
Combined Transcriptome and Metabolome Analyses Reveal Candidate Genes Involved in Tangor (Citrus reticulata × Citrus sinensis) Fruit Development and Quality Formation

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Experimental Design
2.2. Determination of Fruit Firmness and Sugar and Organic Acid Content
2.3. Metabolite Profiling Using UPLC-MS/MS and Data Analysis
2.4. RNA Library Preparation and Sequencing
2.5. Quantification of Gene Transcription Level and Analysis of DEGs
2.6. Quantitative Real-Time PCR (qRT-PCR) Analysis
2.7. Statistical Analysis
3. Results
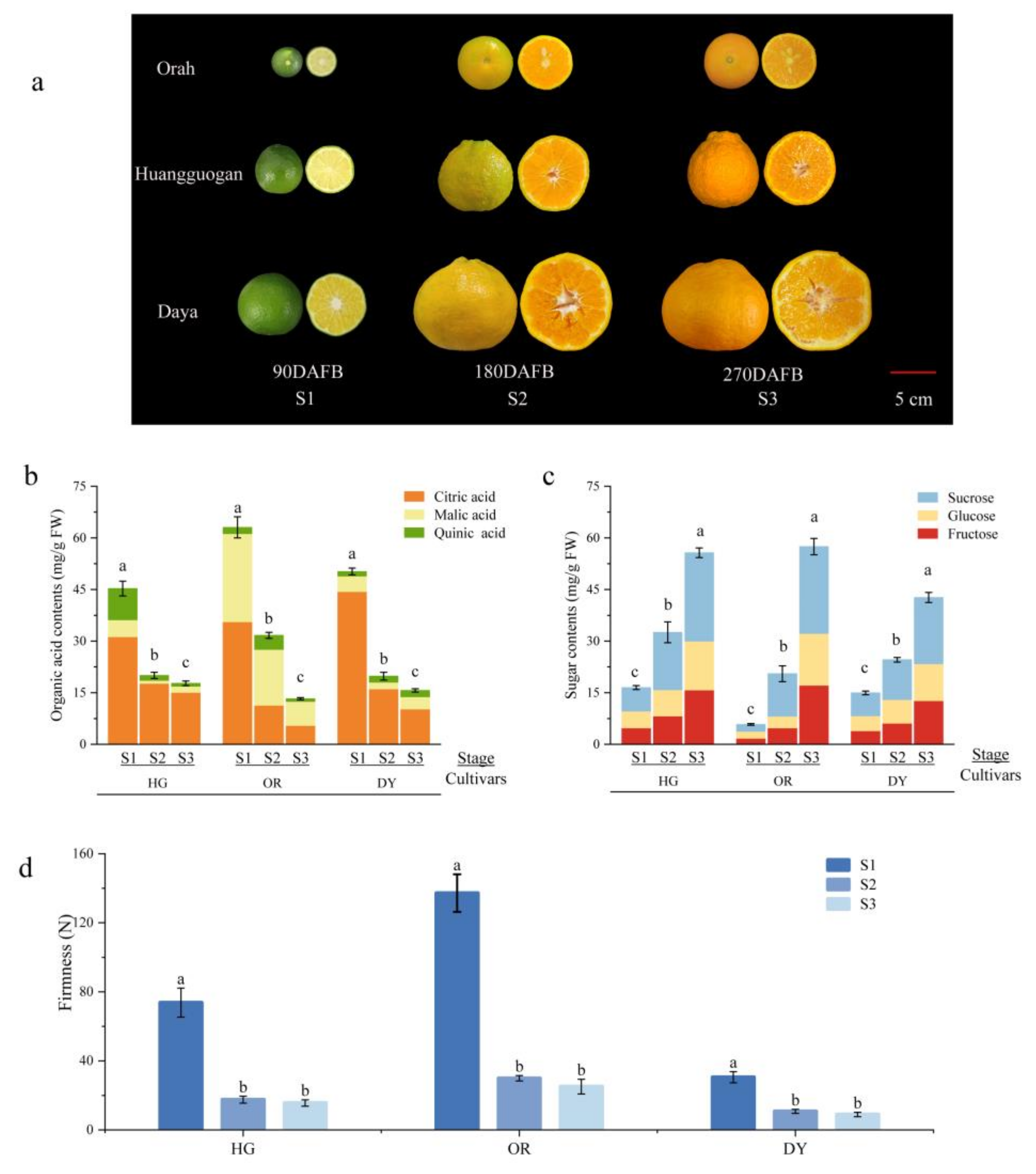
3.1. Changes in Fruit Appearance and Physiological Indicators at Different Developmental Stages
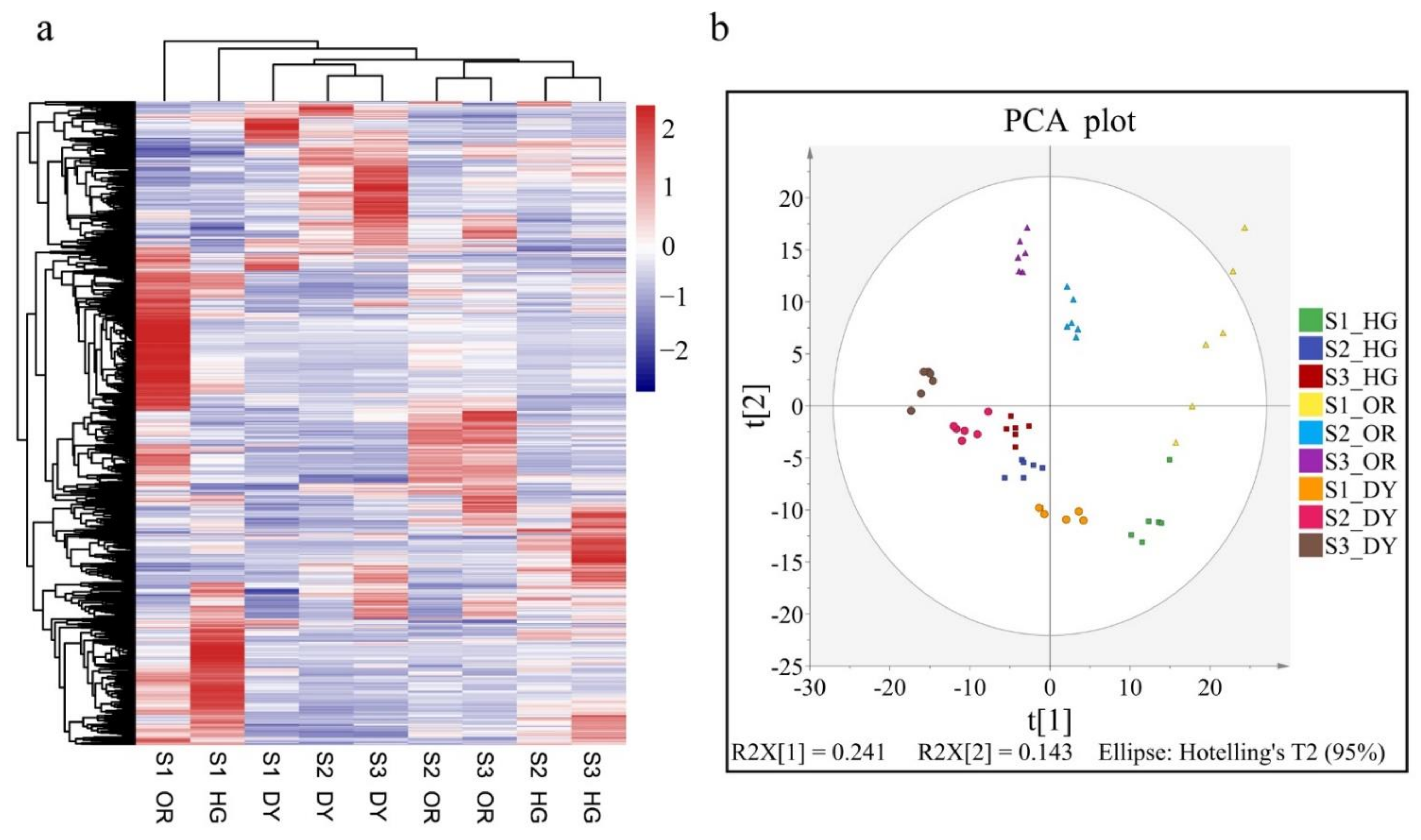
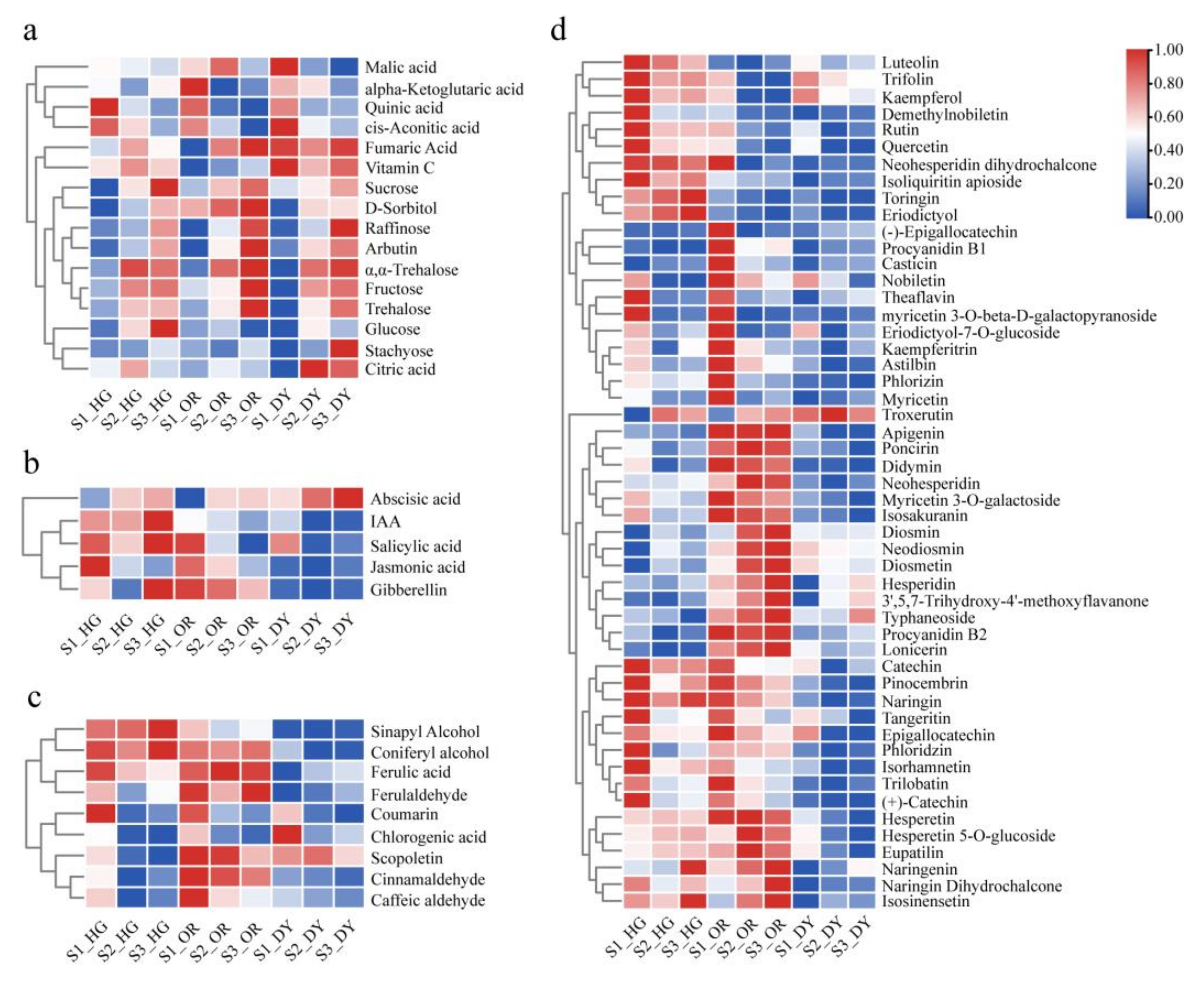
3.2. UPLC-MS/MS-Based Quantitative Metabolomic Analysis of the Citrus Fruits
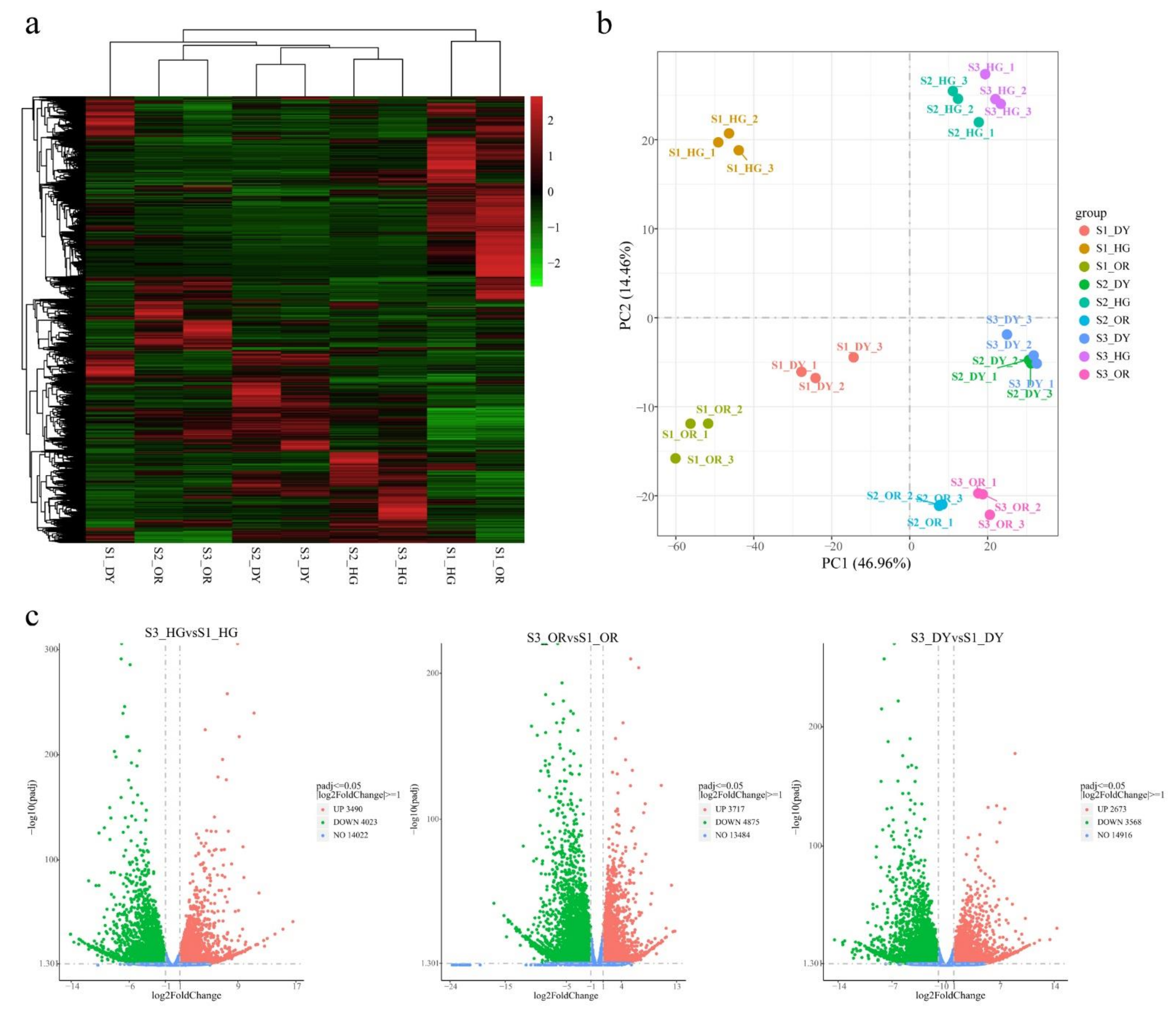
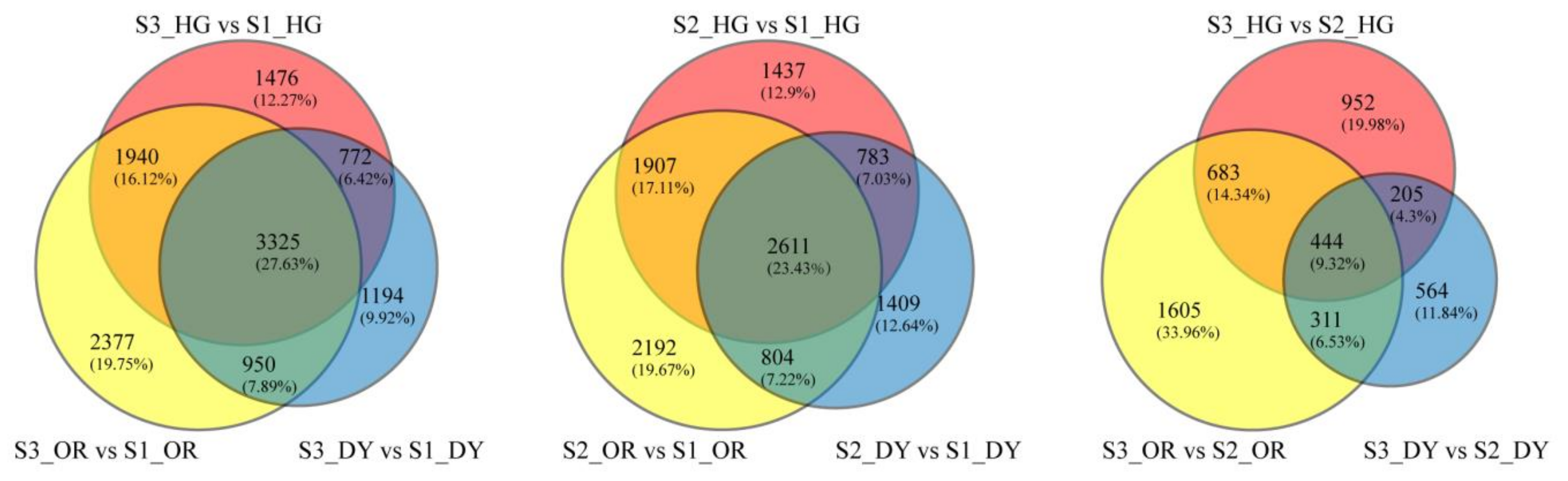
3.3. General Description of the Transcriptome
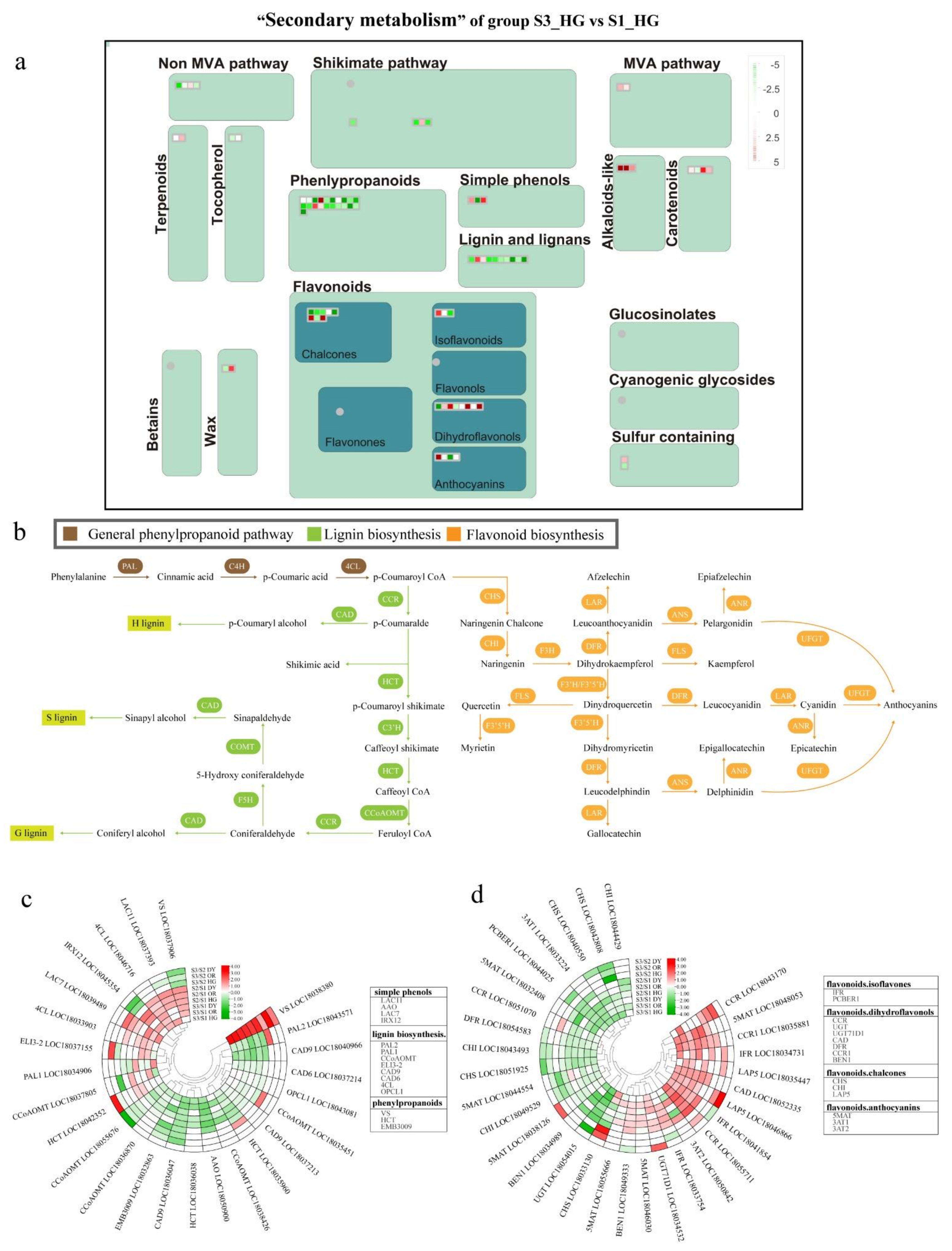
3.4. Differentially Expressed Endogenous Metabolism-Related Genes at Different Developmental Stages
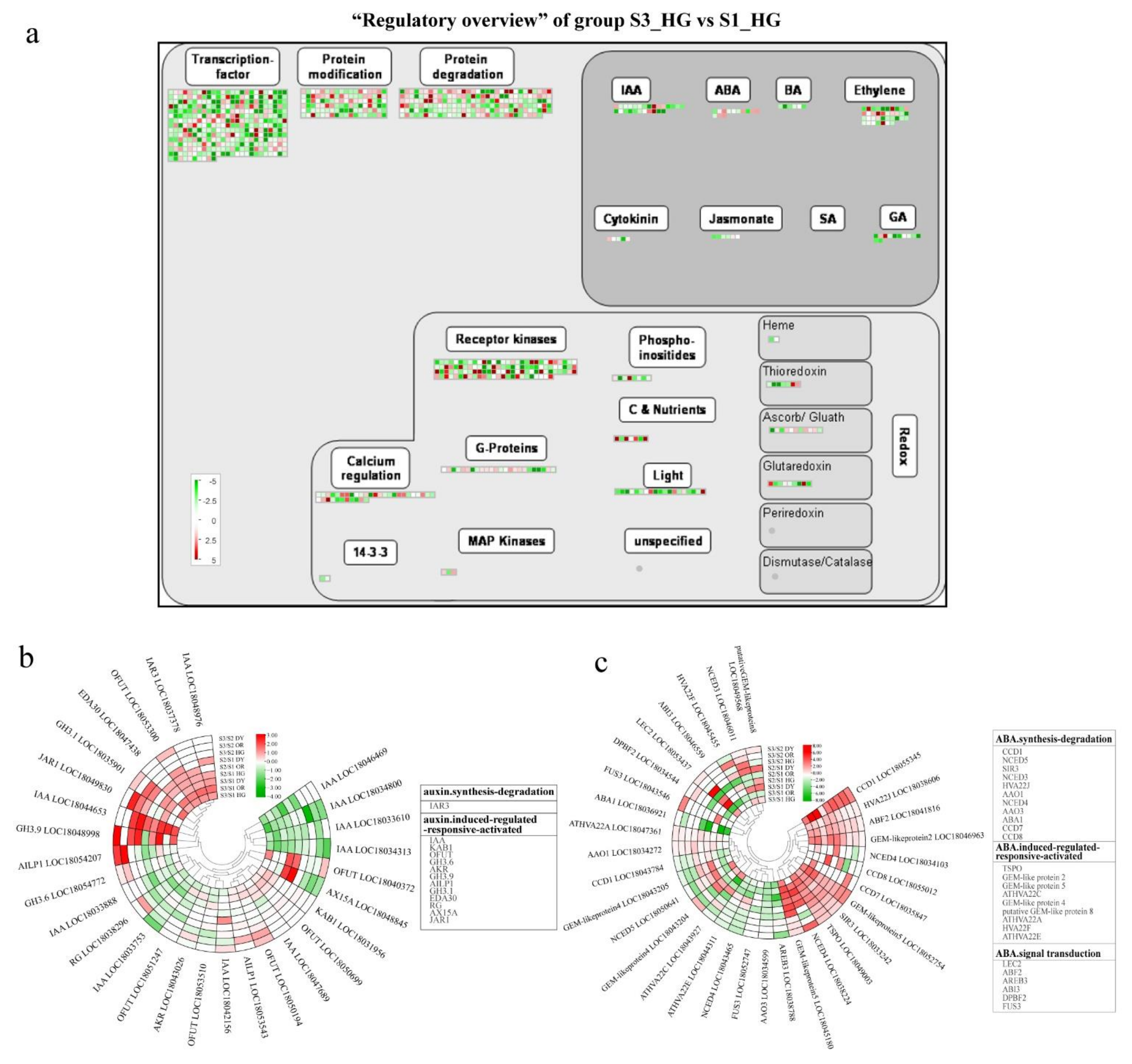
3.5. Plant Hormone-Related DEGs during Fruit Development and Ripening
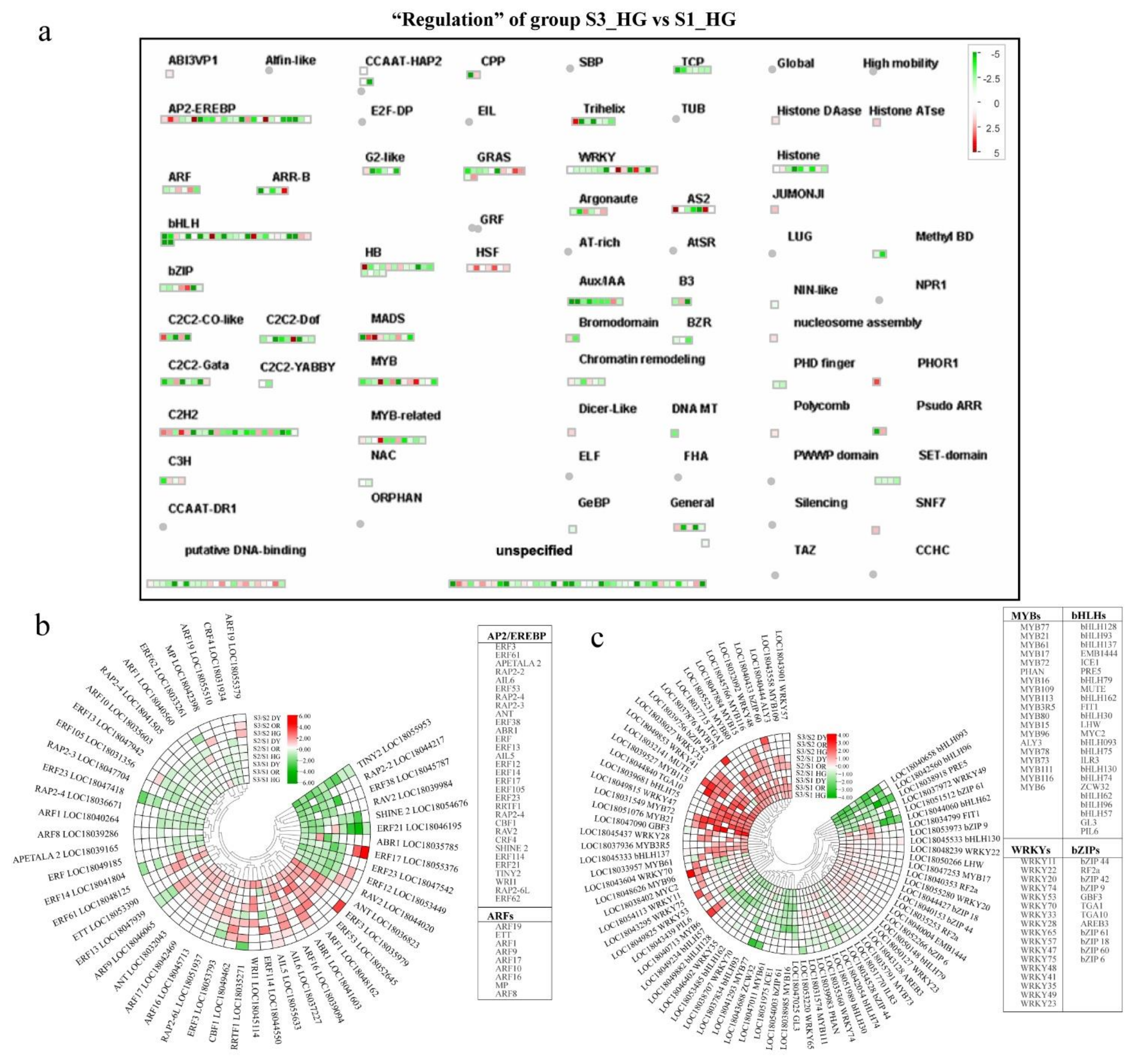
3.6. Differential Expression of Transcriptional Regulatory Genes during Fruit Development and Ripening
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Putnik, P.; Barba, F.J.; Lorenzo, J.M.; Gabrić, D.; Shpigelman, A.; Cravotto, G.; Bursać Kovačević, D. An Integrated Approach to Mandarin Processing: Food Safety and Nutritional Quality, Consumer Preference, and Nutrient Bioaccessibility. Compr. Rev. Food Sci. Food Saf. 2017, 16, 1345–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.A.; Sugimoto, C.; Kinjo, H.; Azama, C.; Mitsube, F.; Talon, M.; Gmitter, F.G., Jr.; Rokhsar, D.S. Diversification of mandarin citrus by hybrid speciation and apomixis. Nat. Commun. 