1. Introduction
Contrary to the “classical view” of protein folding, that describes folding in terms of a defined sequence of states along the reaction coordinate axis, the “new view” of protein folding replaces this single-pathway model with trajectories on a rugged energy landscape [
1,
2,
3,
4]. Upon this energy landscape, the specific trajectory taken by a given molecule is determined by thermodynamic probabilities [
2]. This statistical model predicts that multiple pathways between the folded and unfolded states must exist for all proteins [
3]. Whether all trajectories between states remain possible at the single molecule level, the probability of a particular molecule taking a specific pathway depends on the starting conformation of the polypeptide chain, allowable thermal motion and relative height of energetic barriers.
Nevertheless, there is very little experimental evidence for the existence of such parallel folding pathways [
5,
6,
7,
8,
9,
10,
11]; the most unequivocal experimental evidence was provided by the Ig-like domain I27 (also called I91) of the giant muscle protein Titin [
10,
11]. This is probably because of the lack of techniques allowing a full (or even simplified) description of the folding landscape. Indeed, the full description of a protein folding reaction requires understanding of how the environment of each atom—at least, of each residue—from each individual protein in solution evolves during the reaction. This is far beyond what was achieved with most of the protein folding studies, relying on the measurement of global physical and spectroscopic observables, such as fluorescence, circular dichroism, FTIR or SAXS. These techniques give access to ensemble averages and lack the desired spatial (structural) or temporal resolution. Single-molecule techniques using Förster resonance energy transfer (FRET) [
12] or IR spectroscopy [
13] have enough temporal resolution to describe the heterogeneity of the molecule ensemble, but lack high spatial resolution. In addition, with these techniques, the acquisition of site-specific information at multiple points of the polypeptide chain requires the separate preparation of protein variants modified at each residue (or pair of residues) of interest and a separate set of experiments for each.
Multidimensional NMR spectroscopy is one of the experimental techniques with the potential to contribute to a high-resolution, site-specific, time-resolved description of the protein folding reaction. This is essentially due to (i) the extreme sensitivity of NMR observables to the structural environment, and (ii) the fact that an abundance of site-specific probes can be studied simultaneously in a multidimensional NMR spectrum [
14]. Even if this technique can be combined with the “classical” chemical or temperature perturbations, when combined with high-hydrostatic pressure perturbation it can yield unprecedented details on protein folding pathways [
15,
16,
17,
18,
19,
20]. Thus, the probability of contact between specific residues was readily measured from residue-specific denaturation curves obtained from NMR data, and used to constrain Go-model calculations, allowing the characterization of the structure and energetics of the folding landscape of different proteins and the identification of major folding intermediates [
21,
22]. Nevertheless, in these previous studies, the length of the MD calculations restricted the description of the folding landscape only at a given pressure, precluding the characterization of all the conformers populating the landscape along the full pressure axis (1–2500 bar). Here, we propose an alternative method that allows the full structural description of the conformers populating the folding landscape during the folding/unfolding reaction. Among other things, we replaced the Go-model simulations with
Cyana3 calculations [
23], a popular software commonly used to model the 3D structure of protein from distance restraints derived from NMR data. Working on the dihedral angle space,
Cyana3 allows considerably faster calculations than the Go-model simulations, allowing the complete exploration of the folding landscape within reasonable computational times.
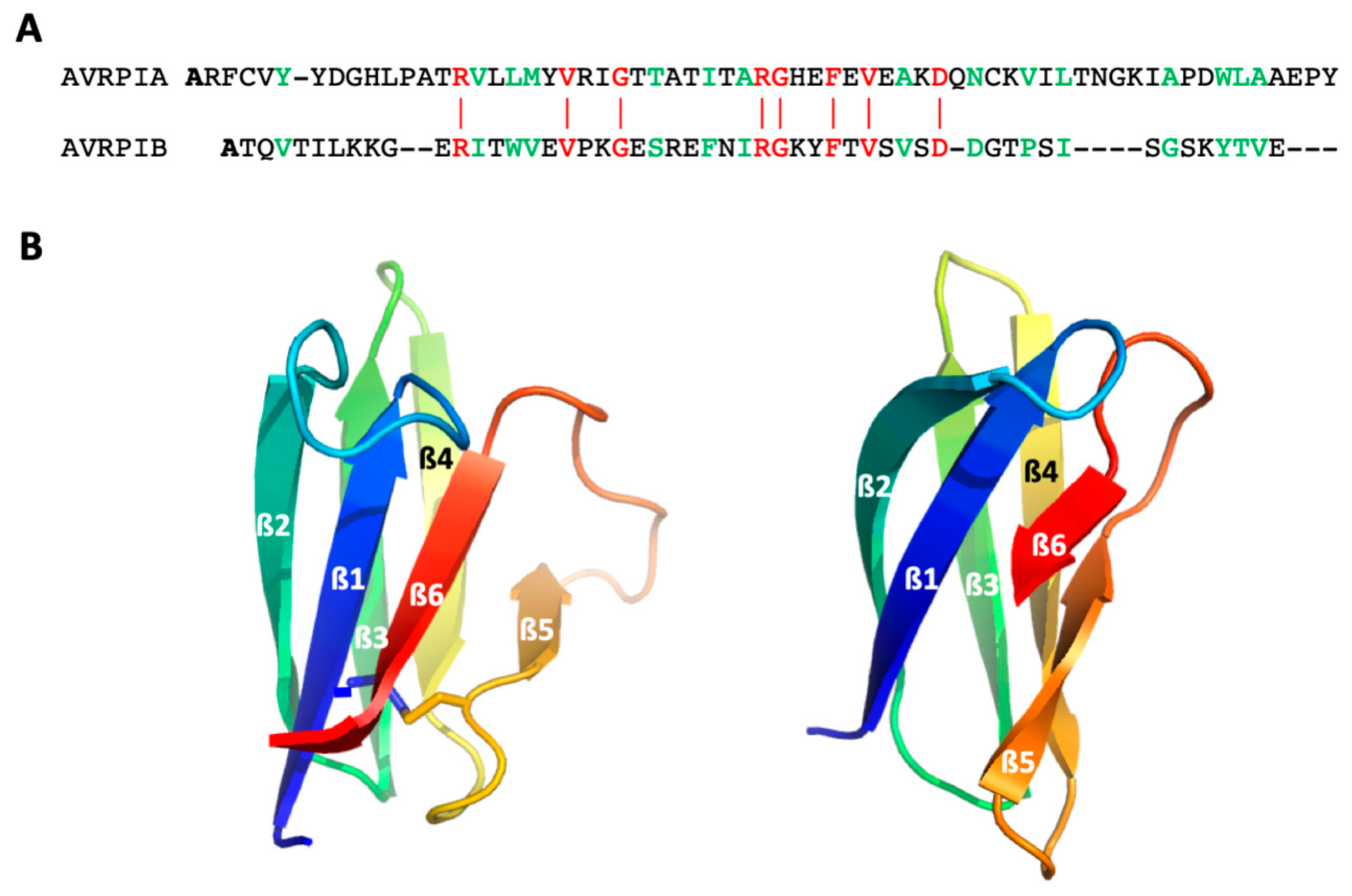
We tested our approach on two small globular model proteins, AVR-Pia and AVR-Pib from the blast fungus
Magnaporthe oryzae, belonging to the structurally conserved but sequence-unrelated MAX (Magnaporthe Avirulence and ToxB-like) effectors superfamily [
24], which play an important role during biotrophic host colonization, and can, in some cases, circumvent host immunity. Despite their low sequence identity (≈12%), these two proteins display a similar 3D fold, characteristic of the MAX effector family: a sandwich of two three-stranded antiparallel ß-sheets, with an identical topology (
Figure 1). Nevertheless, AVR-Pib lacks the usually well-conserved disulfide bond linking the two ß-sheets. In addition, to test our method the full description of the folding landscape for these two proteins may highlight more general questioning, such as whether proteins belonging to a same structural family have a similar folding pathway with similar folding intermediates? The answer to this question is expected to bring some clues about the way that a folding pathway is encoded by the primary sequence.
