
Neurotoxicity Associated with Treatment of Acute Lymphoblastic Leukemia Chemotherapy and Immunotherapy


Abstract
:1. Introduction
2. Neurotoxicity of Conventional Therapy
2.1. Methotrexate
2.2. Vincristine
2.3. L-Asparaginase
2.4. Nelarabine
3. Neurotoxicity of Immunotherapy
3.1. Blinatumomab
3.2. Inotuzumab Ozogamicin
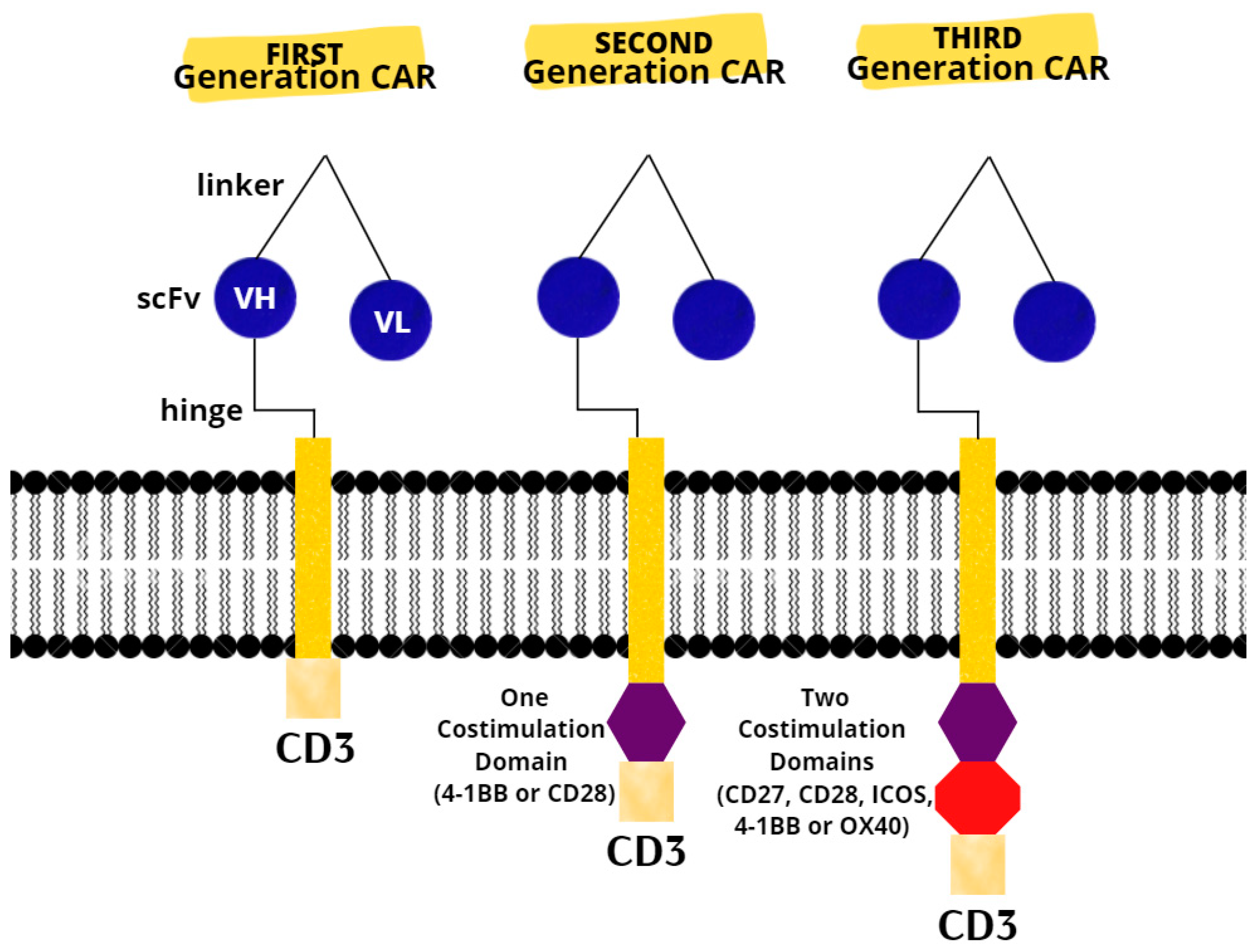
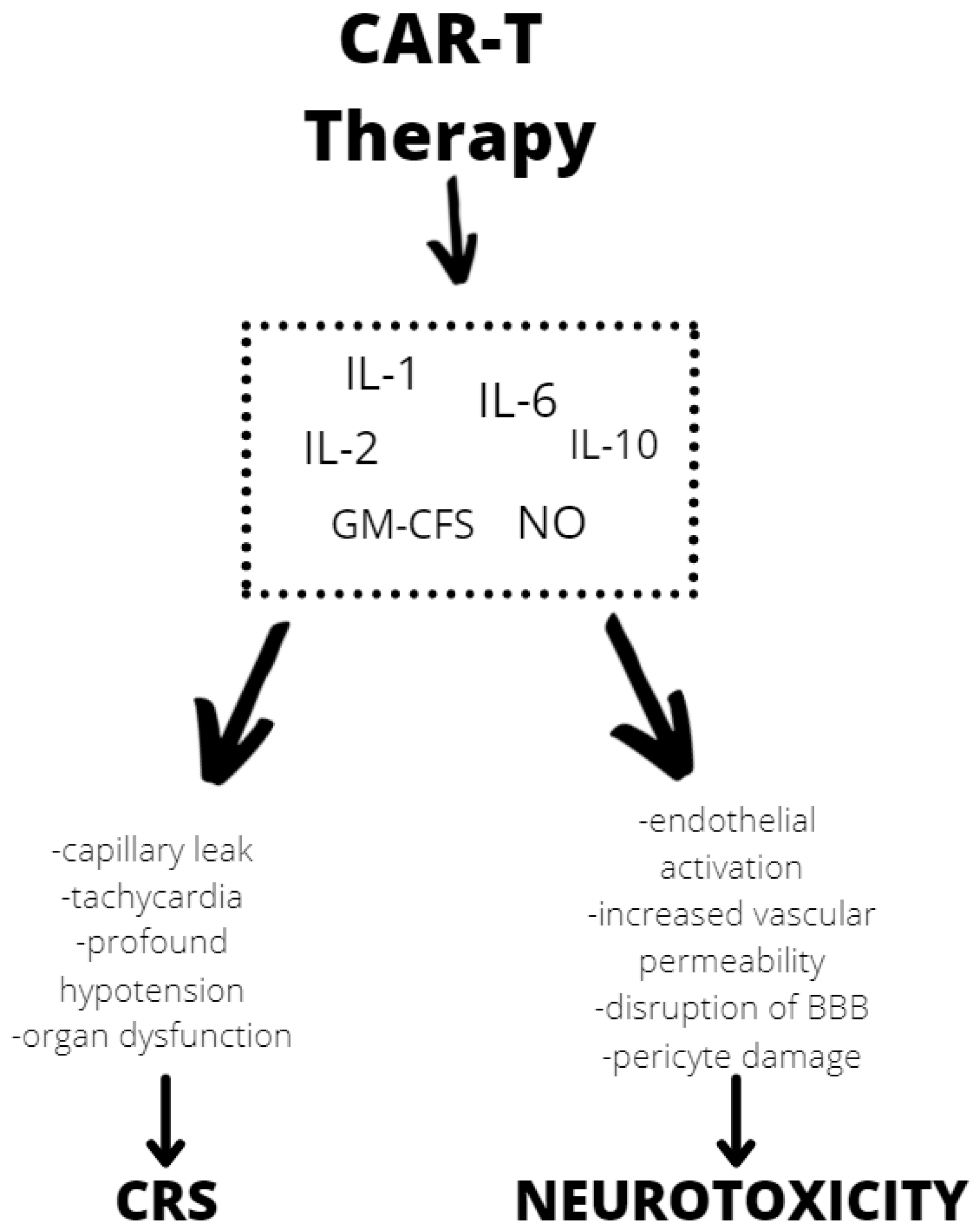
3.3. CAR T-Cell Therapy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABCB1 | ATP Binding Cassette Subfamily B Member 1 |
| ABCB11 | ATP Binding Cassette Subfamily C Member 11 |
| ABCC10 | ATP Binding Cassette Subfamily C Member 10 |
| ABCC2 | ATP Binding Cassette Subfamily C Member 2 |
| ABCC4 | ATP Binding Cassette Subfamily C Member 4 |
| ABCC5 | ATP Binding Cassette Subfamily C Member 1 |
| ADHD | attention deficit hyperactivity disorder |
| ADORA2A | Adenosine A2A Receptor |
| AIEOP | Associazione Italiana di Ematologia e Oncologia Pediatrica |
| ALL | acute lymphoblastic leukemia |
| ALL-BFM-95 | ALL-Berlin–Frankfurt–Münster |
| Ara-G | Nelarabine |
| ara-GTP | ara-G triphosphate |
| Arg27His | amino acid arginine to histidine at codon 27 |
| BBB | blood–brain barrier |
| BiTEs | dual-specific T-cell-binding antibodies |
| CD3 | cluster of differentiation 3 |
| CDA | Cytidine Deaminase |
| CEP72 | Centrosomal Protein 72 |
| CNS | central nervous system |
| COG | Pediatric Oncology Group |
| CR | complete response |
| Cri | a complete response with incomplete cell count recovery |
| CRP | C-reactive protein |
| CRS | cytokine release syndrome |
| CSF | cerebrospinal fluid |
| CTCAE | Common Terminology Criteria for Adverse Events |
| cTNS | clinical Total Neuropathy Score |
| CYP2C8 | Cytochrome P450 Family 2 Subfamily C Member 8 |
| CYP3A4 | Cytochrome P450 Family 3 Subfamily A Member 4 |
| CYP3A5 | Cytochrome P450 Family 3 Subfamily A Member 5 |
| DCK | Deoxycytidine Kinase |
| DHFR19bp | Dihydrofolate Reductase 19 bp polymorphism |
| ENT1 | Equilibrative nucleoside transporter 1 |
| GIT1 | G Protein-Coupled Receptor Kinase Interacting ArfGAP 1 |
| GM-CSF | granulocyte-macrophage colony stimulating factor |
| GRIA1 | Glutamate Ionotropic Receptor AMPA Type Subunit 1 |
| HLA-DRB1 | Major Histocompatibility Complex Class II, DR Beta1 |
| HSCT | allogeneic hematopoietic stem cell transplant (HSCT) |
| I.V. | intravenous |
| ICANS | immune effector cell-associated neurotoxicity syndrome |
| ICE | Immune Effector Cell-Associated Encephalopathy score |
| ICH | intracranial hemorrhage |
| ICOS | Inducible T-cell costimulator |
| ID-MTX | intermediate-dose methotrexate |
| InO | Inotuzumab |
| L-ASP | L-asparaginase |
| MAP4 | Microtubule-Associated Protein 4 |
| MRD | minimal residual disease |
| MTHFR 677C>T | Methylenetetrahydrofolate Reductase polymorphism |
| MTHFR 677TT | Methylenetetrahydrofolate Reductase polymorphism |
| MTX | methotrexate |
| MTX-NT | neurotoxicity associated with methotrexate |
| NO | nitric oxide |
| NT5C2 | 5’-Nucleotidase, Cytosolic II |
| NT5C3, 5’ | Nucleotidase, Cytosolic IIIA |
| R/R | relapsed or refractory |
| RRM1 | Ribonucleotide Reductase Catalytic Subunit M1 |