2021, 12, 4377. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T. Misunderstanding with Regards Citrus Classification and Nomeclature. Bull. Univ. Osaka Prefect. Ser. B Agric. Biol. 1969, 21, 139–145. [Google Scholar]
- Feng, S.; Niu, L.; Suh, J.H.; Hung, W.L.; Wang, Y. Comprehensive Metabolomics Analysis of Mandarins (Citrus reticulata) as a Tool for Variety, Rootstock, and Grove Discrimination. J. Agric. Food Chem. 2018, 66, 10317–10326. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Kim, H.M.; Lee, S. Simultaneous determination of polymethoxyflavones in Citrus species, Kiyomi tangor and Satsuma mandarin, by high performance liquid chromatography. Food Chem. 2012, 134, 1220–1224. [Google Scholar] [CrossRef]
- Cirmi, S.; Navarra, M.; Woodside, J.V.; Cantwell, M.M. Citrus fruits intake and oral cancer risk: A systematic review and meta-analysis. Pharm. Res. 2018, 133, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Li, L.J.; Tan, W.S.; Li, W.J.; Zhu, Y.B.; Cheng, Y.S.; Ni, H. Citrus Taste Modification Potentials by Genetic Engineering. Int. J. Mol. Sci. 2019, 20, 6194. [Google Scholar] [CrossRef] [Green Version]
- Feng, G.; Ai, X.; Yi, H.; Guo, W.; Wu, J. Genomic and transcriptomic analyses of Citrus sinensis varieties provide insights into Valencia orange fruit mastication trait formation. Hortic. Res. 2021, 8, 218. [Google Scholar] [CrossRef]