3. Discussion
To determine experimentally how the folding pathways of a protein differ, how the sub-structures are assembled, was a long-standing challenge. Based on experimental high pressure NMR data, we aimed to resolve whether sub-structures’ formation during folding (or melting during unfolding) could be disentangled from a complex conformational landscape.
High-pressure NMR analysis allowed the determination of residue specific native state probabilities along the range of pressure from 1 to 2500 bars. In this approach, the native state of a residue
i is defined by (i) the inter-atomic distances measured in the X-ray structures between Cα
i and the Cα of all other residues having their Cα atom within a sphere of 9 Å radius centered at Cα
i and (ii) the φ
i, ψ
i values measured in the X-ray structures. The pressure-dependent residue specific native-state probabilities were translated into inter-residue contact probabilities that were filtered and used to restrain the conformer calculations by
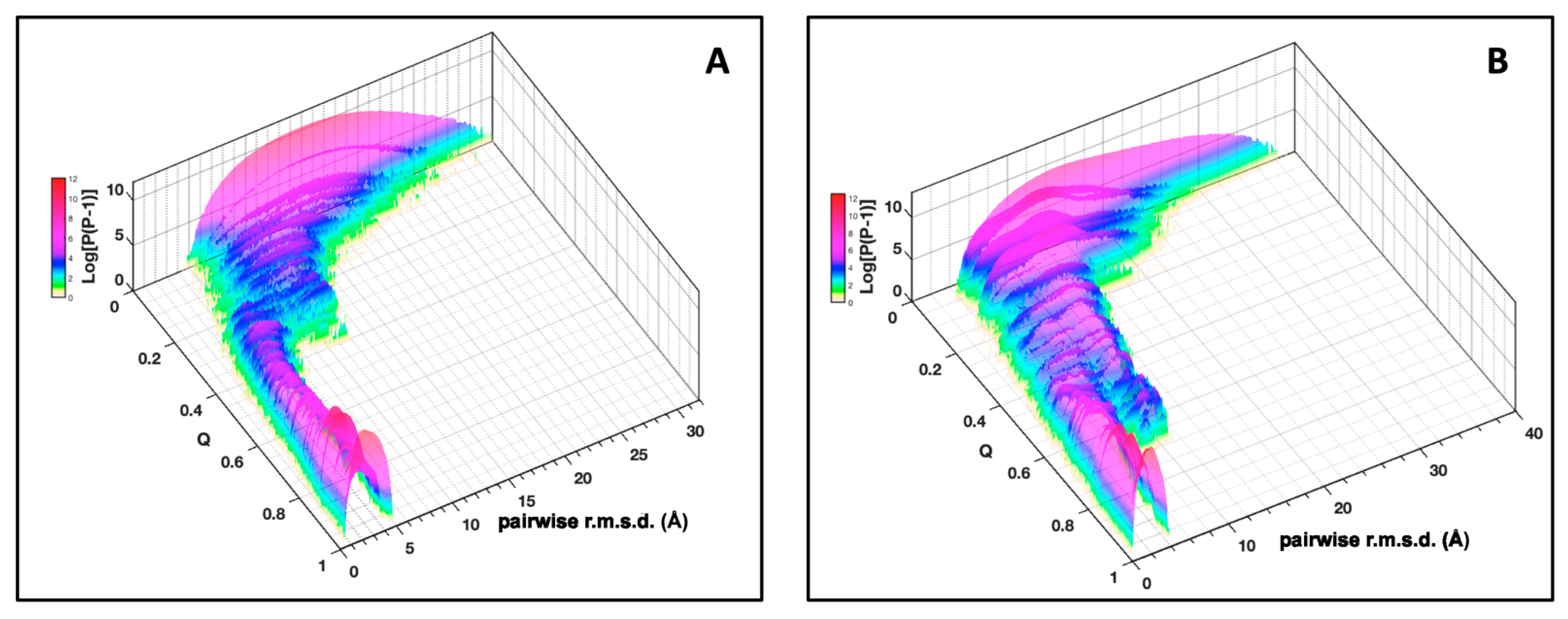
Cyana3. Then, the ensemble of conformers obtained from these calculations was used to build a conformational landscape. The conformers were clustered as a function of the reaction coordinate, Q, as order parameter. This is motivated by the fact that, in a funnel-like energy landscape, the energy of the conformations is reasonably correlated to the degree of
nativeness [
