesity in Zebrafish Leading to Hyperglycemia
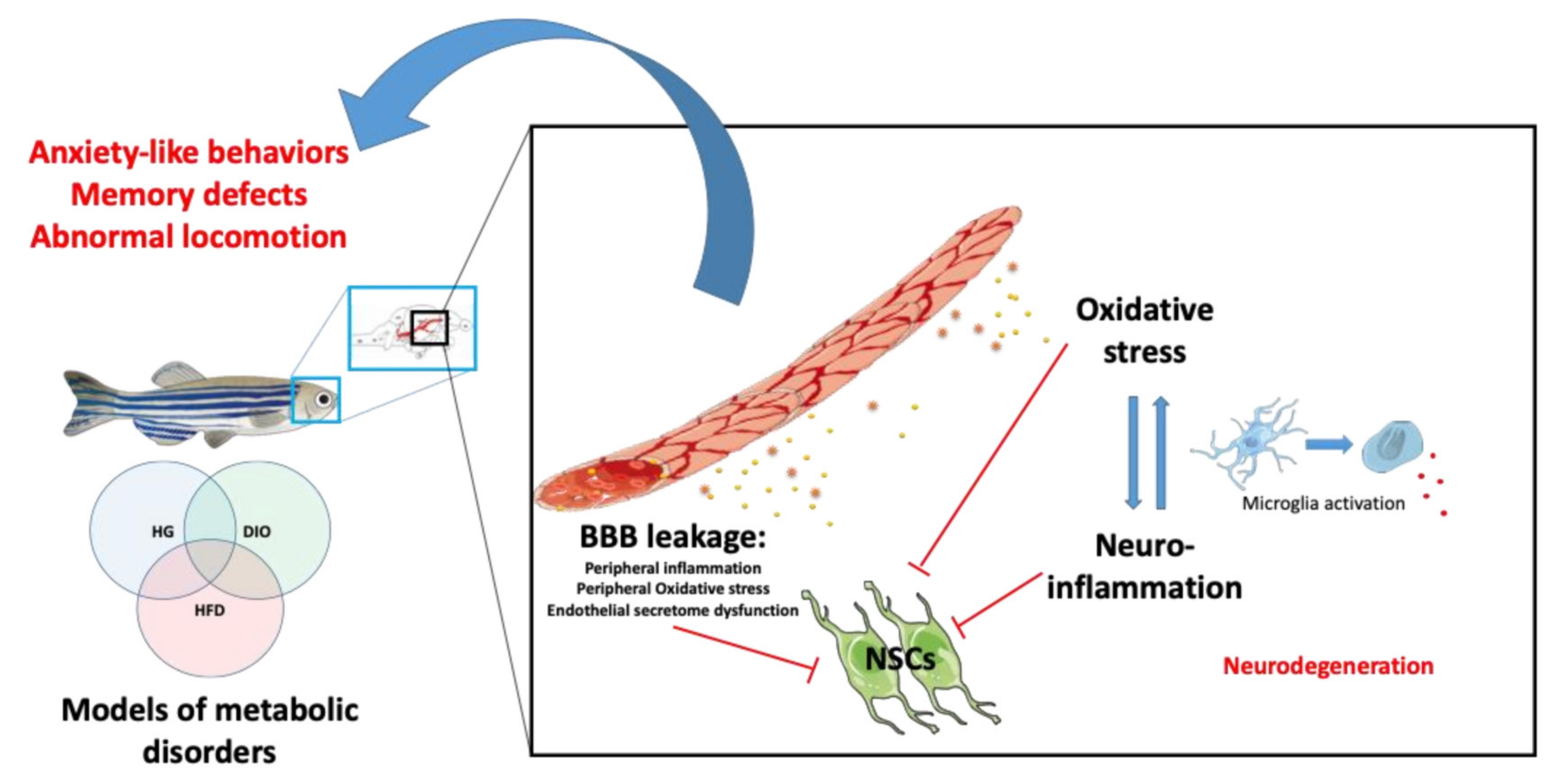
3. The Effects of Metabolic Disorders on Brain Plasticity and Function: Focus on Zebrafish and Comparative Aspects
3.1. Hyperglycemia and Brain Homeostasis in Adult Zebrafish
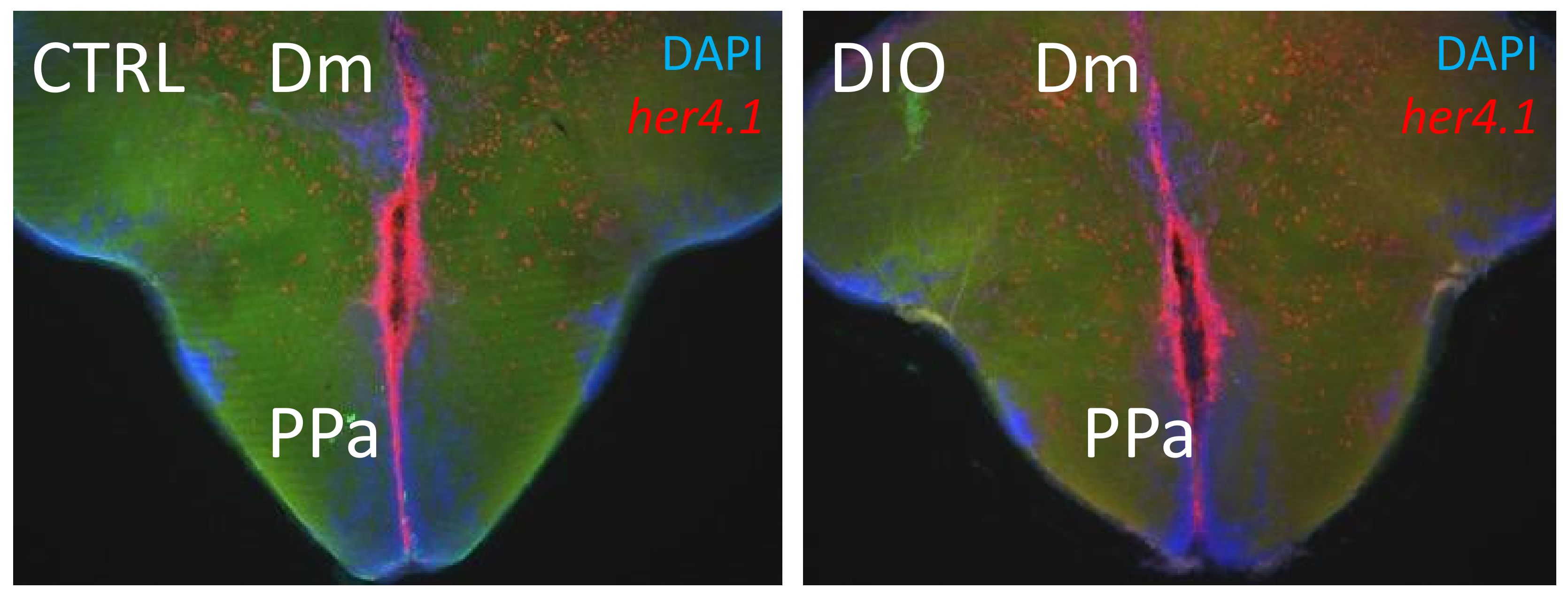
3.2. Obesity and Brain Homeostasis in Zebrafish
3.3. Effects of Hypercholesterolemia on the CNS
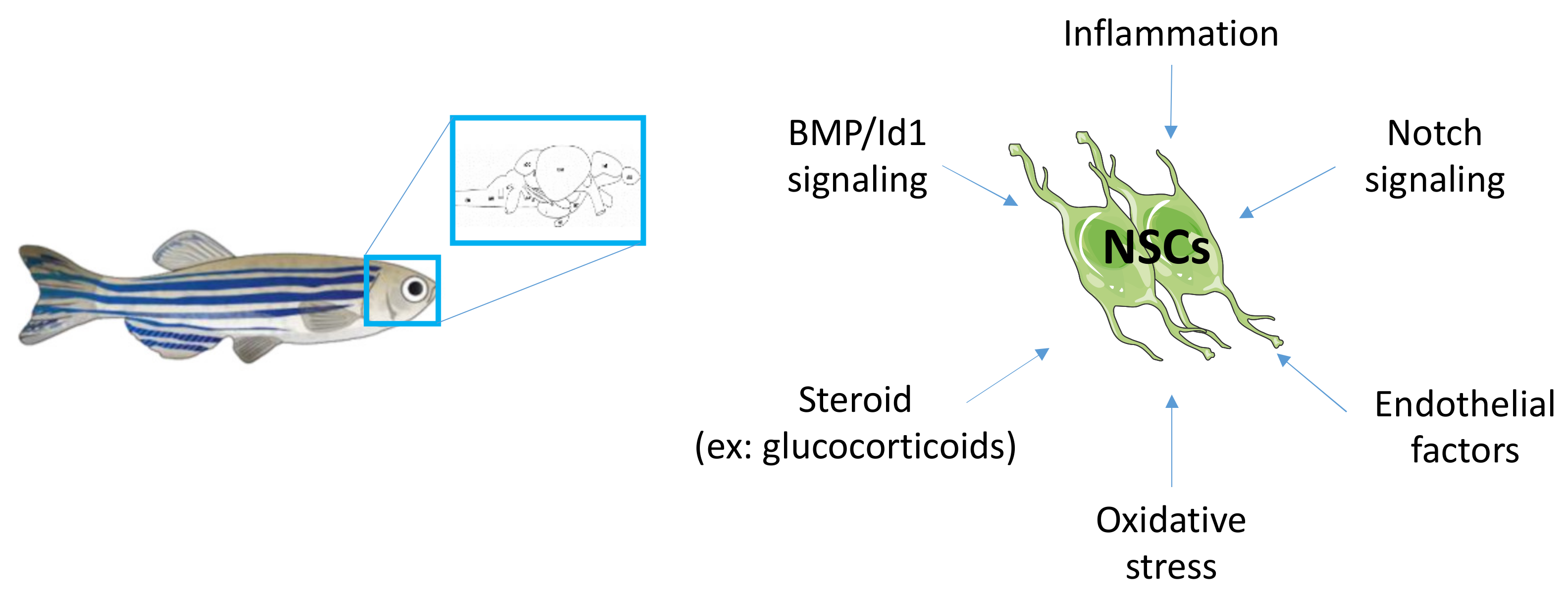
4. Brain Dis-Plasticity and Metabolic Disorders: Molecular Mechanisms to Investigate
4.1. Notch Signaling in Mammal and Zebrafish Neurogenesis
4.2. BMP/Id1 Signaling and Cross-Talk with Notch Signaling in NSCs
4.3. The Role of Stress Hormone Signaling in Neurogenesis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurgel, M.H.C.; Montenegro Junior, R.M.; Melo Ponte, C.M.; Sousa, T.C.S.; Silva, P.G.B.; de Sousa Belem, L.; Furtado, F.L.B.; de Araujo Batista, L.A.; Pereira, A.C.; Santos, R.D. Metabolic syndrome, diabetes and inadequate lifestyle in first-degree relatives of acute myocardial infarction survivors younger than 45 years old. Lipids Health Dis. 2017, 16, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBoer, M.D.; Filipp, S.L.; Sims, M.; Musani, S.K.; Gurka, M.J. Risk of Ischemic Stroke Increases Over the Spectrum of Metabolic Syndrome Severity. Stroke 2020, 51, 2548–2552. [Google Scholar] [CrossRef] [PubMed]
- Katsiki, N.; Anagnostis, P.; Kotsa, K.; Goulis, D.G.; Mikhailidis, D.P. Obesity, Metabolic Syndrome and the Risk of Microvascular Complications in Patients with Diabetes mellitus. Curr. Pharm. Des. 2019, 25, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Bangen, K.J.; Armstrong, N.M.; Au, R.; Gross, A.L. Metabolic Syndrome and Cognitive Trajectories in the Framingham Offspring Study. J. Alzheimers Dis. 2019, 71, 931–943. [Google Scholar] [CrossRef]
- Blumentals, W.A.; Hwu, P.; Kobayashi, N.; Ogura, E. Obesity in hospitalized type 2 diabetes patients: A descriptive study. Med. Sci. Monit. 2013, 19, 359–365. [Google Scholar]
- Ritz, E. Metabolic syndrome and kidney disease. Blood Purif. 2008, 26, 59–62. [Google Scholar] [CrossRef]
- Tune, J.D.; Goodwill, A.G.; Sassoon, D.J.; Mather, K.J. Cardiovascular consequences of metabolic syndrome. Transl. Res. 2017, 183, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Artunc, F.; Schleicher, E.; Weigert, C.; Fritsche, A.; Stefan, N.; Haring, H.U. The impact of insulin resistance on the kidney and vasculature. Nat. Rev. Nephrol. 2016, 12, 721–737. [Google Scholar] [CrossRef] [Green Version]
- Van der Heijden, R.A.; Sheedfar, F.; Morrison, M.C.; Hommelberg, P.P.; Kor, D.; Kloosterhuis, N.J.; Gruben, N.; Youssef, S.A.; de Bruin, A.; Hofker, M.H.; et al. High-fat diet induced obesity primes inflammation in adipose tissue prior to liver in C57BL/6j mice. Aging 2015, 7, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomlinson, J.W. Non-alcoholic fatty liver disease and diabetes. Metabolism 2016, 65, 1096–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre, C.; Dinel, A.L.; Ferreira, G.; Laye, S.; Castanon, N. Diet-induced obesity progressively alters cognition, anxiety-like behavior and lipopolysaccharide-induced depressive-like behavior: Focus on brain indoleamine 2,3-dioxygenase activation. Brain Behav Immun 2014, 41, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.N.N.; Lima-Filho, R.A.S.; De Felice, F.G. Connecting Alzheimer’s disease to diabetes: Underlying mechanisms and potential therapeutic targets. Neuropharmacology 2018, 136, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Roh, H.T.; Cho, S.Y.; So, W.Y. Obesity promotes oxidative stress and exacerbates blood-brain barrier disruption after high-intensity exercise. J. Sport Health Sci. 2017, 6, 225–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, D.Y.; Yim, H.S.; Jung, H.Y.; Nam, S.M.; Kim, J.W.; Choi, J.H.; Seong, J.K.; Yoon, Y.S.; Kim, D.W.; Hwang, I.K. Chronic type 2 diabetes reduces the integrity of the blood-brain barrier by reducing tight junction proteins in the hippocampus. J. Vet. Med. Sci. 2016, 78, 957–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef]
- Miller, A.A.; Spencer, S.J. Obesity and neuroinflammation: A pathway to cognitive impairment. Brain Behav Immun 2014, 42, 10–21. [Google Scholar] [CrossRef]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef]
- Sarparanta, J.; Garcia-Macia, M.; Singh, R. Autophagy and Mitochondria in Obesity and Type 2 Diabetes. Curr Diabetes Rev. 2017, 13, 352–369. [Google Scholar] [CrossRef]
- Bondia-Pons, I.; Ryan, L.; Martinez, J.A. Oxidative stress and inflammation interactions in human obesity. J. Physiol Biochem 2012, 68, 701–711. [Google Scholar] [CrossRef]
- Ghaddar, B.; Lubke, L.; Couret, D.; Rastegar, S.; Diotel, N. Cellular Mechanisms Participating in Brain Repair of Adult Zebrafish and Mammals after Injury. Cells 2021, 10, 391. [Google Scholar] [CrossRef] [PubMed]
- Diotel, N.; Lubke, L.; Strahle, U.; Rastegar, S. Common and Distinct Features of Adult Neurogenesis and Regeneration in the Telencephalon of Zebrafish and Mammals. Front. Neurosci 2020, 14, 568930. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Bou Diab, Z.; Nabha, S.; Fares, Y. Neurogenesis in the adult hippocampus: History, regulation, and prospective roles. Int. J. Neurosci. 2019, 129, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.J.; Kitamura, T.; Saitoh, Y.; Ohkawa, N.; Kondo, T.; Inokuchi, K. Adult Neurogenesis Conserves Hippocampal Memory Capacity. J. Neurosci 2018, 38, 6854–6863. [Google Scholar] [CrossRef] [Green Version]
- Kempermann, G.; Gage, F.H.; Aigner, L.; Song, H.; Curtis, M.A.; Thuret, S.; Kuhn, H.G.; Jessberger, S.; Frankland, P.W.; Cameron, H.A.; et al. Human Adult Neurogenesis: Evidence and Remaining Questions. Cell Stem Cell 2018, 23, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Boldrini, M.; Fulmore, C.A.; Tartt, A.N.; Simeon, L.R.; Pavlova, I.; Poposka, V.; Rosoklija, G.B.; Stankov, A.; Arango, V.; Dwork, A.J.; et al. Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell 2018, 22, 589–599.e5. [Google Scholar] [CrossRef] [Green Version]
- Ernst, A.; Frisen, J. Adult neurogenesis in humans- common and unique traits in mammals. PLoS Biol. 2015, 13, e1002045. [Google Scholar] [CrossRef]
- Lindsey, B.W.; Tropepe, V. A comparative framework for understanding the biological principles of adult neurogenesis. Prog. Neurobiol. 2006, 80, 281–307. [Google Scholar] [CrossRef]
- Kohman, R.A.; Rhodes, J.S. Neurogenesis, inflammation and behavior. Brain Behav. Immun. 2013, 27, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Yirmiya, R.; Goshen, I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 2011, 25, 181–213. [Google Scholar] [CrossRef]
- Ryan, S.M.; Nolan, Y.M. Neuroinflammation negatively affects adult hippocampal neurogenesis and cognition: Can exercise compensate? Neurosci. Biobehav. Rev. 2015, 61, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Covacu, R.; Brundin, L. Effects of Neuroinflammation on Neural Stem Cells. Neuroscientist 2017, 23, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Mishra, A.; Tiwari, V.; Shukla, S. Enhanced neuroinflammation and oxidative stress are associated with altered hippocampal neurogenesis in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine treated mice. Behav. Pharmacol 2019, 30, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Borsini, A.; Zunszain, P.A.; Thuret, S.; Pariante, C.M. The role of inflammatory cytokines as key modulators of neurogenesis. Trends Neurosci. 2015, 38, 145–157. [Google Scholar] [CrossRef] [Green Version]