| scFv | single-chain fragment variable |
| SLC19A1 | Solute Carrier Family 19 Member 1 |
| SLC5A7 | Solute Carrier Family 5 Member 7 |
| SNPs | Single nucleotide polymorphisms |
| SOS | sinusoidal obstruction syndrome |
| T-ALL | T-cell acute lymphoblastic leukemia |
| T-LBL | T-cell lymphoblastic lymphoma |
| TIT | triple intrathecal therapy |
| TUBB1 | Tubulin Beta 1 Class VI |
| TUBB2A | Tubulin Beta 2A Class IIa |
| TUBB2B | Tubulin Beta 2B Class IIb |
| TUBB3 | Tubulin Beta 3 Class III |
| TUBB4A | Tubulin Beta 4A Class Iva |
| TYMS | Thymidylate Synthetase |
| UKALL2003 trial | United Kingdom National Randomised Trial For Children and Young Adults with Acute Lymphoblastic Leukaemia and Lymphoma 2003 |
| VCR | vincristine |
| VH | heavy chain variable gene segment |
| VL | variable region |
| VIPN | vincristine-induced peripheral neuropathy |
| ZBTB1 | Zinc Finger and BTB Domain Containing 1 |
References
- Jeha, S.; Pei, D.; Choi, J.; Cheng, C.; Sandlund, J.T.; Coustan-Smith, E.; Campana, D.; Inaba, H.; Rubnitz, J.E.; Ribeiro, R.C.; et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia without Cranial Irradiation: St Jude Total Therapy Study 16. J. Clin. Oncol. 2019, 37, 3377–3391. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Poppe, M.M.; Hua, C.H.; Marcus, K.J.; Esiashvili, N. Acute lymphoblastic leukemia. Pediatr. Blood Cancer 2021, 68 (Suppl. S2), e28371. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H. Precision medicine in acute lymphoblastic leukemia. Front. Med. 2020, 14, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Malczewska, M.; Kośmider, K.; Bednarz, K.; Ostapińska, K.; Lejman, M.; Zawitkowska, J. Recent Advances in Treatment Options for Childhood Acute Lymphoblastic Leukemia. Cancers 2022, 14, 2021. [Google Scholar] [CrossRef]
- Rahiman, E.A.; Rajendran, A.; Sankhyan, N.; Singh, P.; Muralidharan, J.; Bansal, D.; Trehan, A. Acute neurological complications during acute lymphoblastic leukemia therapy: A single-center experience over 10 years. Indian J. Cancer 2021, 58, 545–552. [Google Scholar]
- Rivera, A.M.; May, S.; Lei, M.; Qualls, S.; Bushey, K.; Rubin, D.B.; Barra, M.E. Associated Neurotoxicity: Current Management and Emerging Treatment Strategies. Crit. Care Nurs. Q. 2020, 43, 191–204. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [Green Version]
- Van de Velde, M.E.; Kaspers, G.L.; Abbink, F.C.H.; Wilhelm, A.J.; Ket, J.C.F.; van den Berg, M.H. Vincristine-induced peripheral neuropathy in children with cancer: A systematic review. Crit. Rev. Oncol. Hematol. 2017, 114, 114–130. [Google Scholar] [CrossRef]
- Soffietti, R.; Trevisan, E.; Rudà, R. Neurologic complications of chemotherapy and other newer and experimental approaches. Handb. Clin. Neurol. 2014, 121, 1199–1218. [Google Scholar]
