
Design of Rational JAK3 Inhibitors Based on the Parent Core Structure of 1,7-Dihydro-Dipyrrolo [2,3-b:3′,2′-e] Pyridine



Abstract
:1. Introduction
2. Results and Discussion
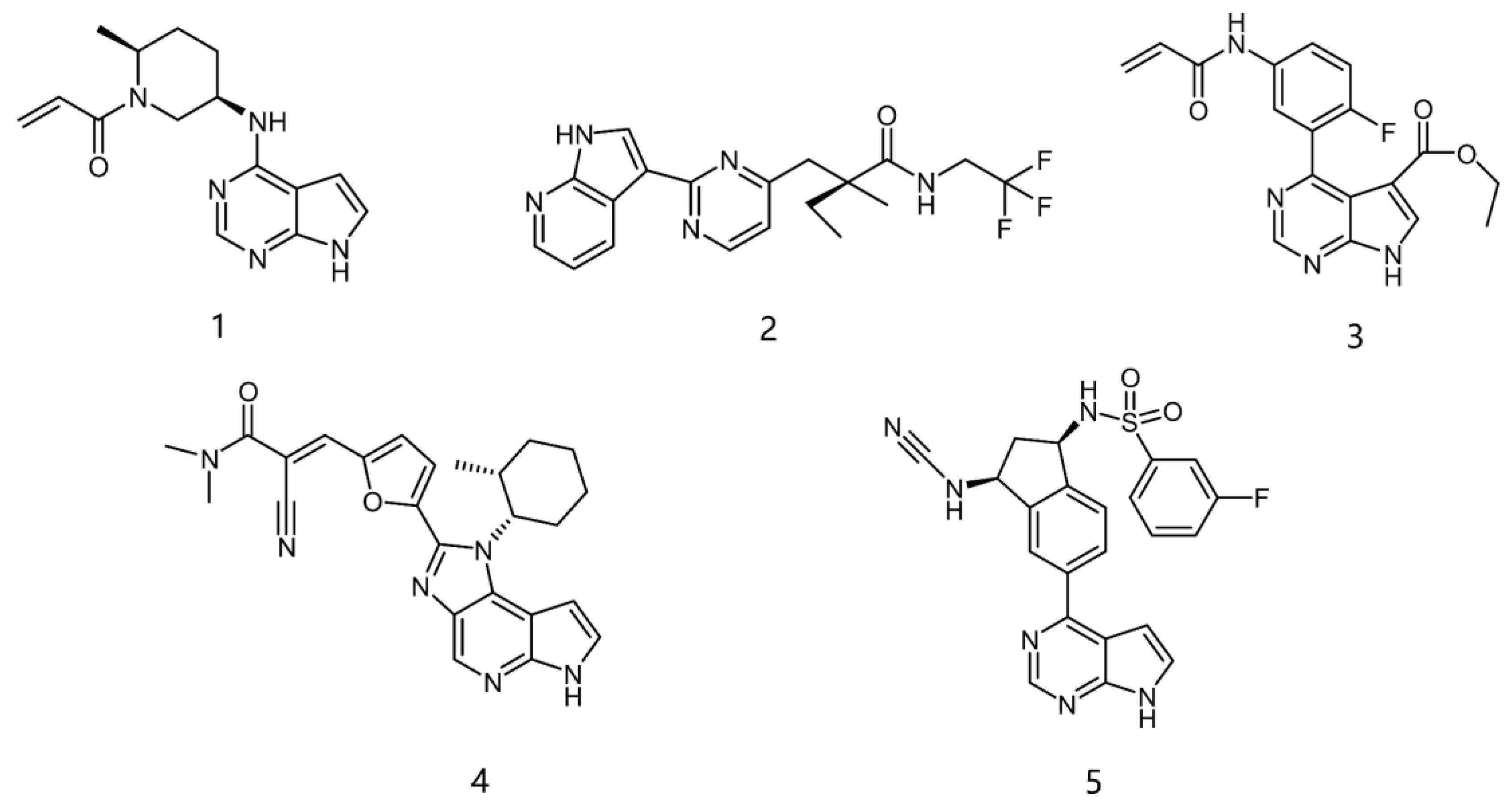
2.1. Structural Design of JAK3 Inhibitors
2.2. Analysis of Activity and Selectivity of the Inhibitors
2.3. ADMET Analysis
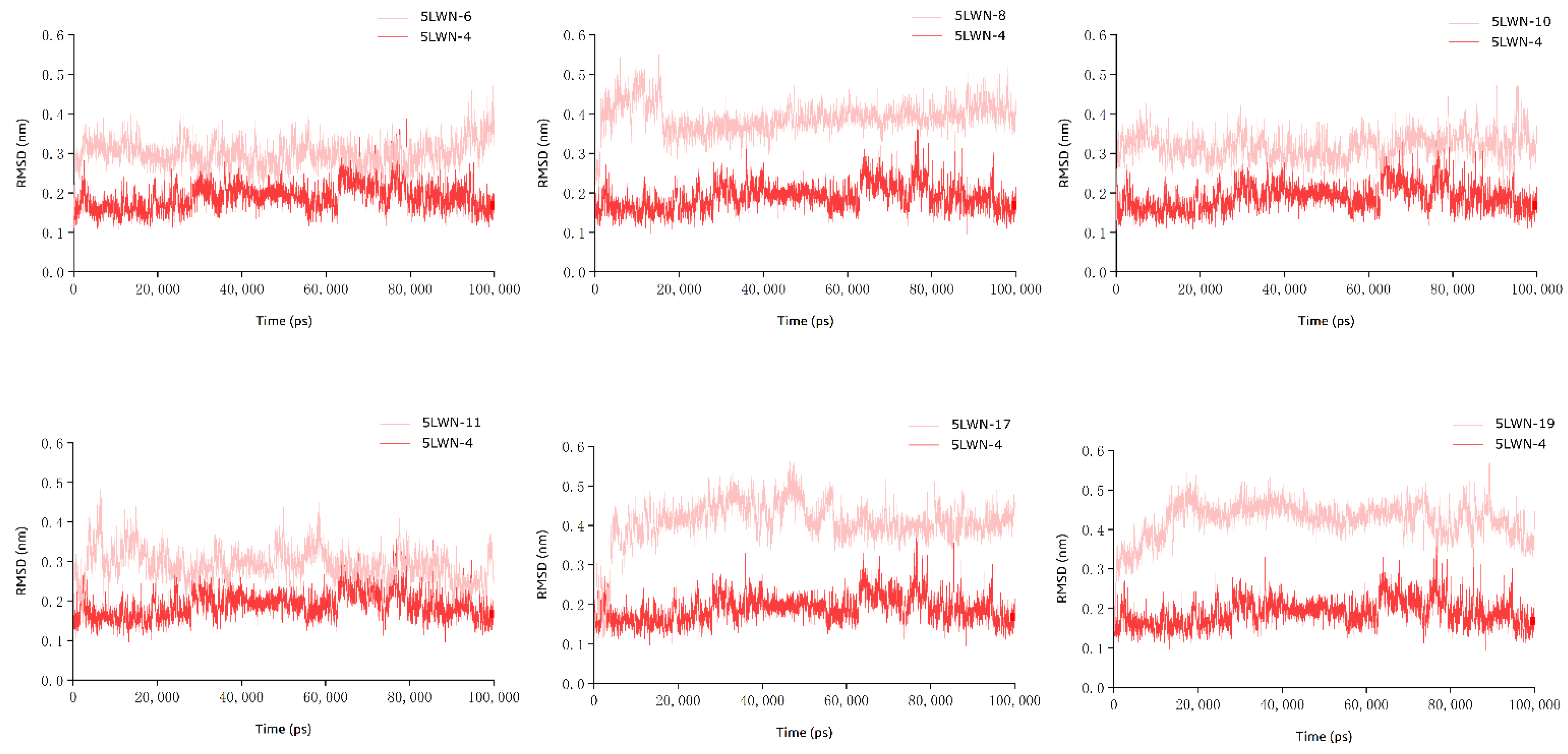
2.4. MD Simulation Analysis
2.5. Evaluation of the Binding Energy
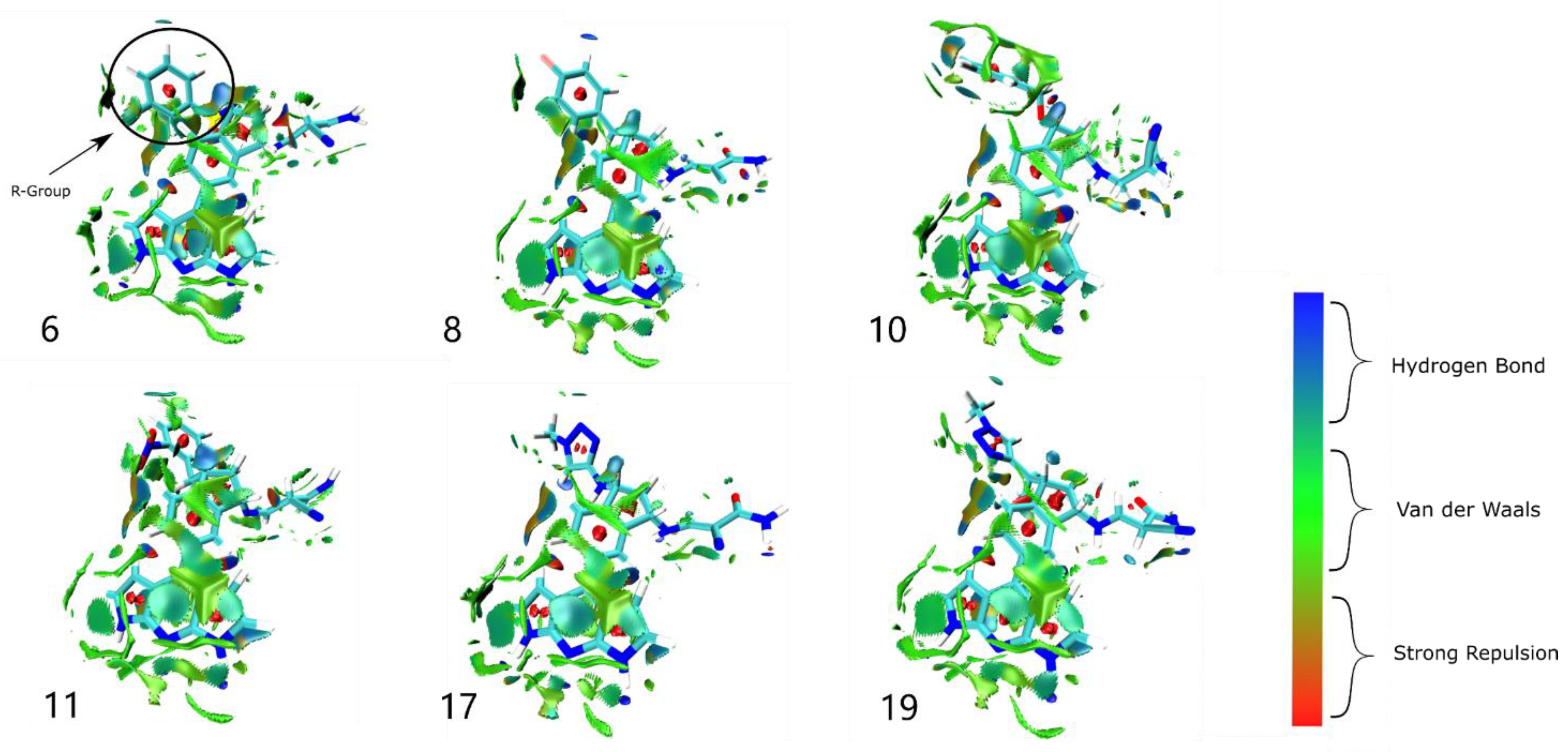
2.6. Weak Interaction Analysis
3. Materials and Methods
3.1. Receptor Preparation
3.2. Scaffold Growth and Ligand Preparation
3.3. ADMET Prediction
3.4. Molecular Docking
3.5. Molecular Dynamic Simulation
3.6. MM−PBSA Free Energy Calculations
3.7. Weak Interaction Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roda, G.; Dal Buono, A.; Argollo, M.; Danese, S. JAK selectivity: More precision less troubles. Expert Rev. Gastroenterol. Hepatol. 2020, 14, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Stabile, H.; Scarno, G.; Fionda, C.; Gismondi, A.; Santoni, A.; Gadina, M.; Sciume, G. JAK/STAT signaling in regulation of innate lymphoid cells: The gods before the guardians. Immunol. Rev. 2018, 286, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, H.T.; Chyuan, I.T.; Lai, J.H. Targeting the JAK-STAT pathway in autoimmune diseases and cancers: A focus on molecular mechanisms and therapeutic potential. Biochem. Pharmacol. 2021, 193, 114760. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Yi, E.H.; Jee, J.G.; Jeong, A.J.; Sandoval, C.; Park, I.C.; Baeg, G.H.; Ye, S.K. Tubulosine selectively inhibits JAK3 signalling by binding to the ATP-binding site of the kinase of JAK3. J. Cell Mol. Med. 2020, 24, 7427–7438. [Google Scholar] [CrossRef]
- Kempson, J.; Ovalle, D.; Guo, J.; Wrobleski, S.T.; Lin, S.; Spergel, S.H.; Duan, J.J.; Jiang, B.; Lu, Z.; Das, J.; et al. Discovery of highly potent, selective, covalent inhibitors of JAK3. Bioorg. Med. Chem. Lett. 2017, 27, 4622–4625. [Google Scholar] [CrossRef]