- Zonis, S.; Ljubimov, V.A.; Mahgerefteh, M.; Pechnick, R.N.; Wawrowsky, K.; Chesnokova, V. p21Cip restrains hippocampal neurogenesis and protects neuronal progenitors from apoptosis during acute systemic inflammation. Hippocampus 2013, 23, 1383–1394. [Google Scholar] [CrossRef] [Green Version]
- Zonis, S.; Pechnick, R.N.; Ljubimov, V.A.; Mahgerefteh, M.; Wawrowsky, K.; Michelsen, K.S.; Chesnokova, V. Chronic intestinal inflammation alters hippocampal neurogenesis. J. Neuroinflamm. 2015, 12, 65. [Google Scholar] [CrossRef] [Green Version]
- Kleinert, M.; Clemmensen, C.; Hofmann, S.M.; Moore, M.C.; Renner, S.; Woods, S.C.; Huypens, P.; Beckers, J.; de Angelis, M.H.; Schurmann, A.; et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2018, 14, 140–162. [Google Scholar] [CrossRef] [Green Version]
- Ghaddar, B.; Veeren, B.; Rondeau, P.; Bringart, M.; Lefebvre d’Hellencourt, C.; Meilhac, O.; Bascands, J.L.; Diotel, N. Impaired brain homeostasis and neurogenesis in diet-induced overweight zebrafish: A preventive role from A. borbonica extract. Sci. Rep. 2020, 10, 14496. [Google Scholar] [CrossRef]
- Landgraf, K.; Schuster, S.; Meusel, A.; Garten, A.; Riemer, T.; Schleinitz, D.; Kiess, W.; Korner, A. Short-term overfeeding of zebrafish with normal or high-fat diet as a model for the development of metabolically healthy versus unhealthy obesity. BMC Physiol. 2017, 17, 4. [Google Scholar] [CrossRef] [Green Version]
- Dorsemans, A.C.; Soule, S.; Weger, M.; Bourdon, E.; Lefebvre d’Hellencourt, C.; Meilhac, O.; Diotel, N. Impaired constitutive and regenerative neurogenesis in adult hyperglycemic zebrafish. J. Comp. Neurol. 2017, 525, 442–458. [Google Scholar] [CrossRef]
- Montalbano, G.; Mania, M.; Guerrera, M.C.; Laura, R.; Abbate, F.; Levanti, M.; Maugeri, A.; Germana, A.; Navarra, M. Effects of a Flavonoid-Rich Extract from Citrus sinensis Juice on a Diet-Induced Obese Zebrafish. Int. J. Mol. Sci. 2019, 20, 5116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capiotti, K.M.; Antonioli, R., Jr.; Kist, L.W.; Bogo, M.R.; Bonan, C.D.; Da Silva, R.S. Persistent impaired glucose metabolism in a zebrafish hyperglycemia model. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2014, 171, 58–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capiotti, K.M.; De Moraes, D.A.; Menezes, F.P.; Kist, L.W.; Bogo, M.R.; Da Silva, R.S. Hyperglycemia induces memory impairment linked to increased acetylcholinesterase activity in zebrafish (Danio rerio). Behav. Brain Res. 2014, 274, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.L.; Spitsbergen, J.M.; Wolterbeek, A.P.; Woutersen, R.A. Normal anatomy and histology of the adult zebrafish. Toxicol. Pathol. 2011, 39, 759–775. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Heckler, K.; Kroll, J. Zebrafish as a Model for the Study of Microvascular Complications of Diabetes and Their Mechanisms. Int. J. Mol. Sci. 2017, 18, 2002. [Google Scholar] [CrossRef] [Green Version]
- Zang, L.; Shimada, Y.; Nishimura, N. Development of a Novel Zebrafish Model for Type 2 Diabetes Mellitus. Sci. Rep. 2017, 7, 1461. [Google Scholar] [CrossRef] [Green Version]
- Wiggenhauser, L.M.; Kroll, J. Vascular Damage in Obesity and Diabetes: Highlighting Links Between Endothelial Dysfunction and Metabolic Disease in Zebrafish and Man. Curr. Vasc. Pharmacol. 2019, 17, 476–490. [Google Scholar] [CrossRef]
- Grandel, H.; Brand, M. Comparative aspects of adult neural stem cell activity in vertebrates. Dev. Genes Evol. 2013, 223, 131–147. [Google Scholar] [CrossRef]
- Grandel, H.; Kaslin, J.; Ganz, J.; Wenzel, I.; Brand, M. Neural stem cells and neurogenesis in the adult zebrafish brain: Origin, proliferation dynamics, migration and cell fate. Dev. Biol. 2006, 295, 263–277. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, E.; Mouriec, K.; Anglade, I.; Menuet, A.; Le Page, Y.; Gueguen, M.M.; Marmignon, M.H.; Brion, F.; Pakdel, F.; Kah, O. Identification of aromatase-positive radial glial cells as progenitor cells in the ventricular layer of the forebrain in zebrafish. J. Comp. Neurol. 2007, 501, 150–167. [Google Scholar] [CrossRef] [PubMed]
- Lucini, C.; D’Angelo, L.; Cacialli, P.; Palladino, A.; de Girolamo, P. BDNF, Brain, and Regeneration: Insights from Zebrafish. Int. J. Mol. Sci. 2018, 19, 3155. [Google Scholar] [CrossRef] [Green Version]
- Zambusi, A.; Ninkovic, J. Regeneration of the central nervous system-principles from brain regeneration in adult zebrafish. World J. Stem Cells 2020, 12, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Lupperger, V.; Buggenthin, F.; Chapouton, P.; Marr, C. Image analysis of neural stem cell division patterns in the zebrafish brain. Cytometry A 2018, 93, 314–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, R.; Beil, T.; Strähle, U.; Rastegar, S. Stab wound injury of the zebrafish adult telencephalon: A method to investigate vertebrate brain neurogenesis and regeneration. J. Vis. Exp. 2014, 90, e51753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsemans, A.C.; Lefebvre d’Hellencourt, C.; Ait-Arsa, I.; Jestin, E.; Meilhac, O.; Diotel, N. Acute and Chronic Models of Hyperglycemia in Zebrafish: A Method to Assess the Impact of Hyperglycemia on Neurogenesis and the Biodistribution of Radiolabeled Molecules. J. Vis. Exp. 2017, 124, e55203. [Google Scholar] [CrossRef] [PubMed]
- Couret, D.; Bourane, S.; Catan, A.; Nativel, B.; Planesse, C.; Dorsemans, A.C.; Ait-Arsa, I.; Cournot, M.; Rondeau, P.; Patche, J.; et al. A hemorrhagic transformation model of mechanical stroke therapy with acute hyperglycemia in mice. J. Comp. Neurol. 2018, 526, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Couret, D.; Planesse, C.; Patche, J.; Diotel, N.; Nativel, B.; Bourane, S.; Meilhac, O. Lack of Neuroprotective Effects of High-Density Lipoprotein Therapy in Stroke under Acute Hyperglycemic Conditions. Molecules 2021, 26, 6365. [Google Scholar] [CrossRef] [PubMed]
- Abookasis, D.; Shemesh, D.; Bokobza, N.; Bloygrund, H.; Franjy-Tal, Y.; Rozenberg, K.; Rosenzweig, T. Evaluation the effect of acute hyperglycemia on cerebral tissue properties with diffuse optical imaging systems. In Proceedings of the Neurophotonics. International Society for Optics and Photonics, Online, 1 April 2020; p. 11360. [Google Scholar] [CrossRef]
- Kinkel, M.D.; Eames, S.C.; Philipson, L.H.; Prince, V.E. Intraperitoneal injection into adult zebrafish. J. Vis. Exp. 2010, 30, 2126. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.B.; Koustubhan, P.; Greenman, M.; Parsons, M.J.; Walter, I.; Moss, L.G. Regeneration of the pancreas in adult zebrafish. Diabetes 2009, 58, 1844–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaspre, F.; Beer, R.L.; Rovira, M.; Huang, W.; Wang, G.; Gee, S.; Vitery Mdel, C.; Wheelan, S.J.; Parsons, M.J. Centroacinar Cells Are Progenitors That Contribute to Endocrine Pancreas Regeneration. Diabetes 2015, 64, 3499–3509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Intine, R.V.; Olsen, A.S.; Sarras, M.P., Jr. A zebrafish model of diabetes mellitus and metabolic memory. J. Vis. Exp. 2013, 28, e50232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benchoula, K.; Khatib, A.; Quzwain, F.M.C.; Che Mohamad, C.A.; Wan Sulaiman, W.M.A.; Abdul Wahab, R.; Ahmed, Q.U.; Abdul Ghaffar, M.; Saiman, M.Z.; Alajmi, M.F.; et al. Optimization of Hyperglycemic Induction in Zebrafish and Evaluation of Its Blood Glucose Level and Metabolite Fingerprint Treated with Psychotria malayana Jack Leaf Extract. Molecules 2019, 24, 1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.S.; Loots du, T. Experimental rodent models of type 2 diabetes: A review. Methods Find. Exp. Clin. Pharmacol. 2009, 31, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Szkudelski, T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol. Res. 2001, 50, 537–546. [Google Scholar] [PubMed]