33]. We also controlled that the geometrical space sampling was strongly correlated with the energetics of the system, by calculating the free energy of the conformers.
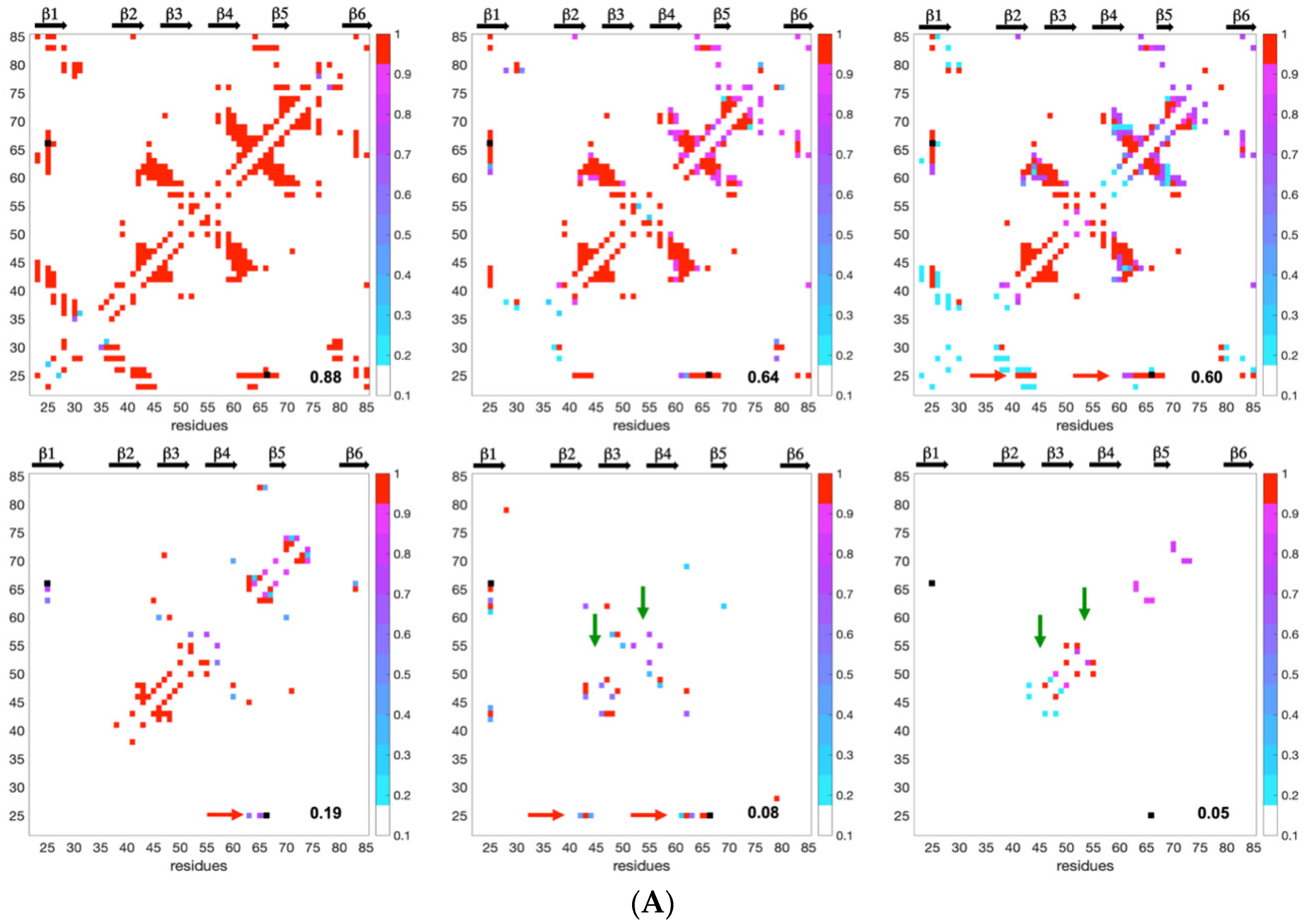
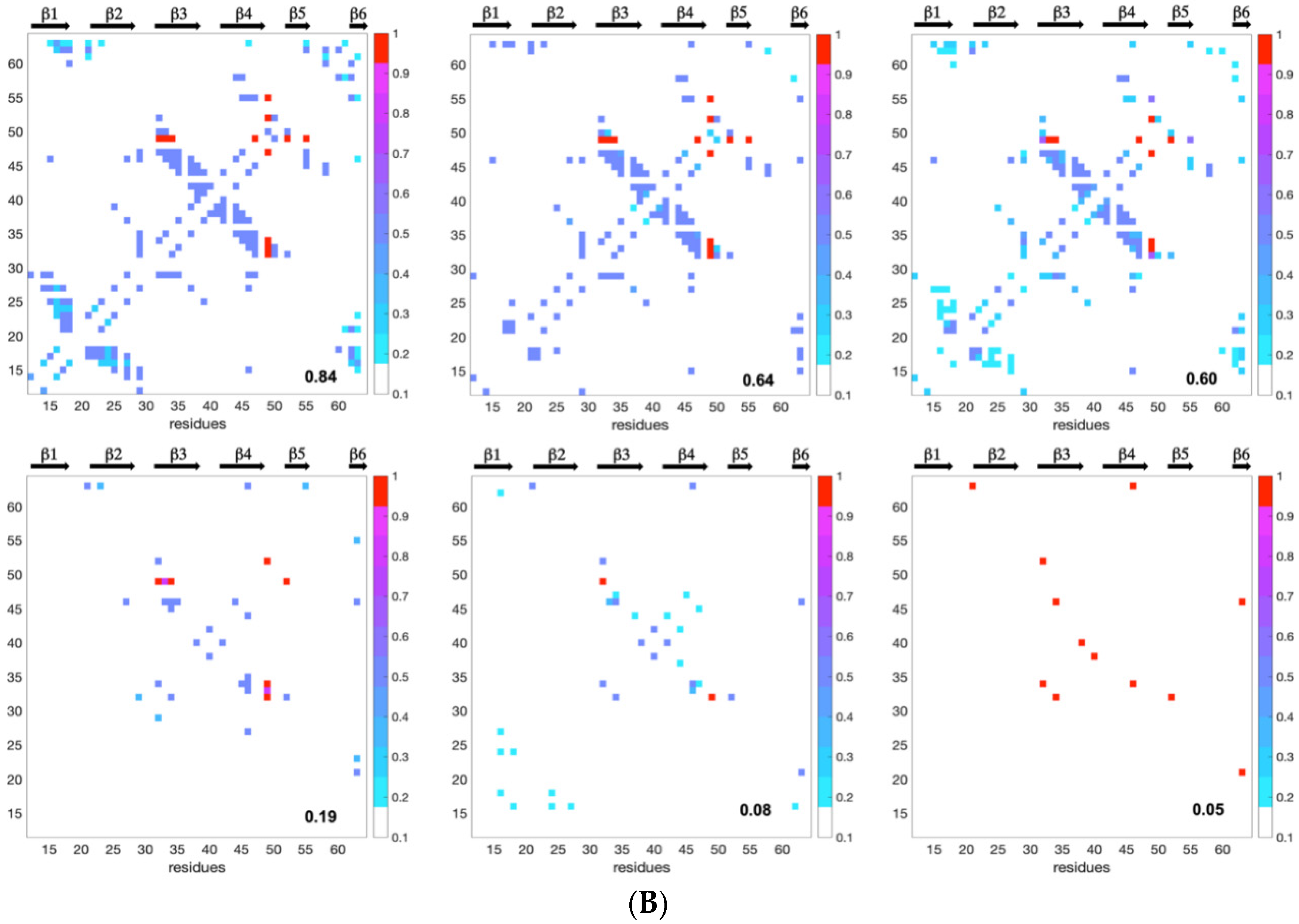
Since the distribution and density of the constraints along the protein sequence were different for the two MAX effectors, we introduced a statistical selection of conformer clusters retaining only the clusters that were statistically different from randomness. Accordingly, analyzing the landscapes was a targeted approach relying on (i) conformer clustering and (ii) statistical α-value rejection. This process allowed us to extract a discontinuous subset of clustered conformations and snapshots of probabilistic contact maps along Q. Through their probabilistic contact maps,
Figure 8 describes the characteristics of inescapable pressure-induced conformers, some of which were used to illustrate
Figure 9.
3.1. Folding Funnels
The agreement between the simulation results and the experimental data supports the idea that energetic frustration is indeed sufficiently reduced and that the protein folding mechanism, at least for small globular proteins, is strongly dependent on topological effects [
33]. Thus, topological techniques were often found to be very powerful and were successfully applied to numerous problems in Physics, from theories of fundamental interactions to models of condensed matter [
34]. Topology plays an important role in protein folding [
35,
36] and dynamics [
37], and, in particular, for self-entanglement [