- Qiu, Q.; Feng, Q.; Tan, X.; Guo, M. JAK3-selective inhibitor peficitinib for the treatment of rheumatoid arthritis. Expert Rev. Clin. Pharmacol. 2019, 12, 547–554. [Google Scholar] [CrossRef]
- Keeling, S.; Maksymowych, W.P. JAK inhibitors, psoriatic arthritis, and axial spondyloarthritis: A critical review of clinical trials. Expert Rev. Clin. Immunol. 2021, 17, 701–715. [Google Scholar] [CrossRef]
- Campanaro, F.; Batticciotto, A.; Zaffaroni, A.; Cappelli, A.; Donadini, M.P.; Squizzato, A. JAK inhibitors and psoriatic arthritis: A systematic review and meta-analysis. Autoimmun. Rev. 2021, 20, 102902. [Google Scholar] [CrossRef]
- McLornan, D.P.; Pope, J.E.; Gotlib, J.; Harrison, C.N. Current and future status of JAK inhibitors. Lancet 2021, 398, 803–816. [Google Scholar] [CrossRef]
- Lu, X.; Smaill, J.B.; Patterson, A.V.; Ding, K. Discovery of Cysteine-targeting covalent protein kinase inhibitors. J. Med. Chem. 2022, 65, 58–83. [Google Scholar] [CrossRef]
- Dai, J.; Yang, L.; Addison, G. Current status in the discovery of covalent janus kinase 3 (JAK3) inhibitors. Mini. Rev. Med. Chem. 2019, 19, 1531–1543. [Google Scholar] [CrossRef]
- Remenyi, J.; Naik, R.J.; Wang, J.; Razsolkov, M.; Verano, A.; Cai, Q.; Tan, L.; Toth, R.; Raggett, S.; Baillie, C.; et al. Generation of a chemical genetic model for JAK3. Sci. Rep. 2021, 11, 10093. [Google Scholar] [CrossRef]
- Thorarensen, A.; Dowty, M.E.; Banker, M.E.; Juba, B.; Jussif, J.; Lin, T.; Vincent, F.; Czerwinski, R.M.; Casimiro-Garcia, A.; Unwalla, R.; et al. Design of a janus kinase 3 (JAK3) specific inhibitor 1-((2S,5R)-5-((7H-pyrrolo [2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one (PF-06651600) allowing for the interrogation of JAK3 signaling in humans. J. Med. Chem. 2017, 60, 1971–1993. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Miller, R.M.; Krishnan, S.; Uchida, K.; Irwin, J.J.; Eidam, O.; Gibold, L.; Cimermancic, P.; Bonnet, R.; Shoichet, B.K.; et al. Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol. 2014, 10, 1066–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forster, M.; Chaikuad, A.; Bauer, S.M.; Holstein, J.; Robers, M.B.; Corona, C.R.; Gehringer, M.; Pfaffenrot, E.; Ghoreschi, K.; Knapp, S.; et al. Selective JAK3 inhibitors with a covalent reversible binding mode targeting a new induced fit binding pocket. Cell Chem. Biol. 2016, 23, 1335–1340. [Google Scholar] [CrossRef] [Green Version]
- Su, Q.; Ioannidis, S.; Chuaqui, C.; Almeida, L.; Alimzhanov, M.; Bebernitz, G.; Bell, K.; Block, M.; Howard, T.; Huang, S.; et al. Discovery of 1-methyl-1H-imidazole derivatives as potent Jak2 inhibitors. J. Med. Chem. 2014, 57, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Telliez, J.B.; Dowty, M.E.; Wang, L.; Jussif, J.; Lin, T.; Li, L.; Moy, E.; Balbo, P.; Li, W.; Zhao, Y.; et al. Discovery of a JAK3-selective inhibitor: Functional differentiation of JAK3-selective inhibition over pan-JAK or JAK1-selective inhibition. ACS Chem. Biol. 2016, 11, 3442–3451. [Google Scholar] [CrossRef]
- Chen, C.; Lu, D.; Sun, T.; Zhang, T. JAK3 inhibitors for the treatment of inflammatory and autoimmune diseases: A patent review (2016-present). Expert Opin. Ther. Pat. 2022, 32, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Chrencik, J.E.; Patny, A.; Leung, I.K.; Korniski, B.; Emmons, T.L.; Hall, T.; Weinberg, R.A.; Gormley, J.A.; Williams, J.M.; Day, J.E.; et al. Structural and thermodynamic characterization of the TYK2 and JAK3 kinase domains in complex with CP-690550 and CMP-6. J. Mol. Biol. 2010, 400, 413–433. [Google Scholar] [CrossRef]
- Soth, M.; Hermann, J.C.; Yee, C.; Alam, M.; Barnett, J.W.; Berry, P.; Browner, M.F.; Frank, K.; Frauchiger, S.; Harris, S.; et al. 3-Amido pyrrolopyrazine JAK kinase inhibitors: Development of a JAK3 vs JAK1 selective inhibitor and evaluation in cellular and in vivo models. J. Med. Chem. 2013, 56, 345–356. [Google Scholar] [CrossRef]