- Olsen, A.S.; Sarras, M.P., Jr.; Intine, R.V. Limb regeneration is impaired in an adult zebrafish model of diabetes mellitus. Wound Repair Regen. 2010, 18, 532–542. [Google Scholar] [CrossRef] [Green Version]
- Nam, Y.H.; Hong, B.N.; Rodriguez, I.; Ji, M.G.; Kim, K.; Kim, U.J.; Kang, T.H. Synergistic Potentials of Coffee on Injured Pancreatic Islets and Insulin Action via KATP Channel Blocking in Zebrafish. J. Agric. Food Chem. 2015, 63, 5612–5621. [Google Scholar] [CrossRef]
- Shin, E.; Na Hong, B.; Ho Kang, T. An optimal establishment of an acute hyperglycemia zebrafish model. Afr. J. Pharm. Pharmacol. 2012, 6, 2922–2928. [Google Scholar] [CrossRef] [Green Version]
- Neelofar, K.; Arif, Z.; Alam, K.; Ahmad, J. Hyperglycemia induced structural and functional changes in human serum albumin of diabetic patients: A physico-chemical study. Mol. Biosyst. 2016, 12, 2481–2489. [Google Scholar] [CrossRef]
- Steele, C.; Hagopian, W.A.; Gitelman, S.; Masharani, U.; Cavaghan, M.; Rother, K.I.; Donaldson, D.; Harlan, D.M.; Bluestone, J.; Herold, K.C. Insulin secretion in type 1 diabetes. Diabetes 2004, 53, 426–433. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, Y.; Gurung, P.; Wang, T.; Li, L.; Zhang, R.; Li, H.; Guo, R.; Han, Q.; Zhang, J.; et al. The relationship between the thickness of glomerular basement membrane and renal outcomes in patients with diabetic nephropathy. Acta. Diabetol. 2018, 55, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Cao, D.; Yu, H.; Yang, D.; Zhuang, X.; Hu, Y.; Li, J.; Yang, J.; Wu, Q.; Liu, B.; et al. Early retinal neurovascular impairment in patients with diabetes without clinically detectable retinopathy. Br. J. Ophthalmol. 2019, 103, 1747–1752. [Google Scholar] [PubMed]
- Patel, S.; Srivastava, S.; Singh, M.R.; Singh, D. Mechanistic insight into diabetic wounds: Pathogenesis, molecular targets and treatment strategies to pace wound healing. Biomed. Pharmacother. 2019, 112, 108615. [Google Scholar] [CrossRef]
- Wang, Q.; Luo, C.; Lu, G.; Chen, Z. Effect of adenosine monophosphate-activated protein kinase-p53-Kruppel-like factor 2a pathway in hyperglycemia-induced cardiac remodeling in adult zebrafish. J. Diabetes Investig. 2021, 12, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Olsen, A.S.; Sarras, M.P., Jr.; Leontovich, A.; Intine, R.V. Heritable transmission of diabetic metabolic memory in zebrafish correlates with DNA hypomethylation and aberrant gene expression. Diabetes 2012, 61, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, X.L.; Liu, K.C.; Sheng, W.L.; Xia, Q.; Wang, R.C.; Chen, X.Q.; Zhang, Y. Effects of streptozotocin on pancreatic islet beta-cell apoptosis and glucose metabolism in zebrafish larvae. Fish. Physiol. Biochem. 2020, 46, 1025–1038. [Google Scholar] [CrossRef]
- Prince, V.E.; Anderson, R.M.; Dalgin, G. Zebrafish Pancreas Development and Regeneration: Fishing for Diabetes Therapies. Curr. Top. Dev. Biol. 2017, 124, 235–276. [Google Scholar]
- Matsuda, H. Zebrafish as a model for studying functional pancreatic beta cells development and regeneration. Dev. Growth Differ. 2018, 60, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Benchoula, K.; Khatib, A.; Jaffar, A.; Ahmed, Q.U.; Sulaiman, W.; Wahab, R.A.; El-Seedi, H.R. The promise of zebrafish as a model of metabolic syndrome. Exp. Anim. 2019, 68, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Connaughton, V.P.; Baker, C.; Fonde, L.; Gerardi, E.; Slack, C. Alternate Immersion in an External Glucose Solution Differentially Affects Blood Sugar Values in Older Versus Younger Zebrafish Adults. Zebrafish 2016, 13, 87–94. [Google Scholar] [CrossRef]
- Gence, L.; Fernezelian, D.; Bringart, M.; Veeren, B.; Christophe, A.; Brion, F.; Meilhac, O.; Bascands, J.L.; Diotel, N. Hypericum lanceolatum Lam. Medicinal Plant: Potential Toxicity and Therapeutic Effects Based on a Q2 Zebrafish Model. Front. Pharmacol. 2022, 13, 832928. [Google Scholar] [CrossRef] [PubMed]
- Alsahli, M.; Gerich, J.E. Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes Res. Clin. Pract. 2017, 133, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schwartsburd, P. Glucose-lowering Strategies in Diabetes: Pharmacological Development of New Antidiabetic Drugs. Curr. Pharm. Des. 2018, 24, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Block, N.E.; Buse, M.G. Effects of hypercortisolemia and diabetes on skeletal muscle insulin receptor function in vitro and in vivo. Am. J. Physiol. 1989, 256, E39–E48. [Google Scholar] [CrossRef] [PubMed]
- Karanth, S.; Chaurasia, B.; Bowman, F.M.; Tippetts, T.S.; Holland, W.L.; Summers, S.A.; Schlegel, A. FOXN3 controls liver glucose metabolism by regulating gluconeogenic substrate selection. Physiol. Rep. 2019, 7, e14238. [Google Scholar] [CrossRef] [PubMed]
- Karanth, S.; Zinkhan, E.K.; Hill, J.T.; Yost, H.J.; Schlegel, A. FOXN3 Regulates Hepatic Glucose Utilization. Cell Rep. 2016, 15, 2745–2755. [Google Scholar] [CrossRef] [Green Version]
- Kimmel, R.A.; Dobler, S.; Schmitner, N.; Walsen, T.; Freudenblum, J.; Meyer, D. Diabetic pdx1-mutant zebrafish show conserved responses to nutrient overload and anti-glycemic treatment. Sci. Rep. 2015, 5, 14241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddison, L.A.; Joest, K.E.; Kammeyer, R.M.; Chen, W. Skeletal muscle insulin resistance in zebrafish induces alterations in beta-cell number and glucose tolerance in an age- and diet-dependent manner. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E662–E669. [Google Scholar] [CrossRef] [Green Version]
- Salehpour, A.; Rezaei, M.; Khoradmehr, A.; Tahamtani, Y.; Tamadon, A. Which Hyperglycemic Model of Zebrafish (Danio rerio) Suites My Type 2 Diabetes Mellitus Research? A Scoring System for Available Methods. Front. Cell Dev. Biol. 2021, 9, 652061. [Google Scholar] [CrossRef]
- Zang, L.; Maddison, L.A.; Chen, W. Zebrafish as a Model for Obesity and Diabetes. Front. Cell Dev. Biol. 2018, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.L.; Carten, J.D.; Farber, S.A. Zebrafish lipid metabolism: From mediating early patterning to the metabolism of dietary fat and cholesterol. Methods Cell Biol. 2011, 101, 111–141. [Google Scholar] [PubMed] [Green Version]
- Meguro, S.; Hasumura, T.; Hase, T. Body fat accumulation in zebrafish is induced by a diet rich in fat and reduced by supplementation with green tea extract. PLoS ONE 2015, 10, e0120142. [Google Scholar] [CrossRef] [PubMed]