- Beziat, G.; Tavitian, S.; Picard, M.; Faguer, S.; Recher, C.; Huguet, F. Multiple Severe Toxicities of L-Asparaginase and Their Innovative Management during Induction Therapy of Acute Lymphoblastic Leukemia in an Adult Patient. Case Rep. Hematol. 2019, 2019, 9086570. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Santillan, M.; Iparraguirre, L.; Martin-Guerrero, I.; Gutierrez-Camino, A.; Garcia-Orad, A. Review of pharmacogenetics studies of L-asparaginase hypersensitivity in acute lymphoblastic leukemia points to variants in the GRIA1 gene. Drug Metab. Pers. Ther. 2017, 32, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Pei, D.; Yang, W.; Cheng, C.; Jeha, S.; Cox, N.J.; Evans, W.E.; Pui, C.H.; Relling, M.V. Genetic variations in GRIA1 on chromosome 5q33 related to asparaginase hypersensitivity. Clin. Pharm. 2010, 88, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maamari, D.; El-Khoury, H.; Saifi, O.; Muwakkit, S.A.; Zgheib, N.K. Implementation of Pharmacogenetics to Individualize Treatment Regimens for Children with Acute Lymphoblastic Leukemia. Pharmgenom. Pers. Med. 2020, 13, 295–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-M.; Sun, L.-L.; Zeng, W.-X.; Wu, W.-S.; Zhang, G.-L. Effects of a microRNA binding site polymorphism in SLC19A1 on methotrexate concentrations in Chinese children with acute lymphoblastic leukemia. Med. Oncol. 2014, 31, 62. [Google Scholar] [CrossRef] [PubMed]
- Yazıcıoğlu, B.; Kaya, Z.; Güntekin Ergun, S.; Perçin, F.; Koçak, Ü.; Yenicesu, İ.; Gürsel, T. Influence of Folate-Related Gene Polymorphisms on High-Dose Methotrexate-Related Toxicity and Prognosis in Turkish Children with Acute Lymphoblastic Leukemia. Turk. J. Haematol. 2017, 34, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Hartford, C.M.; Duan, S.; Delaney, S.M.; Mi, S.; Kistner, E.O.; Lamba, J.K.; Dolan, M.E. Population-specific genetic variants important in susceptibility to cytarabine arabinoside cytotoxicity. Blood 2009, 113, 2145–2153. [Google Scholar] [CrossRef] [Green Version]
- Di Francia, R.; Crisci, S.; De Monaco, A.; Cafiero, C.; Re, A.; Iaccarino, G.; De Filippi, R.; Frigeri, F.; Corazzelli, G.; Micera, A.; et al. Response and Toxicity to Cytarabine Therapy in Leukemia and Lymphoma: From Dose Puzzle to Pharmacogenomic Biomarkers. Cancers 2021, 13, 966. [Google Scholar] [CrossRef]
- Abraham, A.; Varatharajan, S.; Karathedath, S.; Philip, C.; Lakshmi, K.M.; Jayavelu, A.K.; Mohanan, E.; Janet, N.B.; Srivastava, V.M.; Shaji, R.V.; et al. RNA expression of genes involved in cytarabine metabolism and transport predicts cytarabine response in acute myeloid leukemia. Pharmacogenomics 2015, 16, 877–890. [Google Scholar] [CrossRef] [Green Version]
- Lamba, J.K. Genetic factors influencing cytarabine therapy. Pharmacogenomics 2009, 10, 1657–1674. [Google Scholar] [CrossRef] [Green Version]
- Magge, R.S.; DeAngelis, L.M. The double-edged sword: Neurotoxicity of chemotherapy. Blood Rev. 2015, 29, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Millan, N.C.; Pastrana, A.; Guitter, M.R.; Zubizarreta, P.A.; Monges, M.S.; Felice, M.S. Acute and sub-acute neurological toxicity in children treated for acute lymphoblastic leukemia. Leuk. Res. 2018, 65, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Vagace, J.M.; Caceres-Marzal, C.; Jimenez, M.; Casado, M.S.; de Murillo, S.G.; Gervasini, G. Methotrexate-induced subacute neurotoxicity in a child with acute lymphoblastic leukemia carrying genetic polymorphisms related to folate homeostasis. Am. J. Hematol. 