- Goedken, E.R.; Argiriadi, M.A.; Banach, D.L.; Fiamengo, B.A.; Foley, S.E.; Frank, K.E.; George, J.S.; Harris, C.M.; Hobson, A.D.; Ihle, D.C.; et al. Tricyclic covalent inhibitors selectively target Jak3 through an active site thiol. J. Biol. Chem. 2015, 290, 4573–4589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casimiro-Garcia, A.; Trujillo, J.I.; Vajdos, F.; Juba, B.; Banker, M.E.; Aulabaugh, A.; Balbo, P.; Bauman, J.; Chrencik, J.; Coe, J.W.; et al. Identification of cyanamide-based janus kinase 3 (JAK3) covalent inhibitors. J. Med. Chem. 2018, 61, 10665–10699. [Google Scholar] [CrossRef] [PubMed]
- Forster, M.; Chaikuad, A.; Dimitrov, T.; Doring, E.; Holstein, J.; Berger, B.T.; Gehringer, M.; Ghoreschi, K.; Muller, S.; Knapp, S.; et al. Development, Optimization, and structure-activity relationships of covalent-reversible JAK3 inhibitors based on a tricyclic imidazo [5,4-d]pyrrolo [2,3-b]pyridine scaffold. J. Med. Chem. 2018, 61, 5350–5366. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Leszczynski, J. Open access in silico tools to predict the ADMET profiling of drug candidates. Expert Opin. Drug Discov. 2020, 15, 1473–1487. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Lu, A.; et al. ADMETlab 2.0: An integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef]
- Guosheng, W.; Daniel, H.R.; Charles, I.B.; Michal, V. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1550–1562. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Valdes-Tresanco, M.S.; Valdes-Tresanco, M.E.; Valiente, P.A.; Moreno, E. AMDock: A versatile graphical tool for assisting molecular docking with Autodock Vina and Autodock4. Biol. Direct. 2020, 15, 12. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wataru, S.; Masuhiro, M. Rigid-body dynamics in the isothermal-isobaric ensemble: A test on the accuracy and computational efficiency. J. Comput. Chem. 2003, 24, 920–930. [Google Scholar] [CrossRef]