- Lado, J.; Gambetta, G.; Zacarias, L. Key determinants of citrus fruit quality: Metabolites and main changes during maturation. Sci. Hortic. 2018, 233, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, Z.; Ding, F.; Sun, D.; Ma, Z.; Cheng, Y.; Xu, J. Content changes of bitter compounds in ‘Guoqing No.1’ Satsuma mandarin (Citrus unshiu Marc.) during fruit development of consecutive 3 seasons. Food Chem. 2014, 145, 963–969. [Google Scholar] [CrossRef]
- Jing, L.; Lei, Z.; Zhang, G.; Pilon, A.C.; Huhman, D.V.; Xie, R.; Xi, W.; Zhou, Z.; Sumner, L.W. Metabolite profiles of essential oils in citrus peels and their taxonomic implications. Metabolomics 2015, 11, 952–963. [Google Scholar] [CrossRef]
- Zhang, H.; Xie, Y.; Liu, C.; Chen, S.; Hu, S.; Xie, Z.; Deng, X.; Xu, J. Comprehensive comparative analysis of volatile compounds in citrus fruits of different species. Food Chem. 2017, 230, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Liu, H.; Zhang, A.; Wan, Y.; Hu, R.; Li, M. Analysis of volatile components extracted from the peels of four different Chinese pomelos using TDS-GC-MS. J. Sci. Food Agric. 2014, 94, 3248–3254. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.M. Morphological, anatomical, and physiological changes in the developing fruit of the Valencia orange, Citrus sinensis (L.) Osbeck. J. Appl. Anim. Res. 1958, 40, 154–162. [Google Scholar] [CrossRef]
- Mamat, S.F.; Azizan, K.A.; Baharum, S.N.; Noor, N.M.; Aizat, W.M. GC-MS and LC-MS analyses reveal the distribution of primary and secondary metabolites in mangosteen (Garcinia mangostana Linn.) fruit during ripening. Sci. Hortic. 2020, 262, 109004. [Google Scholar] [CrossRef]
- Gapper, N.E.; Giovannoni, J.J.; Watkins, C.B. Understanding development and ripening of fruit crops in an ‘omics’ era. Hortic. Res. 2014, 1, 14034. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Yun, Z.; Wu, Q.; Qu, H.; Duan, X.; Jiang, Y. Combination of Transcriptomic, Proteomic, and Metabolomic Analysis Reveals the Ripening Mechanism of Banana Pulp. Biomolecules 2019, 9, 523. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Deng, B.; Tian, S.; Guo, M.; Liu, H.; Zhao, X. Metabolic and transcriptomic analyses reveal different metabolite biosynthesis profiles between leaf buds and mature leaves in Ziziphus jujuba mill. Food Chem. 2021, 347, 129005. [Google Scholar] [CrossRef]
- Feng, G.; Wu, J.; Xu, Y.; Lu, L.; Yi, H. High-spatiotemporal-resolution transcriptomes provide insights into fruit development and ripening in Citrus sinensis. Plant Biotechnol. J. 2021, 19, 1337–1353. [Google Scholar] [CrossRef]
- Ding, Y.; Chang, J.; Ma, Q.; Chen, L.; Liu, S.; Jin, S.; Han, J.; Xu, R.; Zhu, A.; Guo, J.; et al. Network analysis of postharvest senescence process in citrus fruits revealed by transcriptomic and metabolomic profiling. Plant Physiol. 2015, 168, 357–376. [Google Scholar] [CrossRef]
- Jiang, C.C.; Zhang, Y.F.; Lin, Y.J.; Chen, Y.; Lu, X.K. Illumina((R)) Sequencing Reveals Candidate Genes of Carotenoid Metabolism in Three Pummelo Cultivars (Citrus Maxima) with Different Pulp Color. Int. J. Mol. Sci. 2019, 20, 2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, T.T.; Xiong, B.; Huang, S.J.; Liao, L.; Qiu, X.; Sun, G.C.; He, Y.Z.Z.; Duan, C.W.; Wang, X.J.; Zhang, X.; et al. Investigation of the cause of reduced sugar content in Kiyomi tangor fruit of Ziyang xiangcheng (Citrus junos Sieb. ex Tanaka) rootstock. Sci. Rep. 2019, 9, 19263. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Dong, T.; Qiu, X.; Rong, Y.; Wang, Z.; Zhu, J. Nitrogen nutrition is a key modulator of the sugar and organic acid content in citrus fruit. PLoS ONE 2019, 14, e0223356. [Google Scholar] [CrossRef]