38], as shown in a case study concerning the folding and unfolding of the slip-knotted AFV3-109 protein [
39]. A hybrid strategy using topological simplification and energy calculation was applied to the nucleosome-folding problem [
40] by using biased molecular dynamics, K-means clustering and the finite temperature string method [
41], that can be applied both in the original Cartesian space of the system or in a set of collective variables.
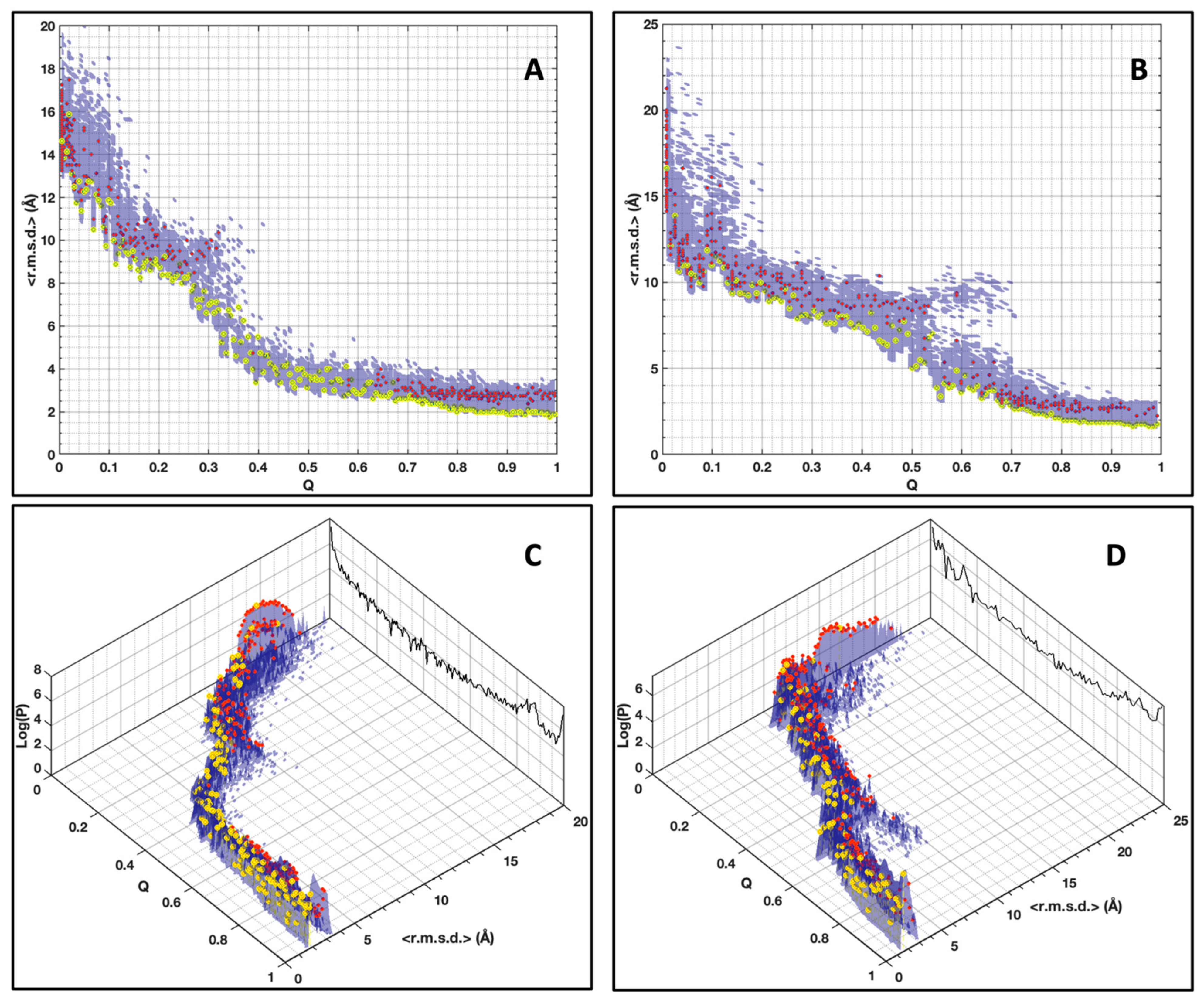
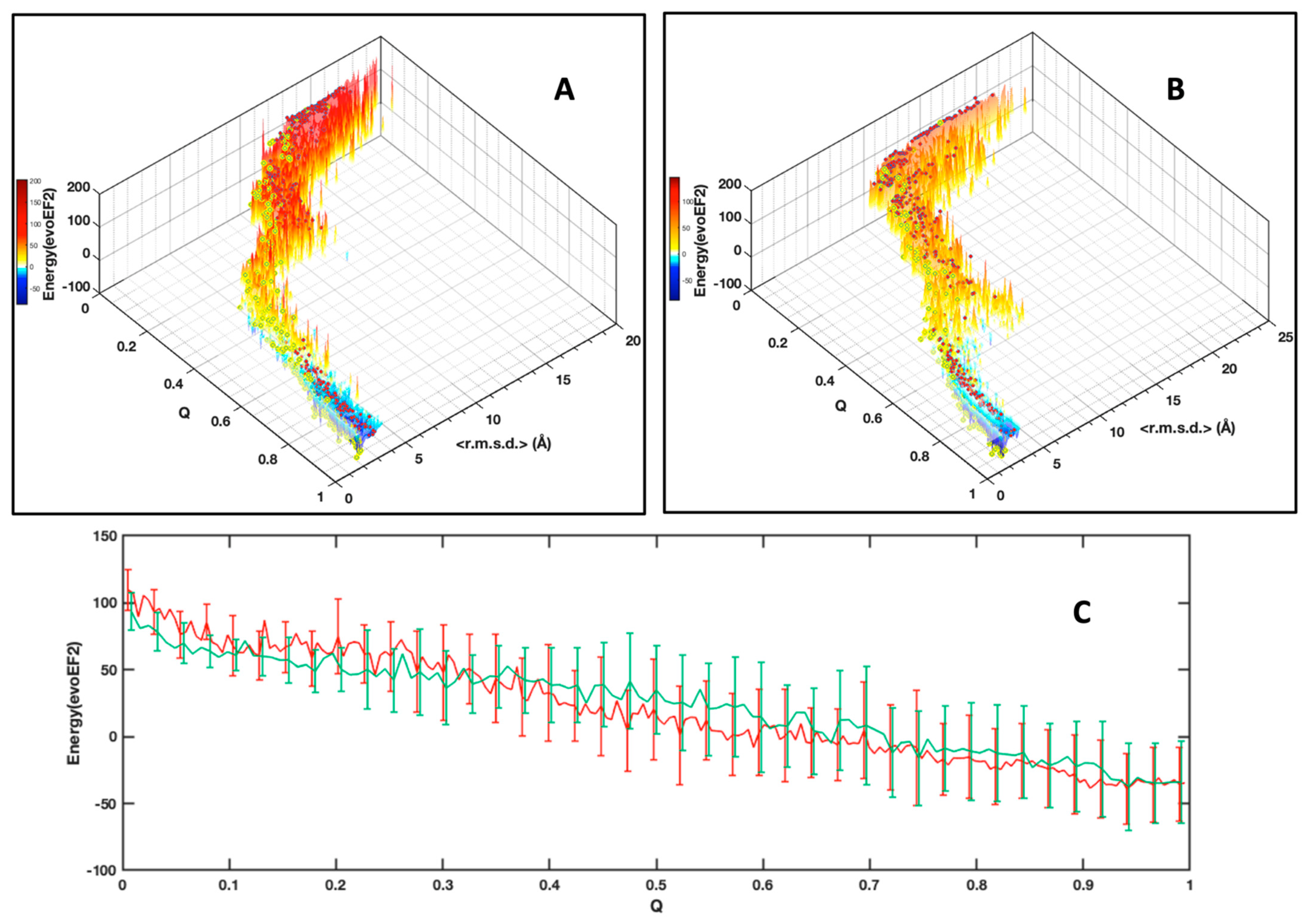
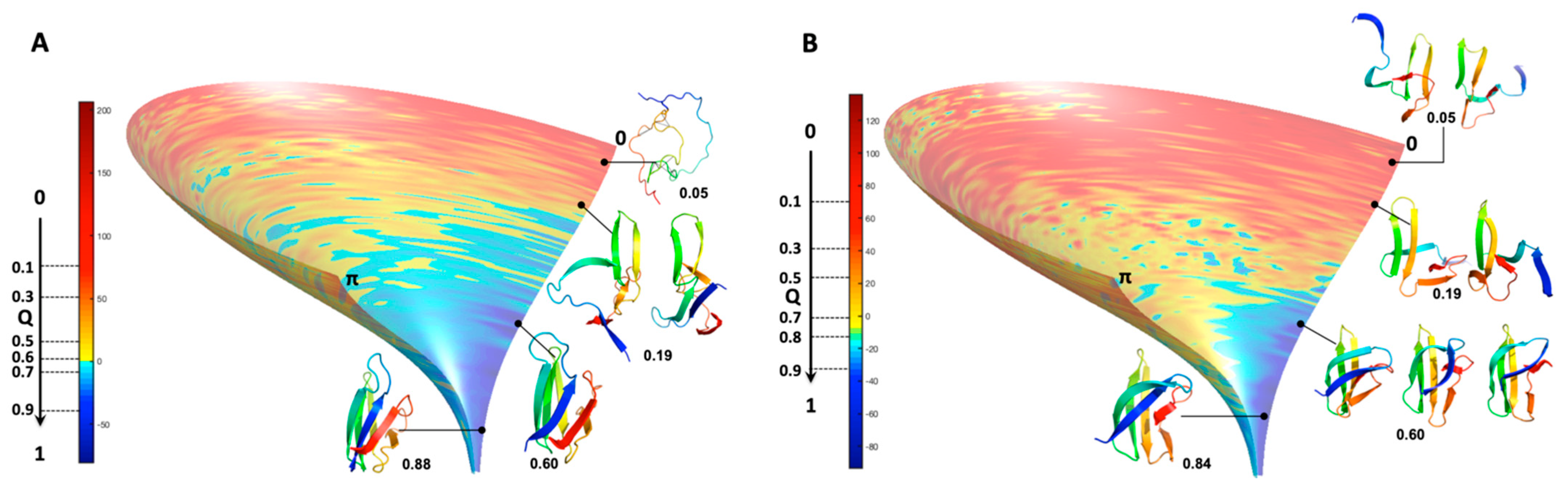
As an alternative to choosing a set of collective variables that lacks generality, and to summarize our results, we simply preferred illustrating the folding funnels for both MAX effectors AVR-Pia and AVR-Pib. We computed an idealized half-funnel-like surface inside a 3D grid having a size of 100 regularly spaced values in the [0 π] interval by progressively constricting the surface with an arbitrary logarithmic function. The energy values shown in
Figure 6B were resampled in the interval of minimum to maximum spreading <r.m.s.d.> at each Q value by applying a discrete cosine function algorithm [
42] to expand and compress the energy matrix to a fixed size (100 × 100). This energy matrix was projected on the surface and used to color the half-funnel cartoon of AVR-Pia and AVR-Pib (
Figure 9). The equivalence between Q axis and energy was taken from the “main populations” (MP) clusters’ average energies (
Figure 6C).