- Montalbano, G.; Mania, M.; Abbate, F.; Navarra, M.; Guerrera, M.C.; Laura, R.; Vega, J.A.; Levanti, M.; Germana, A. Melatonin treatment suppresses appetite genes and improves adipose tissue plasticity in diet-induced obese zebrafish. Endocrine 2018, 62, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Montalbano, G.; Mania, M.; Guerrera, M.C.; Abbate, F.; Laura, R.; Navarra, M.; Vega, J.A.; Ciriaco, E.; Germana, A. Morphological differences in adipose tissue and changes in BDNF/Trkb expression in brain and gut of a diet induced obese zebrafish model. Ann. Anat. 2016, 204, 36–44. [Google Scholar] [CrossRef]
- Ahima, R.S.; Lazar, M.A. Physiology. The health risk of obesity–better metrics imperative. Science 2013, 341, 856–858. [Google Scholar] [CrossRef]
- Chu, C.Y.; Chen, C.F.; Rajendran, R.S.; Shen, C.N.; Chen, T.H.; Yen, C.C.; Chuang, C.K.; Lin, D.S.; Hsiao, C.D. Overexpression of Akt1 enhances adipogenesis and leads to lipoma formation in zebrafish. PLoS ONE 2012, 7, e36474. [Google Scholar]
- Hsu, C.C.; Lai, C.Y.; Lin, C.Y.; Yeh, K.Y.; Her, G.M. MicroRNA-27b Depletion Enhances Endotrophic and Intravascular Lipid Accumulation and Induces Adipocyte Hyperplasia in Zebrafish. Int. J. Mol. Sci. 2017, 19, 93. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Nishimura, Y.; Zang, L.; Hirano, M.; Shimada, Y.; Wang, Z.; Umemoto, N.; Kuroyanagi, J.; Nishimura, N.; Tanaka, T. Diet-induced obesity in zebrafish shares common pathophysiological pathways with mammalian obesity. BMC Physiol. 2010, 10, 21. [Google Scholar] [CrossRef] [Green Version]
- Hasumura, T.; Shimada, Y.; Kuroyanagi, J.; Nishimura, Y.; Meguro, S.; Takema, Y.; Tanaka, T. Green tea extract suppresses adiposity and affects the expression of lipid metabolism genes in diet-induced obese zebrafish. Nutr. Metab. 2012, 9, 73. [Google Scholar] [CrossRef] [Green Version]
- Hiramitsu, M.; Shimada, Y.; Kuroyanagi, J.; Inoue, T.; Katagiri, T.; Zang, L.; Nishimura, Y.; Nishimura, N.; Tanaka, T. Eriocitrin ameliorates diet-induced hepatic steatosis with activation of mitochondrial biogenesis. Sci. Rep. 2014, 4, 3708. [Google Scholar] [CrossRef] [Green Version]
- Forn-Cuni, G.; Varela, M.; Fernandez-Rodriguez, C.M.; Figueras, A.; Novoa, B. Liver immune responses to inflammatory stimuli in a diet-induced obesity model of zebrafish. J. Endocrinol. 2015, 224, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Meguro, S.; Hosoi, S.; Hasumura, T. High-fat diet impairs cognitive function of zebrafish. Sci. Rep. 2019, 9, 17063. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Mueller, O.; Bagwell, J.; Bagnat, M.; Liddle, R.A.; Rawls, J.F. High fat diet induces microbiota-dependent silencing of enteroendocrine cells. Elife 2019, 8, e48479. [Google Scholar] [CrossRef] [PubMed]
- Grisotto, C.; Taile, J.; Planesse, C.; Diotel, N.; Gonthier, M.P.; Meilhac, O.; Couret, D. High-Fat Diet Aggravates Cerebral Infarct, Hemorrhagic Transformation and Neuroinflammation in a Mouse Stroke Model. Int. J. Mol. Sci. 2021, 22, 4571. [Google Scholar] [CrossRef]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, A.M.; Greene, M.W.; Geetha, T.; Babu, J.R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta. Mol. Basis. Dis. 2017, 1863, 499–508. [Google Scholar] [CrossRef]
- Della Vedova, M.C.; Munoz, M.D.; Santillan, L.D.; Plateo-Pignatari, M.G.; Germano, M.J.; Rinaldi Tosi, M.E.; Garcia, S.; Gomez, N.N.; Fornes, M.W.; Gomez Mejiba, S.E.; et al. A Mouse Model of Diet-Induced Obesity Resembling Most Features of Human Metabolic Syndrome. Nutr. Metab. Insights 2016, 9, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Pan, Q.; Dong, H.; Yuan, X.; Li, Y.; Sun, Z.; Dong, X.; Wang, H. Adipose-derived mesenchymal stem cells improve glucose homeostasis in high-fat diet-induced obese mice. Stem. Cell Res. Ther. 2015, 6, 208. [Google Scholar] [CrossRef] [Green Version]
- Carnovali, M.; Luzi, L.; Terruzzi, I.; Banfi, G.; Mariotti, M. Metabolic and bone effects of high-fat diet in adult zebrafish. Endocrine 2018, 61, 317–326. [Google Scholar] [CrossRef]
- Diotel, N.; Rodriguez Viales, R.; Armant, O.; Marz, M.; Ferg, M.; Rastegar, S.; Strahle, U. Comprehensive expression map of transcription regulators in the adult zebrafish telencephalon reveals distinct neurogenic niches. J. Comp. Neurol. 2015, 523, 1202–1221. [Google Scholar] [CrossRef] [Green Version]
- Kizil, C.; Kaslin, J.; Kroehne, V.; Brand, M. Adult neurogenesis and brain regeneration in zebrafish. Dev. Neurobiol. 2012, 72, 429–461. [Google Scholar] [CrossRef]
- Eliceiri, B.P.; Gonzalez, A.M.; Baird, A. Zebrafish model of the blood-brain barrier: Morphological and permeability studies. Methods Mol. Biol. 2011, 686, 371–378. [Google Scholar] [PubMed] [Green Version]
- Chhabria, K.; Vouros, A.; Gray, C.; MacDonald, R.B.; Jiang, Z.; Wilkinson, R.N.; Plant, K.; Vasilaki, E.; Howarth, C.; Chico, T.J.A. Sodium nitroprusside prevents the detrimental effects of glucose on the neurovascular unit and behaviour in zebrafish. Dis. Model. Mech. 2019, 12, dmm039867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhabria, K.; Plant, K.; Bandmann, O.; Wilkinson, R.N.; Martin, C.; Kugler, E.; Armitage, P.A.; Santoscoy, P.L.; Cunliffe, V.T.; Huisken, J.; et al. The effect of hyperglycemia on neurovascular coupling and cerebrovascular patterning in zebrafish. J. Cereb. Blood Flow Metab. 2020, 40, 298–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, M.M.; de Macedo, G.T.; Prestes, A.S.; Ecker, A.; Muller, T.E.; Leitemperger, J.; Fontana, B.D.; Ardisson-Araujo, D.M.P.; Rosemberg, D.B.; Barbosa, N.V. Modulation of redox and insulin signaling underlie the anti-hyperglycemic and antioxidant effects of diphenyl diselenide in zebrafish. Free. Radic. Biol. Med. 2020, 158, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Nellore, J.; Pauline, P.C.; Mohanan, S.P.; Ravikumar, R.; Pillai, R.C. Vinca rosea Normalizes Oxidative Stress and Inhibits Hyperglycemia Induced Increase in VEGF in Zebrafish Retina. Res. J. Pharm. Biol. Chem. Sci. 2013, 4, 927–936. [Google Scholar]
- Oyelaja-Akinsipo, O.B.; Dare, E.O.; Katare, D.P. Protective role of diosgenin against hyperglycaemia-mediated cerebral ischemic brain injury in zebrafish model of type II diabetes mellitus. Heliyon 2020, 6, e03296. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, M.M.; de Macedo, G.T.; Prestes, A.S.; Loro, V.L.; Heidrich, G.M.; Picoloto, R.S.; Rosemberg, D.B.; Barbosa, N.V. Hyperglycemia elicits anxiety-like behaviors in zebrafish: Protective role of dietary diphenyl diselenide. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 85, 128–135. [Google Scholar] [CrossRef]
- Rom, S.; Zuluaga-Ramirez, V.; Gajghate, S.; Seliga, A.; Winfield, M.; Heldt, N.A.; Kolpakov, M.A.; Bashkirova, Y.V.; Sabri, A.K.; Persidsky, Y. Hyperglycemia-Driven Neuroinflammation Compromises BBB Leading to Memory Loss in Both Diabetes Mellitus (DM) Type 1 and Type 2 Mouse Models. Mol. Neurobiol. 2019, 56, 1883–1896. [Google Scholar] [CrossRef]
- Wanrooy, B.J.; Kumar, K.P.; Wen, S.W.; Qin, C.X.; Ritchie, R.H.; Wong, C.H.Y. Distinct contributions of hyperglycemia and high-fat feeding in metabolic syndrome-induced neuroinflammation. J. Neuroinflamm. 2018, 15, 293. [Google Scholar] [CrossRef]
- Alvarez-Nolting, R.; Arnal, E.; Barcia, J.M.; Miranda, M.; Romero, F.J. Protection by DHA of early hippocampal changes in diabetes: Possible role of CREB and NF-kappaB. Neurochem. Res. 2012, 37, 105–115. [Google Scholar] [CrossRef]