- Dai, W.; Xie, D.; Lu, M.; Li, P.; Lv, H.; Yang, C.; Peng, Q.; Zhu, Y.; Guo, L.; Zhang, Y.; et al. Characterization of white tea metabolome: Comparison against green and black tea by a nontargeted metabolomics approach. Food Res. Int. 2017, 96, 40–45. [Google Scholar] [CrossRef]
- Heischmann, S.; Quinn, K.; Cruickshank-Quinn, C.; Liang, L.P.; Reisdorph, R.; Reisdorph, N.; Patel, M. Exploratory Metabolomics Profiling in the Kainic Acid Rat Model Reveals Depletion of 25-Hydroxyvitamin D3 during Epileptogenesis. Sci. Rep. 2016, 6, 31424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Rui, X.; Hao, C.; He, Y. TBtools, a Toolkit for Biologists integrating various HTS-data handling tools with a user-friendly interface. Mol. Plant 2018. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.-X.; Shi, C.-Y.; Liu, X.; Ning, D.-Y.; Jing, L.-F.; Yang, H.; Liu, Y.-Z. Citrate accumulation-related gene expression and/or enzyme activity analysis combined with metabolomics provide a novel insight for an orange mutant. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, L.; Shen, D.; Luo, Y.; Sun, X.; Wang, J.; Luo, T.; Zeng, Y.; Xu, J.; Deng, X.; Cheng, Y. Exogenous γ-aminobutyric acid treatment affects citrate and amino acid accumulation to improve fruit quality and storage performance of postharvest citrus fruit. Food Chem. 2017, 216, 138–145. [Google Scholar] [CrossRef]
- Wang, B.; Li, Z.; Han, Z.; Xue, S.; Bi, Y.; Prusky, D. Effects of nitric oxide treatment on lignin biosynthesis and texture properties at wound sites of muskmelons. Food Chem. 2021, 362, 130193. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Yang, S.; Yuan, Y. Lignin Involvement in Programmed Changes in Peach-Fruit Texture Indicated by Metabolite and Transcriptome Analyses. J. Agric. Food Chem. 2018, 66, 12627–12640. [Google Scholar] [CrossRef]