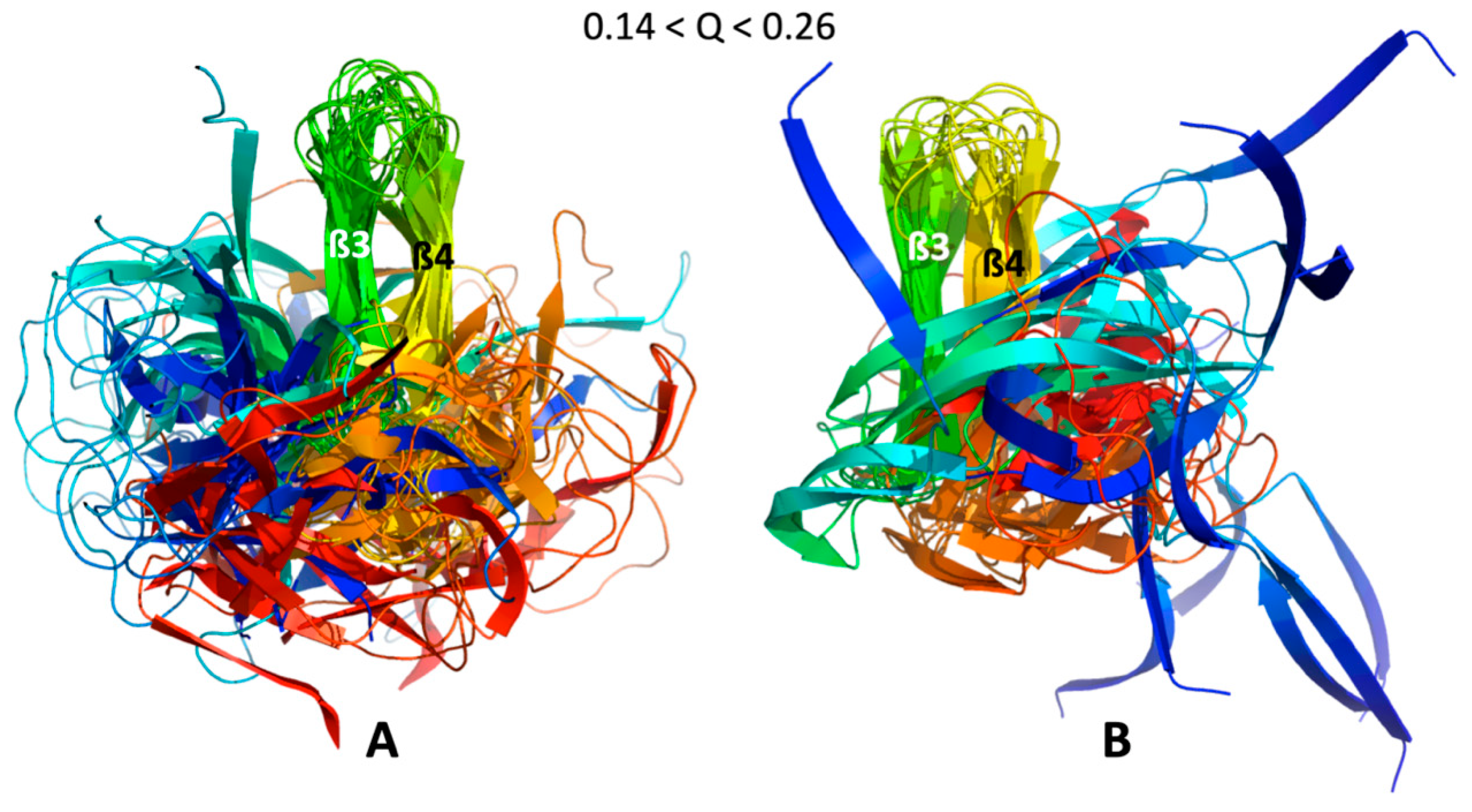
As a consequence of the statistical selection analysis of conformers, we only focused on small parts having high reliability among the whole topology landscape of folding/unfolding under high-pressure. The initial collapse at low Q = 0.05 appears different between the two MAX effectors (short/medium contacts for AVR-Pia and long-range contacts for AVR-Pib), but illustrates only the most probable conformations. In both cases, these early conformations converge to a similar sub-structure folding hub (Q = 0.19) having ß3ß4 anti-parallel strands, while the orientation and position of other strands are not clearly defined. For AVR-Pia, the conformers at Q = 0.6 have an overall topology remarkably similar to the one shown at Q = 0.88. This is far from the case for AVR-Pib, where at Q = 0.6 different orientations are observed for the ß1 strand while ß2 adopts a better-defined orientation and position. In the last stage of the folding reaction (Q = 0.88 and Q = 0.84, for AVR-Pia and AVR-Pib, respectively) the conformers reach almost native topology.
3.2. Early Steps of Folding
Early long-range Cα–Cα contacts are established for AVR-Pib at low Q (0.05) and involve the residues G21, G32, S34, N38, R40, V46, G52 and V63. The limited number of allelic variants (eight variants) within the MAX effector AVR-Pib sub-family precludes drawing conclusions about the conservation patterns from the sequences. However, all of the residues previously listed are conserved, with the exception of V46 that is substituted by Ile in one of the sequences. The two hydrophobic residues, V46 and V63, are in contact, but most of the other interacting residues are glycine or charged/polar residues, and their sequence neighbors, while not strictly conserved, are all also charged or polar with one exception: the flanking neighbor residues of N38 are systematically hydrophobic. This early appearing long-range cohesion does not seem to be uniquely driven by a hydrophobic collapse and highlights the role of transient contacts between polar/charged residues in the early steps of the folding reaction of AVR-Pib. A similar behavior was previously reported for SH3 proteins; unfolding simulations and contact analysis demonstrated that differences in both hydrophobic interactions and side-chain hydrogen bonding interactions drive the folding/unfolding process [
43].
The number of sequence variations is even smaller (five variants) in the case of the AVR-Pia sub-family. Intriguingly, the topological consequences of the conserved disulfide bond C25-C66 were rapidly observed during the folding reaction (Q = 0.08) with a set of Cα–Cα contacts between C25 and residues in the loop between ß4 and ß5. This suggests that the disulfide bond represents a major driving restraint that biases the folding landscape very early, restricting the overall divergence of conformers and simplifying (forcing) convergence to the native topology. Of course, since high-hydrostatic pressure is unable to break this covalent link, the likelihood of the results presented for AVR-Pia in this study depends, at least partially, on the fact that the disulfide bond formation constitutes the very first step of the protein folding, which might be not the case in vivo. It was shown by many experimental and theoretical studies that the pre-formed disulfide bonds can significantly increase the protein stability [
44]. This underlines also some limitations of high-hydrostatic perturbation (as well as many other methods) for the study of protein folding, especially when highly energetic contacts such as covalent bonds are involved. More generally, there should be additional intermediates that were not captured because our analysis of HP-NMR unfolding is only sensitive to structural transitions that bias the topological landscape.
The impact of the guanidine concentration on the folding landscape should not be neglected. Very different sub-denaturant concentrations of guanidinium chloride were used to destabilize the 3D structures of AVR-Pia and AVR-Pib (4.5 M and 1.5 M, respectively) in order to trigger their unfolding reaction in the pressure range allowed by the experimental set-up (1–2500 bar). In a previous study [
29], we showed that guanidine has a limited impact on ∆
V, suggesting that the sub-denaturant concentrations of this harsh denaturant are able to slightly “smooth” the energy landscape by removing high-energy intermediates, while the main low-energy folding intermediates are maintained. The main effect of guanidine was found on the Transition State Ensemble (TSE) populated during protein unfolding. Indeed, P-jump kinetic experiments revealed that, for the same protein, a different sub-denaturing concentration of guanidine yields quantitative (more or less hydrated TSE) and qualitative (regions concerned) differences at the TSE level [
29]. Thus, beside the important difference in their primary structure, the different sub-denaturant concentrations of guanidine used for AVR-Pia and AVR-Pib might impact, to some extent, the folding routes followed by these two proteins during the folding/unfolding reaction.
4. Materials and Methods
4.1. Protein Expression and Purification
The coding genes for AVR-Pia and AVR-Pib proteins were subcloned in pepL, a vector that allows the expression of a periplasm secretion signal peptide, and of a 6xHis-3C fusion protein. Constructs were then transformed into E. coli BL21(DE3) (Stratagene, Amsterdam, The Netherlands).