2011, 86, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, R.; Kaur, H.; Scott, J.X.; Sneha, L.M.; Arun Kumar, G.P.; Srinivasan, A.; Paul, S.F. Pharmacogenetic evaluation of 6-mercaptopurine-mediated toxicity in pediatric acute lymphoblastic leukemia patients from a South Indian population. Pharmacogenomics 2021, 22, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Kotnik, B.F.; Jazbec, J.; Grabar, P.B.; Rodriguez-Antona, C.; Dolzan, V. Association between SLC19A Gene Polymorphism and High Dose Methotrexate Toxicity in Childhood Acute Lymphoblastic Leukaemia and Non-Hodgkin Malignant Lymphoma: Introducing a Haplotype based Approach. Radiol Oncol. 2017, 51, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janik, P.; Berdyński, M.; Safranow, K.; Żekanowski, C. Association of ADORA1 rs2228079 and ADORA2A rs5751876 Polymorphisms with Gilles de la Tourette Syndrome in the Polish Population. PLoS ONE 2015, 10, e0136754. [Google Scholar] [CrossRef]
- Franca, R.; Rebora, P.; Bertorello, N.; Fagioli, F.; Conter, V.; Biondi, A.; Colombini, A.; Micalizzi, C.; Zecca, M.; Parasole, R.; et al. Pharmacogenetics and induction/consolidation therapy toxicities in acute lymphoblastic leukemia patients treated with AIEOP-BFM ALL 2000 protocol. Pharmacogenom. J. 2017, 17, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, S.I.; Yanagimachi, M.; Tanoshima, R.; Urayama, K.Y.; Tanaka, F.; Aida, N.; Goto, H.; Ito, S. Influence of ADORA2A gene polymorphism on leukoencephalopathy risk in MTX-treated pediatric patients affected by hematological malignancies. Pediatr. Blood Cancer 2016, 63, 1983–1989. [Google Scholar] [CrossRef]
- Ramalingam, R.; Kaur, H.; Scott, J.X.; Sneha, L.M.; Arunkumar, G.; Srinivasan, A.; Paul, S.F.D. Evaluation of cytogenetic and molecular markers with MTX-mediated toxicity in pediatric acute lymphoblastic leukemia patients. Cancer Chemother. Pharmacol. 2022, 89, 393–400. [Google Scholar] [CrossRef]
- Vora, A.J.; Goulden, N.; Mitchell, C.D.; Hough, R.; Rowntree, C.; Richards, S.M. UKALL 2003, A Randomised Trial Investigating Treatment Intensification for Children and Young Adults with Minimal Residual Disease Defined High Risk Acute Lymphoblastic Leukaema. Blood 2012, 120, 136. [Google Scholar] [CrossRef]
- Bhojwani, D.; Sabin, N.D.; Pei, D.; Yang, J.J.; Khan, R.B.; Panetta, J.C.; Krull, K.R.; Inaba, H.; Rubnitz, J.E.; Metzger, M.L.; et al. Methotrexate-induced neurotoxicity and leukoencephalopathy in childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2014, 32, 949–959. [Google Scholar] [CrossRef] [Green Version]
- Lavoie Smith, E.M.; Li, L.; Chiang, C.; Thomas, K.; Hutchinson, R.J.; Wells, E.M.; Ho, R.H.; Skiles, J.; Chakraborty, A.; Bridges, C.M.; et al. Patterns and severity of vincristine-induced peripheral neuropathy in children with acute lymphoblastic leukemia. Patterns and severity of vincristine-induced peripheral neuropathy in children with acute lymphoblastic leukemia. J. Peripher. Nerv. Syst. 2015, 20, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Stock, W.; Diouf, B.; Crews, K.R.; Pei, D.; Cheng, C.; Laumann, K.; Mandrekar, S.J.; Luger, S.; Advani, A.; Stone, R.M.; et al. An Inherited Genetic Variant in CEP72 Promoter Predisposes to Vincristine-Induced Peripheral Neuropathy in Adults with Acute Lymphoblastic Leukemia. Clin. Pharmacol. Ther. 2017, 101, 391–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.Y.; Hu, Y.H.; Guo, H.L.; Xia, Y.; Zhang, Y.; Fang, W.R.; Li, Y.M.; Xu, J.; Chen, F.; Wang, Y.R.; et al. Vincristine-Induced Peripheral Neuropathy in Childhood Acute Lymphoblastic Leukemia: Genetic Variation as a Potential Risk Factor. Front. Pharmacol. 2021, 12, 771487. [Google Scholar] [CrossRef] [PubMed]
- Diouf, B.; Wing, C.; Panetta, J.C.; Eddins, D.; Lin, W.; Yang, W.; Fan, Y.; Pei, D.; Cheng, C.; Delaney, S.M.; et al. Identification of small molecules that mitigate vincristine-induced neurotoxicity while sensitizing leukemia cells to vincristine. Clin. Transl. Sci. 2021, 14, 1490–1504. [Google Scholar] [CrossRef]
- Schouten, S.M.; van de Velde, M.E.; Kaspers, G.J.L.; Mokkink, L.B.; van der Sluis, I.M.; van den Bos, C.; Hartman, A.; Abbink, F.C.H.; van den Berg, M.H. Measuring vincristine-induced peripheral neuropathy in children with cancer: Validation of the Dutch pediatric-modified Total Neuropathy Score. Support. Care Cancer 2020, 28, 2867–2873. [Google Scholar] [CrossRef] [Green Version]
- Jain, P.; Gulati, S.; Seth, R.; Bakhshi, S.; Toteja, G.S.; Pandey, R.M. Vincristine-induced neuropathy in childhood ALL (acute lymphoblastic leukemia) survivors: Prevalence and electrophysiological characteristics. J. Child Neurol. 2014, 29, 932–937. [Google Scholar] [CrossRef]
- Balayssac, D.; Ferrier, J.; Descoeur, J.; Ling, B.; Pezet, D.; Eschalier, A.; Authier, N. Chemotherapy-induced peripheral neuropathies: From clinical relevance to preclinical evidence. Expert Opin. Drug Saf. 2011, 10, 407–417. [Google Scholar] [CrossRef]
- Raj, T.A.; Smith, A.M.; Moore, A.S. Vincristine sulfate liposomal injection for acute lymphoblastic leukemia. Int. J. Nanomed. 2013, 8, 4361–4369. [Google Scholar]
- Adil, M.K.; Ali, Z.; Arshad, U.; Fawad, U. Vincristine induced neurotoxicity in children who underwent chemotherapy for acute lymphoblastic leukemia and Wilms tumor. Pak. J. Med. Sci. 2021, 37, 1331–1334. [Google Scholar] [CrossRef]
- Diouf, B.; Crews, K.R.; Lew, G.; Pei, D.; Cheng, C.; Bao, J.; Zheng, J.J.; Yang, W.; Fan, Y.; Wheeler, H.E.; et al. Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA 2015, 313, 815–823. [Google Scholar] [CrossRef]
- Tay, C.G.; Lee, V.W.M.; Ong, L.C.; Goh, K.J.; Ariffin, H.; Fong, C.Y. Vincristine-induced peripheral neuropathy in survivors of childhood acute lymphoblastic leukaemia. Pediatric Blood Cancer 2017, 64, e26471. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, M.; Fiúza, T.; Morais, S.B.; Souza, T.; Trevizani, R. Circumventing the side effects of L-asparaginase. Biomed. Pharmacother. 2021, 139, 111616. [Google Scholar] [CrossRef] [PubMed]