- Tuskan, G.A.; Muchero, W.; Tschaplinski, T.J.; Ragauskas, A.J. Population-level approaches reveal novel aspects of lignin biosynthesis, content, composition and structure. Curr. Opin. Biotechnol. 2019, 56, 250–257. [Google Scholar] [CrossRef]
- Gray, J.; Caparrós-Ruiz, D.; Grotewold, E. Grass phenylpropanoids: Regulate before using! Plant Sci. 2012, 184, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, Z.; Zhang, Y.; Chai, L.; Yi, H.; Deng, X. An integrative analysis of the transcriptome and proteome of the pulp of a spontaneous late-ripening sweet orange mutant and its wild type improves our understanding of fruit ripening in citrus. J. Exp. Bot. 2014, 65, 1651–1671. [Google Scholar] [CrossRef] [Green Version]
- Pattison, R.J.; Csukasi, F.; Catala, C. Mechanisms regulating auxin action during fruit development. Physiol. Plant 2014, 151, 62–72. [Google Scholar] [CrossRef]
- Dietz, K.J.; Vogel, M.O.; Viehhauser, A. AP2/EREBP transcription factors are part of gene regulatory networks and integrate metabolic, hormonal and environmental signals in stress acclimation and retrograde signalling. Protoplasma 2010, 245, 3–14. [Google Scholar] [CrossRef]
- Feng, K.; Hou, X.L.; Xing, G.M.; Liu, J.X.; Duan, A.Q.; Xu, Z.S.; Li, M.Y.; Zhuang, J.; Xiong, A.S. Advances in AP2/ERF super-family transcription factors in plant. Crit. Rev. Biotechnol. 2020, 40, 750–776. [Google Scholar] [CrossRef] [PubMed]
- Li, S.B.; Xie, Z.Z.; Hu, C.G.; Zhang, J.Z. A Review of Auxin Response Factors (ARFs) in Plants. Front. Plant Sci. 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Li, M.; Zhang, C.; Tan, Q.; Yang, X.; Sun, X.; Pan, Z.; Deng, X.; Hu, C. Effects of phosphorus on fruit soluble sugar and citric acid accumulations in citrus. Plant Physiol. Biochem. 2021, 160, 73–81. [Google Scholar] [CrossRef]