Uniform 15N labeling was obtained by growing cells in minimal M9 medium containing 15NH4Cl as the sole source of nitrogen. Protein was expressed overnight at 20 °C after induction with 0.2 mM IPTG. Cells were collected by centrifugation and suspended in 120 mL of cold lysis buffer comprising 200 mM Tris-HCl buffered at pH 8 and containing 500 mM sucrose, to which were added 40 mL of 5 mM EDTA buffered at pH 8, 40 mL of 0.1 mg/mL lysozyme, and 200 mL of TE buffer (200 mM Tris-HCl pH 8 and 0.5 mM EDTA). After 30 min of incubation on ice, 4 mL of MgSO4 were added and cell debris and insoluble materials were removed by centrifugation at 8000 rpm, for 30 min at 6 °C. The supernatant was loaded through a benchtop peristaltic pump onto a COmplete™ His-Tag Purification Column (Roche, Basel, Switzerland) equilibrated with buffer A (50 mM Tris-HCl pH 8, 300 mM NaCl, 0.1 mM benzamidine (and 1 mM DTT for AVR-Pia)). After elution with buffer B (buffer A supplemented with 500 mM imidazole), fractions containing the protein were dialyzed with homemade recombinant His-tagged 3C protease (mixed at 100:1 ratio) overnight at 4 °C in 10 mM Tris-HCl buffered at pH 7 for AVR-Pib and pH 8 for AVR-Pia, 150 mM NaCl (and 1 mM DTT for AVR-Pia). Cleavage was checked with SDS-PAGE and proteins were finally injected through an AKTA system into a Superdex S75 26/60 (GE Healthcare, Buc, France) column, equilibrated with 20 mM Tris-HCl buffered at pH 7 for AVR-Pib and pH 8 for AVR-Pia, 150 mM NaCl. The fractions containing the pure protein were pooled, concentrated to about 1 mM (protein concentration) and dialyzed overnight in 20 mM acetate pH 5.4, 100 mM NaCl for NMR experiments. Samples were then flash-frozen in liquid N2 and stored at −80 °C until NMR analysis.
4.2. Protein Unfolding
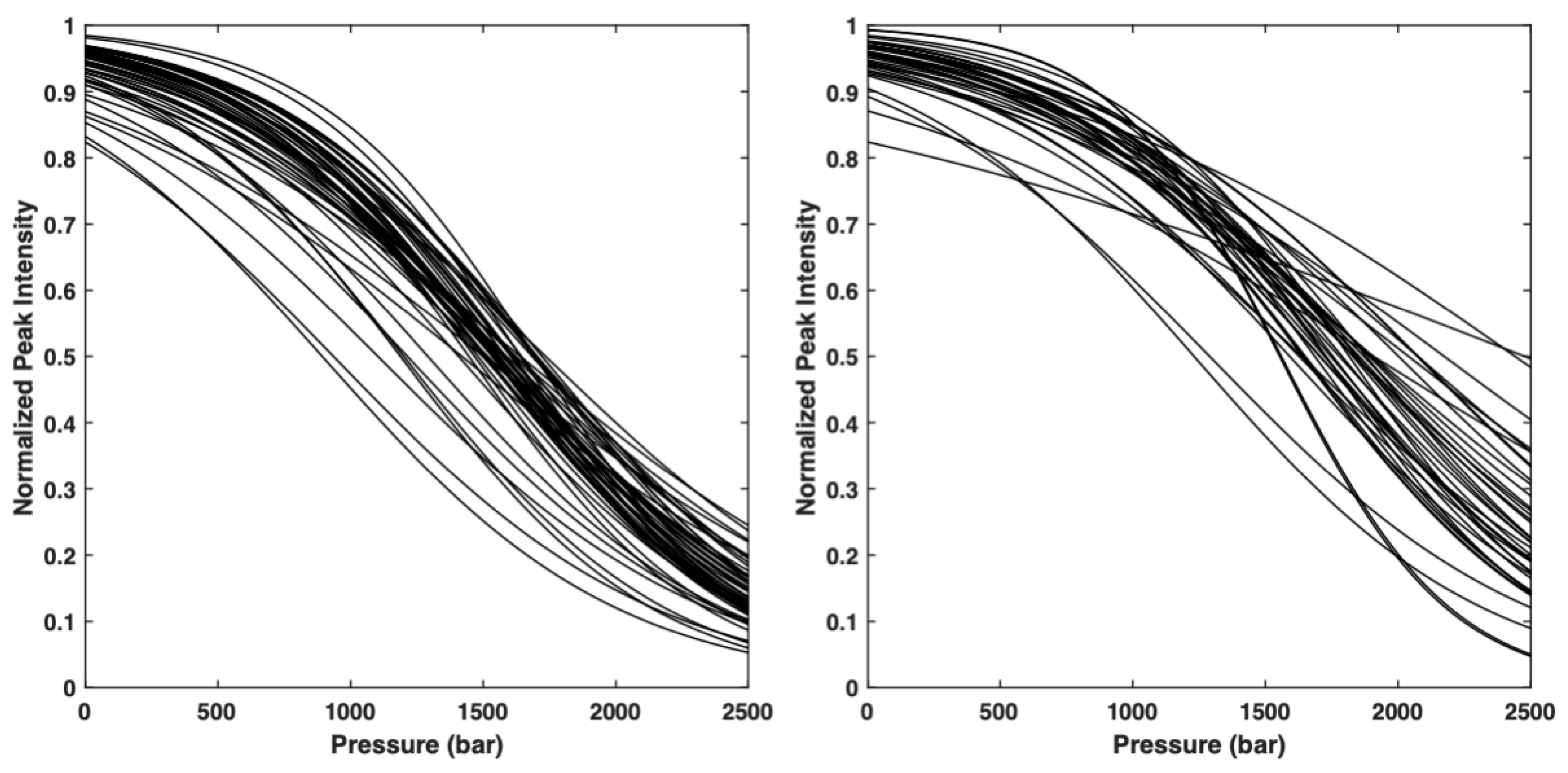
The 2D [1H,15N] HSQC were recorded on a Bruker AVANCE III 600 MHz spectrometer at 20 °C and at 15 different hydrostatic pressures (1, 50, 100, 300, 500, 700, 900, 1100, 1300, 1500, 1700, 1900, 2100, 2300 and 2500 bar). Samples with about 1 mM concentration of 15N-labeled proteins were used on 5 mm o.d. ceramic tubes (330 μL of sample volume) from Daedelus Innovations (Aston, PA, USA). Sub-denaturant concentration of guanidinium chloride (4.5 M and 1.5 M, for AVR-Pia and AVR-Pib, respectively) were added in order to trigger the protein stability into the pressure range allowed by the experimental set-up (1–2500 bar). Hydrostatic pressure was applied to the sample directly within the magnet using the Xtreme Syringe Pump (Daedelus Innovations). Each pressure jump was separated by a 2 h relaxation time, to allow the denaturation reaction to reach full equilibrium. Relaxation times for the folding/unfolding reactions were previously estimated from a series of 1D NMR experiments recorded after 200 bar P-Jump, following the increase in the resonance band corresponding to the methyl groups in the unfolded state of the protein. Note that the 1D spectra recorded at 1 bar on each protein sample before and after the full pressurization process gave strictly the same results, demonstrating the perfect reversibility of the folding/unfolding reaction.
In the case of AVR-Pia, the resonance assignment was already deposited at the BMRB (n° 25460), but under physical and chemical conditions slightly different than those used in the present study (20 mM Citrate pH 5.4, 100 mM NaCl, 4.5 M GuHCl). Since the presence of guanidine in the sample used for the denaturation experiments yields significant shifts in the position of the [
1H,
15N] HSQC cross-peaks, titration experiments were used to re-assign the amide cross-peaks in the conditions of the HP-NMR experiments. For AVR-Pib, we used [
1H,
15N] NOESY-HSQC (mixing time: 150 ms) and [
1H,
15N] TOCSY-HSQC (isotropic mixing time: 60 ms) 3D experiments to assign the amide group resonances in the condition of the denaturation study (20 mM Acetate buffer pH 5.4, 100 mM NaCl, 1.5 M GuHCl), following the classical sequential assignment strategy. Then, the intensities of the amide cross-peaks were measured for the folded species at each pressure and fitted with a two-state model:
In this equation, I is the cross-peak intensity measured at a given pressure, and If and Iu correspond to the cross-peak intensities in the folded state (1 bar) and in the unfolded state (2500 bar), respectively. stands for the residue specific apparent free energy of folding at atmospheric pressure, and corresponds to the residue specific apparent volume of folding for pressure denaturation.