- Belén, L.H.; Lissabet, J.B.; de Oliveira Rangel-Yagui, C.; Effer, B.; Monteiro, G.; Pessoa, A.; Farías Avendaño, J.G. A structural in silico analysis of the immunogenicity of l-asparaginase from Escherichia coli and Erwinia carotovora. Biologicals 2019, 59, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Moghrabi, A.; Levy, D.E.; Asselin, B.; Barr, R.; Clavell, L.; Hurwitz, C.; Samson, Y.; Schorin, M.; Dalton, V.K.; Lipshultz, S.E.; et al. Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood 2007, 109, 896–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, A.M.; Oliveira-Nascimento, L.; Ribeiro, A.; Tairum, C.A.; Breyer, C.A., Jr.; Oliveira, M.A.; Monteiro, G.; Souza-Motta, C.M.; Magalhães, P.O.; Avendaño, J.G.; et al. Therapeutic l-asparaginase: Upstream, downstream and beyond. Crit. Rev. Biotechnol. 2017, 37, 82–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.T.; Wu, K.H.; Lan, S.J.; Tsai, J.J.; Tsai, F.J.; Tsai, C.H. Amino acid concentrations in cerebrospinal fluid in children with acute lymphoblastic leukemia undergoing chemotherapy. Eur. J. Cancer 2005, 41, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, S.E.; Chen, E.C.; Sandhu, M.; Deshpande, K.; Dhaher, R.; Hersey, D.; Eid, T. Branched-Chain Amino Acids and Seizures: A Systematic Review of the Literature. CNS Drugs 2019, 33, 755–770. [Google Scholar] [CrossRef]
- Hamdan, M.Y.; Frenkel, E.P.; Bick, R. L-asparaginase-provoked seizures as singular expression of central nervous toxicity. Clin. Appl. Thromb. Hemost. 2000, 6, 234–238. [Google Scholar] [CrossRef]
- Tong, W.H.; Pieters, R.; Kaspers, G.J.; te Loo, D.M.; Bierings, M.B.; van den Bos, C.; Kollen, W.J.; Hop, W.C.; Lanvers-Kaminsky, C.; Relling, M.V.; et al. A prospective study on drug monitoring of PEGasparaginase and Erwinia asparaginase and asparaginase antibodies in pediatric acute lymphoblastic leukemia. Blood 2014, 123, 2026–2033. [Google Scholar] [CrossRef] [Green Version]
- Hijiya, N.; van der Sluis, I.M. Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leuk. Lymphoma 2016, 57, 748–757. [Google Scholar] [CrossRef]
- Feinberg, W.M.; Swenson, M.R. Cerebrovascular complications of L-asparaginase therapy. Neurology 1988, 38, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Kieslich, M.; Porto, L.; Lanfermann, H.; Jacobi, G.; Schwabe, D.; Böhles, H. Cerebrovascular complications of L-asparaginase in the therapy of acute lymphoblastic leukemia. J. Pediatr. Hematol. Oncol. 2003, 25, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Aldoss, I.; Douer, D. How I treat the toxicities of pegasparaginase in adults with acute lymphoblastic leukemia. Blood 2020, 135, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Göttl, U.; Heinecke, A.; von Kries, R.; Nürnberger, W.; Münchow, N.; Junker, R. Thrombotic events revisited in children with acute lymphoblastic leukemia: Impact of concomitant Escherichia coli asparaginase/prednisone administration. Thromb. Res. 2001, 103, 165–172. [Google Scholar] [CrossRef]