- Piazza, F.V.; Pinto, G.V.; Trott, G.; Marcuzzo, S.; Gomez, R.; Fernandes Mda, C. Enriched environment prevents memory deficits in type 1 diabetic rats. Behav. Brain Res. 2011, 217, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Balu, D.T.; Hodes, G.E.; Hill, T.E.; Ho, N.; Rahman, Z.; Bender, C.N.; Ring, R.H.; Dwyer, J.M.; Rosenzweig-Lipson, S.; Hughes, Z.A.; et al. Flow cytometric analysis of BrdU incorporation as a high-throughput method for measuring adult neurogenesis in the mouse. J. Pharmacol. Toxicol. Methods 2009, 59, 100–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauquis, J.; Roig, P.; Homo-Delarche, F.; De Nicola, A.; Saravia, F. Reduced hippocampal neurogenesis and number of hilar neurones in streptozotocin-induced diabetic mice: Reversion by antidepressant treatment. Eur. J. Neurosci. 2006, 23, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Ho, N.; Sommers, M.S.; Lucki, I. Effects of diabetes on hippocampal neurogenesis: Links to cognition and depression. Neurosci. Biobehav. Rev. 2013, 37, 1346–1362. [Google Scholar] [CrossRef] [Green Version]
- Corem, N.; Anzi, S.; Gelb, S.; Ben-Zvi, A. Leptin receptor deficiency induces early, transient and hyperglycaemia-independent blood-brain barrier dysfunction. Sci. Rep. 2019, 9, 2884. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, R.; Chiba, Y.; Nakagawa, T.; Nishi, N.; Murakami, R.; Matsumoto, K.; Kawauchi, M.; Yamamoto, T.; Ueno, M. Albumin microvascular leakage in brains with diabetes mellitus. Microsc. Res. Tech. 2016, 79, 833–837. [Google Scholar] [CrossRef]
- Nerurkar, P.V.; Johns, L.M.; Buesa, L.M.; Kipyakwai, G.; Volper, E.; Sato, R.; Shah, P.; Feher, D.; Williams, P.G.; Nerurkar, V.R. Momordica charantia (bitter melon) attenuates high-fat diet-associated oxidative stress and neuroinflammation. J. Neuroinflamm. 2011, 8, 64. [Google Scholar] [CrossRef]
- Muriach, M.; Flores-Bellver, M.; Romero, F.J.; Barcia, J.M. Diabetes and the brain: Oxidative stress, inflammation, and autophagy. Oxid. Med. Cell. Longev. 2014, 2014, 102158. [Google Scholar] [CrossRef] [Green Version]
- Ghaddar, B.; Bringart, M.; Lefebvre d’Hellencourt, C.; Meilhac, O.; Diotel, N. Deleterious Effects of Overfeeding on Brain Homeostasis and Plasticity in Adult Zebrafish. Zebrafish 2021, 18, 190–206. [Google Scholar] [CrossRef]
- Imperatore, R.; Tunisi, L.; Mavaro, I.; D’Angelo, L.; Attanasio, C.; Safari, O.; Motlagh, H.A.; De Girolamo, P.; Cristino, L.; Varricchio, E.; et al. Immunohistochemical Analysis of Intestinal and Central Nervous System Morphology in an Obese Animal Model (Danio rerio) Treated with 3,5-T2: A Possible Farm Management Practice? Animals 2020, 10, 1131. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Lai, K.; Zhong, Q.; Demin, K.A.; Kalueff, A.V.; Song, C. High-glucose/high-cholesterol diet in zebrafish evokes diabetic and affective pathogenesis: The role of peripheral and central inflammation, microglia and apoptosis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 96, 109752. [Google Scholar] [CrossRef] [PubMed]
- Luzio, A.; Figueiredo, M.; Matos, M.M.; Coimbra, A.M.; Alvaro, A.R.; Monteiro, S.M. Effects of short-term exposure to genistein and overfeeding diet on the neural and retinal progenitor competence of adult zebrafish (Danio rerio). Neurotoxicol. Teratol. 2021, 88, 107030. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, A.J.; McGowan, E.M.; Yu, L.; Zhdanova, I.V. Impaired Sleep, Circadian Rhythms and Neurogenesis in Diet-Induced Premature Aging. Int. J. Mol. Sci. 2017, 18, 2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weger, M.; Diotel, N.; Dorsemans, A.C.; Dickmeis, T.; Weger, B.D. Stem cells and the circadian clock. Dev. Biol. 2017, 431, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Weger, M.; Weger, B.D.; Diotel, N.; Rastegar, S.; Hirota, T.; Kay, S.A.; Strahle, U.; Dickmeis, T. Real-time in vivo monitoring of circadian E-box enhancer activity: A robust and sensitive zebrafish reporter line for developmental, chemical and neural biology of the circadian clock. Dev. Biol. 2013, 380, 259–273. [Google Scholar] [CrossRef] [Green Version]
- Ogata, S.; Ito, S.; Masuda, T.; Ohtsuki, S. Changes of Blood-Brain Barrier and Brain Parenchymal Protein Expression Levels of Mice under Different Insulin-Resistance Conditions Induced by High-Fat Diet. Pharm. Res. 2019, 36, 141. [Google Scholar] [CrossRef]
- Salameh, T.S.; Mortell, W.G.; Logsdon, A.F.; Butterfield, D.A.; Banks, W.A. Disruption of the hippocampal and hypothalamic blood-brain barrier in a diet-induced obese model of type II diabetes: Prevention and treatment by the mitochondrial carbonic anhydrase inhibitor, topiramate. Fluids Barriers CNS 2019, 16, 1. [Google Scholar] [CrossRef] [Green Version]
- Valcarcel-Ares, M.N.; Tucsek, Z.; Kiss, T.; Giles, C.B.; Tarantini, S.; Yabluchanskiy, A.; Balasubramanian, P.; Gautam, T.; Galvan, V.; Ballabh, P.; et al. Obesity in Aging Exacerbates Neuroinflammation, Dysregulating Synaptic Function-Related Genes and Altering Eicosanoid Synthesis in the Mouse Hippocampus: Potential Role in Impaired Synaptic Plasticity and Cognitive Decline. J. Gerontol A Biol. Sci Med. Sci. 2019, 74, 290–298. [Google Scholar] [CrossRef]
- Hersey, M.; Woodruff, J.L.; Maxwell, N.; Sadek, A.T.; Bykalo, M.K.; Bain, I.; Grillo, C.A.; Piroli, G.G.; Hashemi, P.; Reagan, L.P. High-fat diet induces neuroinflammation and reduces the serotonergic response to escitalopram in the hippocampus of obese rats. Brain Behav. Immun. 2021, 96, 63–72. [Google Scholar] [CrossRef]
- Ly, M.; Raji, C.A.; Yu, G.Z.; Wang, Q.; Wang, Y.; Schindler, S.E.; An, H.; Samara, A.; Eisenstein, S.A.; Hershey, T.; et al. Obesity and White Matter Neuroinflammation Related Edema in Alzheimer’s Disease Dementia Biomarker Negative Cognitively Normal Individuals. J. Alzheimers Dis. 2021, 79, 1801–1811. [Google Scholar] [CrossRef]
- Nam, S.M.; Kim, J.W.; Kwon, H.J.; Yoo, D.Y.; Jung, H.Y.; Kim, D.W.; Hwang, I.K.; Seong, J.K.; Yoon, Y.S. Differential Effects of Low- and High-dose Zinc Supplementation on Synaptic Plasticity and Neurogenesis in the Hippocampus of Control and High-fat Diet-fed Mice. Neurochem. Res. 2017, 42, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Suarez, J.; Rivera, P.; Aparisi Rey, A.; Perez-Martin, M.; Arrabal, S.; Rodriguez de Fonseca, F.; Ruiz de Azua, I.; Lutz, B. Adipocyte cannabinoid CB1 receptor deficiency alleviates high fat diet-induced memory deficit, depressive-like behavior, neuroinflammation and impairment in adult neurogenesis. Psychoneuroendocrinology 2019, 110, 104418. [Google Scholar] [CrossRef] [PubMed]
- Wongchitrat, P.; Lansubsakul, N.; Kamsrijai, U.; Sae-Ung, K.; Mukda, S.; Govitrapong, P. Melatonin attenuates the high-fat diet and streptozotocin-induced reduction in rat hippocampal neurogenesis. Neurochem. Int. 2016, 100, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Saiyasit, N.; Chunchai, T.; Prus, D.; Suparan, K.; Pittayapong, P.; Apaijai, N.; Pratchayasakul, W.; Sripetchwandee, J.; Chattipakorn, M.D.P.D.N.; Chattipakorn, S.C. Gut dysbiosis develops before metabolic disturbance and cognitive decline in high-fat diet-induced obese condition. Nutrition 2020, 69, 110576. [Google Scholar] [CrossRef]
- Durrington, P. Dyslipidaemia. Lancet 2003, 362, 717–731. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar]