4.3. Topological Space Analysis
Let n Cα atoms, and di,j the distance between the i and j Cα atoms, where i = 1, 2,…, n and j = 1, 2,…, n.
The native constraint list
Cc, for any
dc cut-off, is given by filtering:
with
.
The filtered constraint list
for a filter
for Cα atoms
i and
j, is given by:
is the probability of a contact between Cα atoms
i and
j, given by the geometric mean [
19] expressed as:
where
and
are the statistical translations of the fraction (short cut as “the folded state probability” in the following) that each of the two residues
i and
j is in the folded state at a given pressure.
Let Q
T and Q
f, the number of constraints in the
and
list, respectively, we define the fraction of native constraints for a filtered constraint list
to be Q = Q
f/Q
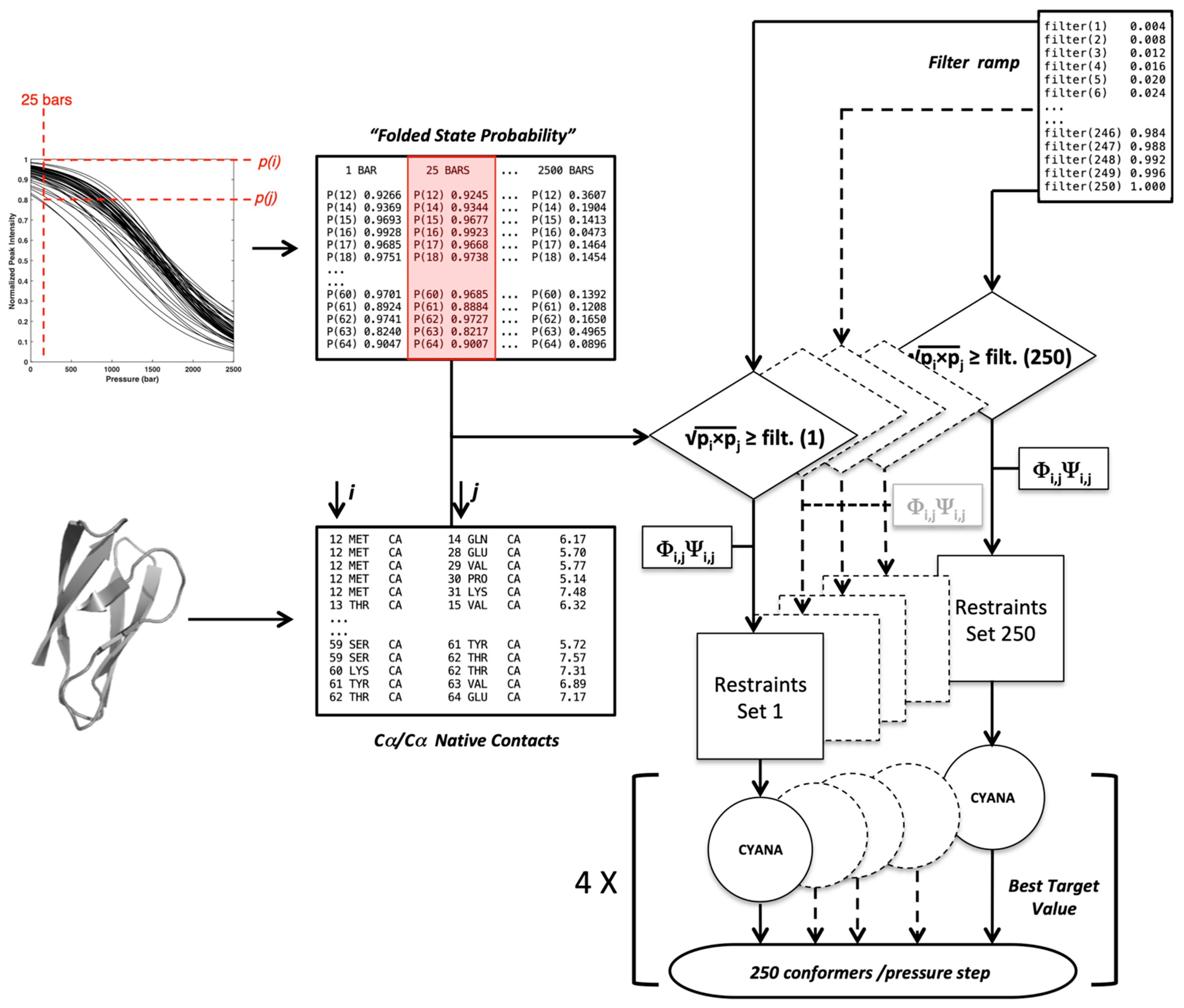
T. The flowchart schematized below (
Figure 10) gives the protocol used to generate a set of conformers at each pressure filtered by a selection
filter ramp. The population of conformers is then sorted by the fraction of native constraints per conformer (Q).
This flowchart crosses information coming from two main lists:
- -
A list of Cα–Cα distance upper bounds with a cutoff of 9 Å generated from the PDB structure 6Q76 (B chain), 5Z1V (A chain) for AVR-Pia and AVR-Pib, respectively. In addition, lists of backbone dihedral restraints (Φ/Ψ at ±10°) were also derived from the structures;
- -
A list containing the probability to find a residue i in a folded state, obtained from the normalized experimental residue-specific denaturation curve obtained for residue i. These curves are obtained from the fit of the intensity decrease with the pressure of HSQC cross-peak of residue i with Equation (1).