- Hussain, S.B.; Shi, C.-Y.; Guo, L.-X.; Kamran, H.M.; Sadka, A.; Liu, Y.-Z. Recent Advances in the Regulation of Citric Acid Metabolism in Citrus Fruit. Crit. Rev. Plant Sci. 2017, 36, 241–256. [Google Scholar] [CrossRef]
- Lu, X.; Zhao, C.; Shi, H.; Liao, Y.; Xu, F.; Du, H.; Xiao, H.; Zheng, J. Nutrients and bioactives in citrus fruits: Different citrus varieties, fruit parts, and growth stages. Crit. Rev. Food Sci. Nutr. 2021, 1–24. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.-J.; Chen, J.-B.; Cao, J.-P.; Li, X.; Sun, C.-D. Citrus flavonoids and their antioxidant evaluation. Crit. Rev. Food Sci. Nutr. 2020, 1–22. [Google Scholar] [CrossRef]
- Figueroa, C.R.; Rosli, H.G.; Civello, P.M.; Martinez, G.A.; Herrera, R.; Moya-Leon, M.A. Changes in cell wall polysaccharides and cell wall degrading enzymes during ripening of Fragaria chiloensis and Fragaria xananassa fruits. Sci. Hortic. 2010, 124, 454–462. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, M.; Cheng, F.; Dai, C.; Sun, Y.; Lu, J.; Huang, Y.; Li, M.; He, Y.; Wang, F.; et al. Identification of microRNAs correlated with citrus granulation based on bioinformatics and molecular biology analysis. Postharvest Biol. Technol. 2016, 118, 59–67. [Google Scholar] [CrossRef]
- Rademacher, E.H.; Lokerse, A.S.; Schlereth, A.; Llavata-Peris, C.I.; Bayer, M.; Kientz, M.; Freire Rios, A.; Borst, J.W.; Lukowitz, W.; Jürgens, G.; et al. Different auxin response machineries control distinct cell fates in the early plant embryo. Dev. Cell 2012, 22, 211–222. [Google Scholar] [CrossRef] [Green Version]