- Berk, H. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics poisson-boltzmann surface area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Wu, P.; Chaudret, R.; Hu, X.; Yang, W. Noncovalent interaction analysis in fluctuating environments. J. Chem. Theory Comput. 2013, 9, 2226–2234. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Tian, L. Multiwfn: A Multifunctional Wavefunction Analyzer; Beijing Kein Research Center for Natural Sciences: Beijing, China, 2022; pp. 276–277. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bai, Q.; Yao, X. Investigation of allosteric modulation mechanism of metabotropic glutamate receptor 1 by molecular dynamics simulations, free energy and weak interaction analysis. Sci. Rep. 2016, 6, 21763. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitors | R | Affinity (kcal/mol) | Estimated Ki (nm) | H-Bond Interaction | Hydrophobic Interaction | Halogen Interaction |
|---|---|---|---|---|---|---|
| 6 |  | −10.1 | 39.51 | GLU903, LEU905, ARG911, ARG953 | LEU828, VAL836, ALA853, MET902, LEU956 | ASP967 |
| 7 | 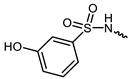 | −10.8 | 12.12 | GLU903, LEU905, AGR953, ASN954 | LEU828, ALA853, VAL884, VAL836, MET902, LEU956, ALA966, ASP967 | - |
| 8 | 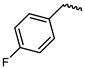 | −11.8 | 2.24 | LYS855, GLU903, LEU905, ASP912, AGR953 | LEY828, VAL836, ALA853, LEU956, MET902, ASP967 | ASP967 |
| 9 | 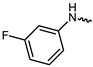 | −11.1 | 7.31 | LYS855, GLU871, GLU903, LEU905, ASP967 | LEU828, VAL836, ALA853, MET902, LEU956 | - |
| 10 | 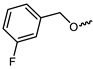 | −11.0 | 8.65 | GLU903, LEU905, ASP912, AGR953 | LEU828, LYS830, VAL836, ALA853, MET902, LEU956 | ASP967 |
| 11 | 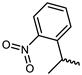 | −11.1 | 7.31 | GLU903, LEU905, AGR911, AGR953 | LEU828, VAL836, ALA853, MET902, LEU956, ASP967 | - |