- Grace, R.F.; Dahlberg, S.E.; Neuberg, D.; Sallan, S.E.; Connors, J.M.; Neufeld, E.J.; DeAngelo, D.J.; Silverman, L.B. The frequency and management of asparaginase-related thrombosis in paediatric and adult patients with acute lymphoblastic leukaemia treated on Dana-Farber Cancer Institute consortium protocols. Br. J. Haematol. 2011, 152, 452–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caruso, V.; Iacoviello, L.; Di Castelnuovo, A.; Storti, S.; Mariani, G.; de Gaetano, G.; Donati, M.B. Thrombotic complications in childhood acute lymphoblastic leukemia: A meta-analysis of 17 prospective studies comprising 1752 pediatric patients. Blood 2006, 108, 2216–2222. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.H.; Johnson, J.R.; Justice, R.; Pazdur, R. FDA Drug Approval Summary: Nelarabine (Arranon®) for the Treatment of T-Cell Lymphoblastic Leukemia/Lymphoma. Oncologist 2008, 13, 709–714. [Google Scholar] [CrossRef]
- Hayashi, R.J.; Winter, S.S.; Dunsmore, K.P.; Devidas, M.; Chen, Z.; Wood, B.L.; Hermiston, M.L.; Teachey, D.T.; Perkins, S.L.; Miles, R.R. Successful Outcomes of Newly Diagnosed T Lymphoblastic Lymphoma: Results from Children’s Oncology Group AALL0434. J. Clin. Oncol. 2020, 38, 3062–3070. [Google Scholar] [CrossRef]
- Dunsmore, K.P.; Winter, S.S.; Devidas, M.; Wood, B.L.; Esiashvili, N.; Chen, Z.; Eisenberg, N.; Briegel, N.; Hayashi, R.J.; Gastier-Foster, J.M.; et al. Children’s Oncology Group AALL0434: A Phase III Randomized Clinical Trial Testing Nelarabine in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2020, 38, 3282–3293. [Google Scholar] [CrossRef]
- Kumamoto, T.; Goto, H.; Ogawa, C.; Hori, T.; Deguchi, T.; Araki, T.; Saito, A.M.; Manabe, A.; Horibe, H.; Toyoda, H. FLEND (nelarabine, fludarabine, and etoposide) for relapsed T-cell acute lymphoblastic leukemia in children: A report from Japan Children’s Cancer Group. Int. J. Hematol. 2020, 112, 720–724. [Google Scholar] [CrossRef]
- Kuhlen, M.; Bleckmann, K.; Möricke, A.; Schrappe, M.; Vieth, S.; Escherich, G.; Bronsema, A.; Vonalt, A.; Queudeville, M.; Zwaan, C.M.; et al. Neurotoxic side effects in children with refractory or relapsed T-cell malignancies treated with nelarabine based therapy. Br. J. Haematol. 2017, 179, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Przepiorka, D.; Ko, C.W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topp, M.S.; Gökbuget, N.; Stein, A.S.; Zugmaier, G.; O’Brien, S.; Bargou, R.C.; Dombret, H.; Fielding, A.K.; Heffner, L.; Larson, R.A.; et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: A multicentre, single-arm, phase 2 study. Lancet Oncol. 2015, 16, 57–66. [Google Scholar] [CrossRef]
- Nagorsen, D.; Kufer, P.; Baeuerle, P.A.; Bargou, R. Blinatumomab: A historical perspective. Pharmacol. Ther. 2012, 136, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Zugmaier, G.; Mergen, N.; Bader, P.; Jeha, S.; Schlegel, P.G.; Bourquin, J.P.; Handgretinger, R.; Brethon, B.; Rössig, C. Blinatumomab in pediatric relapsed/refractory B-cell acute lymphoblastic leukemia: RIALTO expanded access study final analysis. Blood Adv. 2022, 6, 1004–1014. [Google Scholar] [CrossRef] [PubMed]