- Grundy, S.M. Does Dietary Cholesterol Matter? Curr. Atheroscler. Rep. 2016, 18, 68. [Google Scholar] [CrossRef]
- Vincent, M.J.; Allen, B.; Palacios, O.M.; Haber, L.T.; Maki, K.C. Meta-regression analysis of the effects of dietary cholesterol intake on LDL and HDL cholesterol. Am. J. Clin. Nutr. 2019, 109, 7–16. [Google Scholar] [CrossRef]
- Bhatnagar, D.; Soran, H.; Durrington, P.N. Hypercholesterolaemia and its management. BMJ 2008, 337, a993. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, J.; Engel, D.F.; de Paula, G.C.; Dos Santos, D.B.; Lopes, J.B.; Farina, M.; Moreira, E.L.G.; de Bem, A.F. High Cholesterol Diet Exacerbates Blood-Brain Barrier Disruption in LDLr-/- Mice: Impact on Cognitive Function. J. Alzheimers Dis. 2020, 78, 97–115. [Google Scholar] [CrossRef]
- Czuba, E.; Steliga, A.; Lietzau, G.; Kowianski, P. Cholesterol as a modifying agent of the neurovascular unit structure and function under physiological and pathological conditions. Metab. Brain Dis. 2017, 32, 935–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirumangalakudi, L.; Prakasam, A.; Zhang, R.; Bimonte-Nelson, H.; Sambamurti, K.; Kindy, M.S.; Bhat, N.R. High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J. Neurochem. 2008, 106, 475–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, D.F.; Grzyb, A.N.; de Oliveira, J.; Potzsch, A.; Walker, T.L.; Brocardo, P.S.; Kempermann, G.; de Bem, A.F. Impaired adult hippocampal neurogenesis in a mouse model of familial hypercholesterolemia: A role for the LDL receptor and cholesterol metabolism in adult neural precursor cells. Mol. Metab. 2019, 30, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Singh, C.P. Histopathological changes in different tissues of T. fasciatus under the acute impact of BHC. Toxicol. Lett. 1982, 14, 151–156. [Google Scholar] [CrossRef]
- Zambon, D.; Quintana, M.; Mata, P.; Alonso, R.; Benavent, J.; Cruz-Sanchez, F.; Gich, J.; Pocovi, M.; Civeira, F.; Capurro, S.; et al. Higher incidence of mild cognitive impairment in familial hypercholesterolemia. Am. J. Med. 2010, 123, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Kim, J.H.; Choi, K.H.; Jang, Y.J.; Bae, S.S.; Choi, B.T.; Shin, H.K. Hypercholesterolemia accelerates amyloid beta-induced cognitive deficits. Int J. Mol. Med. 2013, 31, 577–582. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Zhai, Y.; Liang, X.; Chen, W.; Lin, R.; Ma, L.; Huang, Y.; Zhao, D.; Liang, Y.; Zhao, W.; et al. Connecting the Dots Between Hypercholesterolemia and Alzheimer’s Disease: A Potential Mechanism Based on 27-Hydroxycholesterol. Front. Neurosci. 2022, 16, 842814. [Google Scholar] [CrossRef]
- Maron, D.J.; Fazio, S.; Linton, M.F. Current perspectives on statins. Circulation 2000, 101, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Van Vliet, P. Cholesterol and late-life cognitive decline. J. Alzheimers Dis. 2012, 30 (Suppl. S2), S147–S162. [Google Scholar] [CrossRef]
- Swiger, K.J.; Manalac, R.J.; Blumenthal, R.S.; Blaha, M.J.; Martin, S.S. Statins and cognition: A systematic review and meta-analysis of short- and long-term cognitive effects. Mayo Clin. Proc. 2013, 88, 1213–1221. [Google Scholar] [CrossRef]
- Wagstaff, L.R.; Mitton, M.W.; Arvik, B.M.; Doraiswamy, P.M. Statin-associated memory loss: Analysis of 60 case reports and review of the literature. Pharmacotherapy 2003, 23, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Mandas, A.; Congiu, M.G.; Abete, C.; Dessi, S.; Manconi, P.E.; Musio, M.; Columbu, S.; Racugno, W. Cognitive decline and depressive symptoms in late-life are associated with statin use: Evidence from a population-based study of Sardinian old people living in their own home. Neurol Res. 2014, 36, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Hottman, D.; Cheng, S.; Gram, A.; LeBlanc, K.; Yuan, L.L.; Li, L. Systemic or Forebrain Neuron-Specific Deficiency of Geranylgeranyltransferase-1 Impairs Synaptic Plasticity and Reduces Dendritic Spine Density. Neuroscience 2018, 373, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Giampietro, C.; Lionetti, M.C.; Costantini, G.; Mutti, F.; Zapperi, S.; La Porta, C.A. Cholesterol impairment contributes to neuroserpin aggregation. Sci. Rep. 2017, 7, 43669. [Google Scholar] [CrossRef] [Green Version]
- Fabbro, S.; Seeds, N.W. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J. Neurochem. 2009, 109, 303–315. [Google Scholar] [CrossRef]
- Mollazadeh, H.; Tavana, E.; Fanni, G.; Bo, S.; Banach, M.; Pirro, M.; von Haehling, S.; Jamialahmadi, T.; Sahebkar, A. Effects of statins on mitochondrial pathways. J. Cachexia Sarcopenia Muscle 2021, 12, 237–251. [Google Scholar] [CrossRef]
- Urbano, F.; Bugliani, M.; Filippello, A.; Scamporrino, A.; Di Mauro, S.; Di Pino, A.; Scicali, R.; Noto, D.; Rabuazzo, A.M.; Averna, M.; et al. Atorvastatin but Not Pravastatin Impairs Mitochondrial Function in Human Pancreatic Islets and Rat beta-Cells. Direct Effect of Oxidative Stress. Sci. Rep. 2017, 7, 11863. [Google Scholar] [CrossRef] [Green Version]
- Stoletov, K.; Fang, L.; Choi, S.H.; Hartvigsen, K.; Hansen, L.F.; Hall, C.; Pattison, J.; Juliano, J.; Miller, E.R.; Almazan, F.; et al. Vascular lipid accumulation, lipoprotein oxidation, and macrophage lipid uptake in hypercholesterolemic zebrafish. Circ. Res. 2009, 104, 952–960. [Google Scholar] [CrossRef]
- Babin, P.J.; Vernier, J.M. Plasma lipoproteins in fish. J. Lipid Res. 1989, 30, 467–489. [Google Scholar] [CrossRef]
- Fang, L.; Liu, C.; Miller, Y.I. Zebrafish models of dyslipidemia: Relevance to atherosclerosis and angiogenesis. Transl. Res. 2014, 163, 99–108. [Google Scholar] [CrossRef] [Green Version]
- Holtta-Vuori, M.; Salo, V.T.; Nyberg, L.; Brackmann, C.; Enejder, A.; Panula, P.; Ikonen, E. Zebrafish: Gaining popularity in lipid research. Biochem. J. 2010, 429, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifton, J.D.; Lucumi, E.; Myers, M.C.; Napper, A.; Hama, K.; Farber, S.A.; Smith, A.B., III; Huryn, D.M.; Diamond, S.L.; Pack, M. Identification of novel inhibitors of dietary lipid absorption using zebrafish. PLoS ONE 2010, 5, e12386. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.S.; Fang, L.; Li, A.C.; Miller, Y.I. Ezetimibe and simvastatin reduce cholesterol levels in zebrafish larvae fed a high-cholesterol diet. Cholesterol 2012, 2012, 564705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, J. Orchestrating transcriptional control of adult neurogenesis. Genes Dev. 2012, 26, 1010–1021. [Google Scholar] [CrossRef] [Green Version]
- Beckervordersandforth, R.; Zhang, C.L.; Lie, D.C. Transcription-Factor-Dependent Control of Adult Hippocampal Neurogenesis. Cold Spring Harb. Perspect. Biol. 2015, 7, a018879. [Google Scholar] [CrossRef]
- Faigle, R.; Song, H. Signaling mechanisms regulating adult neural stem cells and neurogenesis. Biochim. Biophys. Acta 2013, 1830, 2435–2448. [Google Scholar] [CrossRef] [Green Version]
- Jurisch-Yaksi, N.; Yaksi, E.; Kizil, C. Radial glia in the zebrafish brain: Functional, structural, and physiological comparison with the mammalian glia. Glia 2020, 68, 2451–2470. [Google Scholar] [CrossRef]
- Chapouton, P.; Jagasia, R.; Bally-Cuif, L. Adult neurogenesis in non-mammalian vertebrates. Bioessays 2007, 29, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Labusch, M.; Mancini, L.; Morizet, D.; Bally-Cuif, L. Conserved and Divergent Features of Adult Neurogenesis in Zebrafish. Front. Cell Dev. Biol. 2020, 8, 525. [Google Scholar] [CrossRef]