| 12 | 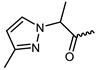 | −11.0 | 8.65 | LEU905, GLU903, AGR911, AGR953 | LEU828, VAL836, ALA853, MET902, LEU956, GLY908, ASP967 | - |
| 13 |  | −11.2 | 6.17 | LYS855, GLU903, LEU905, AGR911, ASP912, AGR953 | LEU828, VAL836, ALA853, MET902, LEU956 | - |
| 14 | 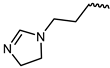 | −10.8 | 12.12 | LYS855, GLU903, LEU905, AGR911, ASP912, AGR953, | LEU828, VAL836, ALA853, MET902, GLY908, LEU956 | - |
| 15 | 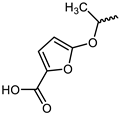 | −10.7 | 14.35 | LYS855, GLU903, LEU905, CYS909, AGR911, AGR953 | LEU828, LYS830, VAL836, ALA853, VAL884, MET902, LEU956, ALA966 | - |
| 16 | 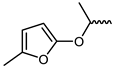 | −11.8 | 2.24 | LEU905, GLU903, AGR953, ASP912 | LEU828, VAL836, ALA853, MET902, LEU956, ASP967 | - |
| 17 |  | −11.3 | 5.21 | GLU903, LEU905, ASP912, AGR953 | LEU828, VAL836, ALA853, MET902, LEU956, ASP967 | - |
| 18 |  | −11.3 | 5.21 | GLU903, LEU905, ASP91, 2AGR953 | LEU828, VAL836, ALA853, LYS855, MET902, LEU956, ASP967 | - |
| 19 |  | −11.6 | 3.14 | GLU903, LEU905, ASP912, AGR953 | LEU828, VAL836, ALA853, MET902, LEU956, ASP967 | - |
| Inhibitors | Lipinski Rules a | HIA b | PPB c | BBB Permeant | CYP2D6 Inhibitor | hERG Blockers d | Carcinogenicity e |
|---|---|---|---|---|---|---|---|
| 6 | Accepted | 0.012 | 95.78% | No | No | 0.158 | 0.052 |
| 8 | Accepted | 0.005 | 97.90% | No | No | 0.175 | 0.055 |
| 10 | Rejected | 0.007 | 96.23% | No | No | 0.27 | 0.039 |
| 11 | Rejected | 0.009 | 98.23% | No | No | 0.209 | 0.091 |
| 17 | Accepted | 0.014 | 89.07% | No | No | 0.201 | 0.169 |
| 19 | Accepted | 0.006 | 93.07% | No | No | 0.323 | 0.179 |
| Inhibitors | SASA Energy (kJ/mol) | Polar Solvation Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | van der Waal Energy (kJ/mol) | Binding Free Energy (kJ/mol) |
|---|---|---|---|---|---|
| 4 | −21.532 +/− 1.219 | 192.312 +/− 18.531 | −34.305 +/− 11.398 | −202.871 +/− 14.174 | −66.395 +/− 14.892 |