- Allan, A.C.; Espley, R.V. MYBs Drive Novel Consumer Traits in Fruits and Vegetables. Trends Plant Sci. 2018, 23, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Yan, J.Y.; Ren, J.Y.; Sun, L.; Xu, C.; Li, G.X.; Ding, Z.J.; Zheng, S.J. A WRKY transcription factor confers aluminum tolerance via regulation of cell wall modifying genes. J. Integr. Plant Biol. 2020, 62, 1176–1192. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bi, X.; Liao, L.; Deng, L.; Jin, Z.; Huang, Z.; Sun, G.; Xiong, B.; Wang, Z. Combined Transcriptome and Metabolome Analyses Reveal Candidate Genes Involved in Tangor (Citrus reticulata × Citrus sinensis) Fruit Development and Quality Formation. Int. J. Mol. Sci. 2022, 23, 5457. https://doi.org/10.3390/ijms23105457
Bi X, Liao L, Deng L, Jin Z, Huang Z, Sun G, Xiong B, Wang Z. Combined Transcriptome and Metabolome Analyses Reveal Candidate Genes Involved in Tangor (Citrus reticulata × Citrus sinensis) Fruit Development and Quality Formation. International Journal of Molecular Sciences. 2022; 23(10):5457. https://doi.org/10.3390/ijms23105457
Chicago/Turabian StyleBi, Xiaoyi, Ling Liao, Lijun Deng, Zhenghua Jin, Zehao Huang, Guochao Sun, Bo Xiong, and Zhihui Wang. 2022. "Combined Transcriptome and Metabolome Analyses Reveal Candidate Genes Involved in Tangor (Citrus reticulata × Citrus sinensis) Fruit Development and Quality Formation" International Journal of Molecular Sciences 23, no. 10: 5457. https://doi.org/10.3390/ijms23105457
APA StyleBi, X., Liao, L., Deng, L., Jin, Z., Huang, Z., Sun, G., Xiong, B., & Wang, Z. (2022). Combined Transcriptome and Metabolome Analyses Reveal Candidate Genes Involved in Tangor (Citrus reticulata × Citrus sinensis) Fruit Development and Quality Formation. International Journal of Molecular Sciences, 23(10), 5457. https://doi.org/10.3390/ijms23105457