- Aujla, P.K.; Naratadam, G.T.; Xu, L.; Raetzman, L.T. Notch/Rbpjkappa signaling regulates progenitor maintenance and differentiation of hypothalamic arcuate neurons. Development 2013, 140, 3511–3521. [Google Scholar] [CrossRef] [Green Version]
- Chapouton, P.; Webb, K.J.; Stigloher, C.; Alunni, A.; Adolf, B.; Hesl, B.; Topp, S.; Kremmer, E.; Bally-Cuif, L. Expression of hairy/enhancer of split genes in neural progenitors and neurogenesis domains of the adult zebrafish brain. J. Comp. Neurol. 2011, 519, 1748–1769. [Google Scholar] [CrossRef] [PubMed]
- Chapouton, P.; Skupien, P.; Hesl, B.; Coolen, M.; Moore, J.C.; Madelaine, R.; Kremmer, E.; Faus-Kessler, T.; Blader, P.; Lawson, N.D.; et al. Notch activity levels control the balance between quiescence and recruitment of adult neural stem cells. J. Neurosci. 2010, 30, 7961–7974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sueda, R.; Kageyama, R. Regulation of active and quiescent somatic stem cells by Notch signaling. Dev. Growth Differ. 2020, 62, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Engler, A.; Taylor, V. Notch: An interactive player in neurogenesis and disease. Cell Tissue Res. 2018, 371, 73–89. [Google Scholar] [CrossRef]
- Alunni, A.; Bally-Cuif, L. A comparative view of regenerative neurogenesis in vertebrates. Development 2016, 143, 741–753. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira-Carlos, V.; Ganz, J.; Hans, S.; Kaslin, J.; Brand, M. Notch receptor expression in neurogenic regions of the adult zebrafish brain. PLoS ONE 2013, 8, e73384. [Google Scholar] [CrossRef] [Green Version]
- Ables, J.L.; Decarolis, N.A.; Johnson, M.A.; Rivera, P.D.; Gao, Z.; Cooper, D.C.; Radtke, F.; Hsieh, J.; Eisch, A.J. Notch1 is required for maintenance of the reservoir of adult hippocampal stem cells. J. Neurosci. 2010, 30, 10484–10492. [Google Scholar] [CrossRef] [Green Version]
- Gaiano, N.; Nye, J.S.; Fishell, G. Radial glial identity is promoted by Notch1 signaling in the murine forebrain. Neuron 2000, 26, 395–404. [Google Scholar] [CrossRef] [Green Version]
- Piccin, D.; Yu, F.; Morshead, C.M. Notch signaling imparts and preserves neural stem characteristics in the adult brain. Stem Cells Dev. 2013, 22, 1541–1550. [Google Scholar] [CrossRef]
- Kishimoto, N.; Shimizu, K.; Sawamoto, K. Neuronal regeneration in a zebrafish model of adult brain injury. Dis. Models Mech. 2012, 5, 200–209. [Google Scholar] [CrossRef] [Green Version]
- Mendes-da-Silva, C.; Lemes, S.F.; Baliani Tda, S.; Versutti, M.D.; Torsoni, M.A. Increased expression of Hes5 protein in Notch signaling pathway in the hippocampus of mice offspring of dams fed a high-fat diet during pregnancy and suckling. Int. J. Dev. Neurosci. 2015, 40, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Jiang, M.; Yang, C.; Wu, Y.; Liu, Y.; Cui, Y.; Huang, G. Maternal high-fat diet affects Msi/Notch/Hes signaling in neural stem cells of offspring mice. J. Nutr. Biochem. 2014, 25, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tang, Y.; Cai, D. IKKbeta/NF-kappaB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat. Cell Biol. 2012, 14, 999–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Tay, S.S.; Ling, E.A.; Dheen, S.T. High glucose alters the expression of genes involved in proliferation and cell-fate specification of embryonic neural stem cells. Diabetologia 2006, 49, 1027–1038. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, D.C.; Harvey, E.M.; Suraj, R.; Erickson, S.L.; Mohammad, L.; Ren, M.; Liu, H.; He, G.; Kaplan, D.R.; Ellis, J.; et al. Methylglyoxal couples metabolic and translational control of Notch signalling in mammalian neural stem cells. Nat. Commun. 2020, 11, 2018. [Google Scholar] [CrossRef] [Green Version]
- Miyazono, K.; Kamiya, Y.; Morikawa, M. Bone morphogenetic protein receptors and signal transduction. J. Biochem 2010, 147, 35–51. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Hollnagel, A.; Oehlmann, V.; Heymer, J.; Ruther, U.; Nordheim, A. Id genes are direct targets of bone morphogenetic protein induction in embryonic stem cells. J. Biol. Chem. 1999, 274, 19838–19845. [Google Scholar] [CrossRef] [Green Version]
- Ling, F.; Kang, B.; Sun, X.H. Id proteins: Small molecules, mighty regulators. Curr. Top. Dev. Biol. 2014, 110, 189–216. [Google Scholar]
- Diotel, N.; Beil, T.; Strahle, U.; Rastegar, S. Differential expression of id genes and their potential regulator znf238 in zebrafish adult neural progenitor cells and neurons suggests distinct functions in adult neurogenesis. Gene Expr. Patterns 2015, 19, 1–13. [Google Scholar] [CrossRef]
- Nam, H.S.; Benezra, R. High levels of Id1 expression define B1 type adult neural stem cells. Cell Stem. Cell 2009, 5, 515–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Lubke, L.; Chen, F.; Beil, T.; Takamiya, M.; Diotel, N.; Strahle, U.; Rastegar, S. Neuron-Radial Glial Cell Communication via BMP/Id1 Signaling Is Key to Long-Term Maintenance of the Regenerative Capacity of the Adult Zebrafish Telencephalon. Cells 2021, 10, 2794. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ferg, M.; Lubke, L.; Takamiya, M.; Beil, T.; Gourain, V.; Diotel, N.; Strahle, U.; Rastegar, S. Bone morphogenetic protein signaling regulates Id1-mediated neural stem cell quiescence in the adult zebrafish brain via a phylogenetically conserved enhancer module. Stem Cells 2020, 38, 875–889. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Viales, R.; Diotel, N.; Ferg, M.; Armant, O.; Eich, J.; Alunni, A.; Marz, M.; Bally-Cuif, L.; Rastegar, S.; Strahle, U. The helix-loop-helix protein id1 controls stem cell proliferation during regenerative neurogenesis in the adult zebrafish telencephalon. Stem Cells 2015, 33, 892–903. [Google Scholar] [CrossRef]
- Jensen, G.S.; Leon-Palmer, N.E.; Townsend, K.L. Bone morphogenetic proteins (BMPs) in the central regulation of energy balance and adult neural plasticity. Metab. Clin. Exp. 2021, 123, 154837. [Google Scholar] [CrossRef]
- Baboota, R.K.; Bluher, M.; Smith, U. Emerging Role of Bone Morphogenetic Protein 4 in Metabolic Disorders. Diabetes 2021, 70, 303–312. [Google Scholar] [CrossRef]
- Townsend, K.L.; Suzuki, R.; Huang, T.L.; Jing, E.; Schulz, T.J.; Lee, K.; Taniguchi, C.M.; Espinoza, D.O.; McDougall, L.E.; Zhang, H.; et al. Bone morphogenetic protein 7 (BMP7) reverses obesity and regulates appetite through a central mTOR pathway. FASEB J. 2012, 26, 2187–2196. [Google Scholar] [CrossRef] [Green Version]
- Townsend, K.L.; Madden, C.J.; Blaszkiewicz, M.; McDougall, L.; Tupone, D.; Lynes, M.D.; Mishina, Y.; Yu, P.; Morrison, S.F.; Tseng, Y.H. Reestablishment of Energy Balance in a Male Mouse Model With POMC Neuron Deletion of BMPR1A. Endocrinology 2017, 158, 4233–4245. [Google Scholar] [CrossRef] [Green Version]
- Vegiopoulos, A.; Herzig, S. Glucocorticoids, metabolism and metabolic diseases. Mol. Cell Endocrinol 2007, 275, 43–61. [Google Scholar] [CrossRef] [Green Version]
- Cruz-Topete, D.; Cidlowski, J.A. One hormone, two actions: Anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation 2015, 22, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Nussinovitch, U.; de Carvalho, J.F.; Pereira, R.M.; Shoenfeld, Y. Glucocorticoids and the cardiovascular system: State of the art. Curr. Pharm. Des. 2010, 16, 3574–3585. [Google Scholar] [CrossRef] [PubMed]