| 6 | −22.127 +/− 1.023 | 248.181 +/− 22.567 | −73.232 +/− 13.944 | −204.047 +/− 13.286 | −51.225 +/− 16.394 |
| 8 | −20.542 +/− 0.875 | 202.732 +/− 11.996 | −54.868 +/− 8.139 | −197.608 +/− 12.352 | −70.286 +/− 11.390 |
| 10 | −20.840 +/− 1.026 | 248.490 +/− 23.490 | −82.609 +/− 12.429 | −186.017 +/− 13.120 | −40.975 +/− 17.830 |
| 11 | −21.146+/− 1.106 | 128.534 +/− 60.574 | 23.361 +/− 31.773 | −195.273 +/− 14.230 | −64.523 +/− 30.463 |
| 17 | −20.403 +/− 0.974 | 244.416 +/− 19.635 | −64.784 +/− 10.452 | −202.051 +/− 12.772 | −42.822 +/− 15.484 |
| 19 | −20.165 +/− 1.000 | 245.281 +/− 23.425 | −65.409 +/− 11.797 | −199.462 +/− 13.097 | −39.754 +/− 18.304 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Meng, D.; Xie, J.; Li, R.; Wang, Z.; Li, J.; Mou, L.; Deng, X.; Deng, P. Design of Rational JAK3 Inhibitors Based on the Parent Core Structure of 1,7-Dihydro-Dipyrrolo [2,3-b:3′,2′-e] Pyridine. Int. J. Mol. Sci. 2022, 23, 5437. https://doi.org/10.3390/ijms23105437
Li Y, Meng D, Xie J, Li R, Wang Z, Li J, Mou L, Deng X, Deng P. Design of Rational JAK3 Inhibitors Based on the Parent Core Structure of 1,7-Dihydro-Dipyrrolo [2,3-b:3′,2′-e] Pyridine. International Journal of Molecular Sciences. 2022; 23(10):5437. https://doi.org/10.3390/ijms23105437
Chicago/Turabian StyleLi, Yihao, Dan Meng, Jiali Xie, Ruoyu Li, Zifan Wang, Jinlong Li, Lin Mou, Xinhao Deng, and Ping Deng. 2022. "Design of Rational JAK3 Inhibitors Based on the Parent Core Structure of 1,7-Dihydro-Dipyrrolo [2,3-b:3′,2′-e] Pyridine" International Journal of Molecular Sciences 23, no. 10: 5437. https://doi.org/10.3390/ijms23105437
APA StyleLi, Y., Meng, D., Xie, J., Li, R., Wang, Z., Li, J., Mou, L., Deng, X., & Deng, P. (2022). Design of Rational JAK3 Inhibitors Based on the Parent Core Structure of 1,7-Dihydro-Dipyrrolo [2,3-b:3′,2′-e] Pyridine. International Journal of Molecular Sciences, 23(10), 5437. https://doi.org/10.3390/ijms23105437