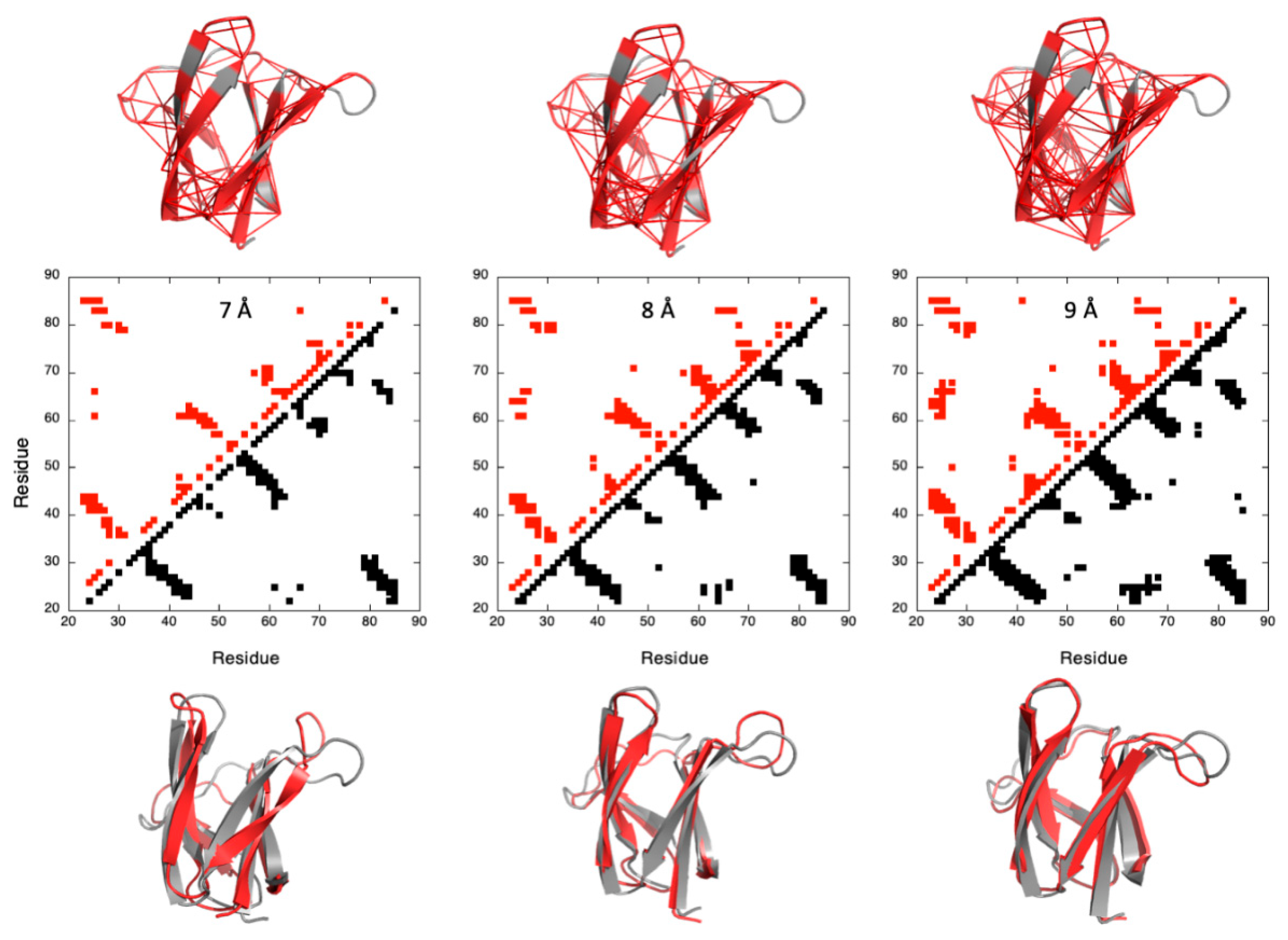
For a given pressure, 250 lists of contacts were established through filtering each native Cα–Cα contact by increasing cut-off values (
) obtained from a ramp (
= 0.004 to
= 1): a native contact between residues
i and
j is included in the list for a given f value if (according to Equation (4)):
Constraint lists () having zero or all of the native contacts were discarded. The Cαi–Cαj distance measured in the X-ray structures of the two MAX effectors was used as the upper bound limit to restrain the distance between residue i and j in the Cyana3 calculations. In addition, the backbone Φi/Ψi and Φj/Ψj dihedral angles measured in the crystal structures were used as the constraints (±10°) only for the residues in the ß-strands to further restrain the available conformational space of the residues involved in contacts during calculations. This procedure was repeated at each pressure, from 1 to 2500 bar, with 25 bar steps.
4.4. Cyana Calculations
The torsion angle dynamics in Cyana3, restrained by the Cα–Cα upper bounds derived from each and backbone dihedral restraints, were used to generate one conformer (selected as having the best target function over 100 calculations) per pressure (from 1 to 2500 bar, with 25 bar steps) and per ramp cut-off (). Cyana3 implements the angle dynamics with minimization of a target function involving optimal geometry defined in a residue library that included Van der Waals atomic radii giving fine, well-defined Ramachandran conditions for the conformers. All calculations were repeated four times with different random conditions. All the models were sorted according to the fraction of native upper bound constraints (Q = Qf/QT).
4.5. Energy Calculations
The internal energy of each conformer was calculated by evoEF2 software [
32]. The conformer was first submitted with the
RepairStructure option that optimizes the energy of the provided model by combinatorial exploration of the side-chain rotamers. Then, the resulting model was submitted to evoEF2 with the
ComputeStability option to calculate the free energy (evoEF2 arbitrary units), and more specific contribution terms (Van der Waals and H-Bonds energies) of the model. The complete EvoEF2 energy function is written as:
Here, E
VDW, E
ELEC, E
HB, E
DESOLV and E
REF represent the total Van der Waals, electrostatic, hydrogen bonding, de-solvation and reference energy terms for a protein system, respectively. The protein reference energy term, E
REF, is used to model the energy of the protein in the unfolded state and it is calculated as the sum of amino acid-specific reference energy values [
29]. In the four additional terms, E
SS describes the disulfide-bonding interactions, E
AAPP represents the energy for calculating amino acid propensities at given backbone (Φ/ψ) angles, E
RAMA is the Ramachandran term for choosing specific backbone angles (Φ /ψ) given a particular amino acid and E
ROT is the energy term for modeling the rotamer probabilities from the rotamer library. Difference in free energy expressed in kcal mol
−1 is easily calculated from evoEF2 energy differences.
4.6. Clustering
We used the MaxCluster (
http://www.sbg.bio.ic.ac.uk/~maxcluster/download.html, accessed on 15 May 2019) software that is fast enough for computing pairs of r.m.s.d. for a large set of structures (in the order of 104 structures). The Nearest Neighbor (NN) clustering algorithm in MaxCluster is based on the method of Shortle et al. [
31]. The clustering must perform two goals: (i) global shape recognition and clustering at low Q values; and (ii) more local r.m.s.d. clustering at high Q values. Accordingly, we adapted MaxCluster parameters that perform well with these two tasks. Since we performed 4
Cyana3 calculations for each constraint list, yielding four conformers per list, we choose the minimum number of conformers to form a cluster to be five (default value).
The ceiling threshold (Tm) was let at the default value (8 Å) in order to be able to cluster conformers, according to their global shapes. The minimum threshold was set to 2.5 Å, with the possibility to regress by 1 Å, which has the effect of slightly increasing the number of clusters at high Q values and discriminating clusters having small local structure differences.
4.7. Contact Maps Statistics
Statistical tests assume a null hypothesis of no relationship or no difference between groups. Then, they determine whether the observed data fall outside of the range of values predicted by the null hypothesis.
The computational flowchart for generating randomly scrambled conformer populations (null hypothesis) is essentially identical to
Figure 10, except that at each pressure and at each filter value (
f) the residue specific data (
) were scrambled before generating the constraint files. That has the effect of de-correlating the pressure to sequence specificity, if any. In other words, it disconnects the pressure and structure. To perform the statistical analysis, we pooled these randomly scrambled conformer populations to the original ‘pressure’ data, and the obtained merged data at each Q were re-clustered with the default minimum cluster size or with minimum size of 10. Under such null hypothesis assumption, the statistical significance of any cluster was assessed by the alpha value threshold α < 0.05, which means that in a given cluster less than 5% of the conformers could be generated by random-scrambling. Accordingly, any cluster having more than 95% of conformers generated from the original ‘pressure’ dataset was selected to be statistically relevant. Fractional contact maps were computed from the upper bound restraint files used to generate these conformers. In these maps, any factional contact corresponds to the normalized frequency (0 to 1) of observing that contact in the selected cluster(s) of the original ‘pressure’ dataset.
Molecular structure Figures and Videos were executed with PyMOL v.1.6. [
45]. The other Figures and Videos were executed with the MatLab 2019 package.
5. Conclusions
In this study, we demonstrate that the identification of folding intermediate conformations is feasible from a targeted geometrical sampling under control of HP-NMR data. The evolution of secondary structure formation or melting during folding/unfolding can be monitored at a residue level and furnishes insights into possible folding scenarios. In previous studies, it was already shown that a description of the conformational landscape of a protein at a given pressure was attainable by combining HP-NMR and molecular dynamic (Go-models) simulations [
21,
22]. Here, based on similar HP-NMR data, we succeeded in describing the full ensemble of conformers populated along the entire pressure axis, from 1 bar (native structure) to 2500 bar (unfolded structures). Among other things, this was made possible by replacing the Go-model simulations by faster
Cyana3 distance-geometry calculations. It should be stressed that, although our approach brings considerably more complete information, the calculation time needed remains reasonable and similar to that used in the previous method (2232 min. for the calculation of the full ensemble of conformers, with 10 CPU Intel Xeon E5-2660 @ 2.20 GHz).
Our strategy can be applied to other proteins of small/moderate sizes that are amenable to HP-NMR measurements. We believe that by identifying sub-structure hubs and intermediate conformations our approach will be valuable to drive meta-dynamics simulations upon pressure unfolding [
46]. For example, it would be possible to investigate multiple pathways by combining individual contact maps’ information that would provide a better understanding of the energetics and kinetics of the pressure-induced unfolding of proteins.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