- Fowden, A.L.; Forhead, A.J. Glucocorticoids as regulatory signals during intrauterine development. Exp. Physiol. 2015, 100, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Taves, M.D.; Gomez-Sanchez, C.E.; Soma, K.K. Extra-adrenal glucocorticoids and mineralocorticoids: Evidence for local synthesis, regulation, and function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E11–E24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEwen, B.S. Physiology and neurobiology of stress and adaptation: Central role of the brain. Physiol. Rev. 2007, 87, 873–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vierhapper, H.; Nowotny, P.; Waldhausl, W. Production rates of cortisol in obesity. Obes. Res. 2004, 12, 1421–1425. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.M.; Boulton, A.; Kumar, S.; Clark, P.M.; Shackleton, C.H. Cortisol metabolism in human obesity: Impaired cortisone-->cortisol conversion in subjects with central adiposity. J. Clin. Endocrinol. Metab. 1999, 84, 1022–1027. [Google Scholar] [CrossRef]
- Akalestou, E.; Genser, L.; Rutter, G.A. Glucocorticoid Metabolism in Obesity and Following Weight Loss. Front. Endocrinol. 2020, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Stomby, A.; Andrew, R.; Walker, B.R.; Olsson, T. Tissue-specific dysregulation of cortisol regeneration by 11betaHSD1 in obesity: Has it promised too much? Diabetologia 2014, 57, 1100–1110. [Google Scholar] [CrossRef] [Green Version]
- Rebuffe-Scrive, M.; Bronnegard, M.; Nilsson, A.; Eldh, J.; Gustafsson, J.A.; Bjorntorp, P. Steroid hormone receptors in human adipose tissues. J. Clin. Endocrinol. Metab. 1990, 71, 1215–1219. [Google Scholar] [CrossRef]
- Chiodini, I.; Adda, G.; Scillitani, A.; Coletti, F.; Morelli, V.; Di Lembo, S.; Epaminonda, P.; Masserini, B.; Beck-Peccoz, P.; Orsi, E.; et al. Cortisol secretion in patients with type 2 diabetes: Relationship with chronic complications. Diabetes Care 2007, 30, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, M.S.; Roy, A.; Brown, S. Increased urinary-free cortisol outputs in diabetic patients. J. Diabetes Complicat. 1998, 12, 24–27. [Google Scholar] [CrossRef]
- Sidibeh, C.O.; Pereira, M.J.; Abalo, X.M.; Boersma, J.G.; Skrtic, S.; Lundkvist, P.; Katsogiannos, P.; Hausch, F.; Castillejo-Lopez, C.; Eriksson, J.W. FKBP5 expression in human adipose tissue: Potential role in glucose and lipid metabolism, adipogenesis and type 2 diabetes. Endocrine 2018, 62, 116–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, C.P.; Hazlehurst, J.M.; Tomlinson, J.W. Glucocorticoids and non-alcoholic fatty liver disease. J. Steroid Biochem. Mol. Biol. 2015, 154, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, Y.; Wu, W.; Xu, T.; Yin, Y.; Zhang, J.; Huang, D.; Li, W. Chronic glucocorticoid exposure activates BK-NLRP1 signal involving in hippocampal neuron damage. J. Neuroinflamm. 2017, 14, 139. [Google Scholar] [CrossRef] [PubMed]
- Odaka, H.; Numakawa, T.; Yoshimura, A.; Nakajima, S.; Adachi, N.; Ooshima, Y.; Inoue, T.; Kunugi, H. Chronic glucocorticoid exposure suppressed the differentiation and survival of embryonic neural stem/progenitor cells: Possible involvement of ERK and PI3K/Akt signaling in the neuronal differentiation. Neurosci. Res. 2016, 113, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Morais, M.; Santos, P.A.; Mateus-Pinheiro, A.; Patricio, P.; Pinto, L.; Sousa, N.; Pedroso, P.; Almeida, S.; Filipe, A.; Bessa, J.M. The effects of chronic stress on hippocampal adult neurogenesis and dendritic plasticity are reversed by selective MAO-A inhibition. J. Psychopharmacol. 2014, 28, 1178–1183. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Rodrigues, A.J.; Silva, J.M.; Tronche, F.; Almeida, O.F.; Sousa, N.; Sotiropoulos, I. Chronic Stress and Glucocorticoids: From Neuronal Plasticity to Neurodegeneration. Neural. Plast. 2016, 2016, 6391686. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Oomen, C.; van Dam, A.M.; Wester, J.; Zhou, J.N.; Joels, M.; Lucassen, P.J. A single-day treatment with mifepristone is sufficient to normalize chronic glucocorticoid induced suppression of hippocampal cell proliferation. PLoS ONE 2012, 7, e46224. [Google Scholar] [CrossRef]
- Caruso, A.; Nicoletti, F.; Mango, D.; Saidi, A.; Orlando, R.; Scaccianoce, S. Stress as risk factor for Alzheimer’s disease. Pharmacol. Res. 2018, 132, 130–134. [Google Scholar] [CrossRef]
- Dey, A.; Hao, S.; Wosiski-Kuhn, M.; Stranahan, A.M. Glucocorticoid-mediated activation of GSK3beta promotes tau phosphorylation and impairs memory in type 2 diabetes. Neurobiol. Aging 2017, 57, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Wendelaar Bonga, S.E. The stress response in fish. Physiol. Rev. 1997, 77, 591–625. [Google Scholar] [CrossRef] [PubMed]
- Weger, M.; Diotel, N.; Weger, B.D.; Beil, T.; Zaucker, A.; Eachus, H.L.; Oakes, J.A.; do Rego, J.L.; Storbeck, K.H.; Gut, P.; et al. Expression and activity profiling of the steroidogenic enzymes of glucocorticoid biosynthesis and the fdx1 co-factors in zebrafish. J. Neuroendocrinol. 2018, 30, e12586. [Google Scholar] [CrossRef] [PubMed]
- Diotel, N.; Do Rego, J.L.; Anglade, I.; Vaillant, C.; Pellegrini, E.; Vaudry, H.; Kah, O. The brain of teleost fish, a source, and a target of sexual steroids. Front. Neurosci. 2011, 5, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diotel, N.; Do Rego, J.L.; Anglade, I.; Vaillant, C.; Pellegrini, E.; Gueguen, M.M.; Mironov, S.; Vaudry, H.; Kah, O. Activity and expression of steroidogenic enzymes in the brain of adult zebrafish. Eur. J. Neurosci. 2011, 34, 45–56. [Google Scholar] [CrossRef]
- Sakamoto, H.; Ukena, K.; Tsutsui, K. Activity and localization of 3beta-hydroxysteroid dehydrogenase/Delta5-Delta4-isomerase in the zebrafish central nervous system. J. Comp. Neurol. 2001, 439, 291–305. [Google Scholar] [CrossRef]
- Than-Trong, E.; Kiani, B.; Dray, N.; Ortica, S.; Simons, B.; Rulands, S.; Alunni, A.; Bally-Cuif, L. Lineage hierarchies and stochasticity ensure the long-term maintenance of adult neural stem cells. Sci. Adv. 2020, 6, eaaz5424. [Google Scholar] [CrossRef]
- Baumgart, E.V.; Barbosa, J.S.; Bally-Cuif, L.; Gotz, M.; Ninkovic, J. Stab wound injury of the zebrafish telencephalon: A model for comparative analysis of reactive gliosis. Glia 2012, 60, 343–357. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghaddar, B.; Diotel, N. Zebrafish: A New Promise to Study the Impact of Metabolic Disorders on the Brain. Int. J. Mol. Sci. 2022, 23, 5372. https://doi.org/10.3390/ijms23105372
Ghaddar B, Diotel N. Zebrafish: A New Promise to Study the Impact of Metabolic Disorders on the Brain. International Journal of Molecular Sciences. 2022; 23(10):5372. https://doi.org/10.3390/ijms23105372
Chicago/Turabian StyleGhaddar, Batoul, and Nicolas Diotel. 2022. "Zebrafish: A New Promise to Study the Impact of Metabolic Disorders on the Brain" International Journal of Molecular Sciences 23, no. 10: 5372. https://doi.org/10.3390/ijms23105372
APA StyleGhaddar, B., & Diotel, N. (2022). Zebrafish: A New Promise to Study the Impact of Metabolic Disorders on the Brain. International Journal of Molecular Sciences, 23(10), 5372. https://doi.org/10.3390/ijms23105372