- Queudeville, M.; Schlegel, P.; Heinz, A.T.; Lenz, T.; Döring, M.; Holzer, U.; Hartmann, U.; Kreyenberg, H.; von Stackelberg, A.; Schrappe, M.; et al. Blinatumomab in pediatric patients with relapsed/refractory B-cell precursor acute lymphoblastic leukemia. Eur. J. Haematol. 2021, 106, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.S.; Schiller, G.; Benjamin, R.; Jia, C.; Zhang, A.; Zhu, M.; Zimmerman, Z.; Topp, M.S. Neurologic adverse events in patients with relapsed/refractory acute lymphoblastic leukemia treated with blinatumomab: Management and mitigating factors. Ann. Hematol. 2019, 98, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Locatelli, F.; Zugmaier, G.; Rizzari, C.; Morris, J.D.; Gruhn, B.; Klingebiel, T.; Parasole, R.; Linderkamp, C.; Flotho, C.; Petit, A. Effect of Blinatumomab vs Chemotherapy on Event-Free Survival Among Children with High-risk First-Relapse B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 2021, 325, 843–854. [Google Scholar] [CrossRef]
- Beneduce, G.; De Matteo, A.; Stellato, P.; Testi, A.M.; Bertorello, N.; Colombini, A.; Putti, M.C.; Rizzari, C.; Cesaro, S.; Cellini, M.; et al. Blinatumomab in Children and Adolescents with Relapsed/Refractory B Cell Precursor Acute Lymphoblastic Leukemia: A Real-Life Multicenter Retrospective Study in Seven AIEOP (Associazione Italiana di Ematologia e Oncologia Pediatrica) Centers. Cancers 2022, 14, 426. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.M.; Jabbour, E.; Wang, T.; Liang White, J.; et al. Final report and long-term survival follow-up from the randomized, phase 3 INO-VATE study. Cancer 2019, 125, 2474–2487. [Google Scholar] [CrossRef] [Green Version]
- Lamb, Y.N. Inotuzumab Ozogamicin: First Global Approval. Drugs 2017, 77, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Morley, N.J.; Marks, D.I. Inotuzumab ozogamicin in the management of acute lymphoblastic leukaemia. Expert Rev. Anticancer Ther. 2016, 16, 159–164. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.J.; Advani, A.S.; Marks, D.I.; Stelljes, M.; Liedtke, M.; Stock, W.; Gökbuget, N.; Jabbour, E.; Merchant, A.; Wang, T.; et al. Inotuzumab ozogamicin for relapsed/refractory acute lymphoblastic leukemia: Outcomes by disease burden. Blood Cancer J. 2020, 10, 81. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Hucks, G.; Rheingold, S.R. The journey to CAR T cell therapy: The pediatric and young adult experience with relapsed or refractory B-ALL. Blood Cancer J. 2019, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Kotch, C.; Barrett, D.; Teachey, D.T. Tocilizumab for the treatment of chimeric antigen receptor T cell-induced cytokine release syndrome. Expert Rev. Clin. Immunol. 2019, 15, 813–822. [Google Scholar] [CrossRef]
- Iriguchi, S.; Kaneko, S. Toward the development of true “off-the-shelf” synthetic T-cell immunotherapy. Cancer Sci. 2019, 110, 16–22. [Google Scholar] [CrossRef]
- Hunter, B.D.; Jacobson, C.A. CAR T-Cell Associated Neurotoxicity: Mechanisms, Clinicopathologic Correlates, and Future Directions. J. Natl. Cancer Inst. 2019, 111, 646–654. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, T.; Litzow, M.R. No free rides: Management of toxicities of novel immunotherapies in ALL, including financial. Blood Adv. 2018, 2, 3393–3403. [Google Scholar] [CrossRef] [PubMed]
- Möhn, N.; Bonda, V.; Grote-Levi, L.; Panagiota, V.; Fröhlich, T.; Schultze-Florey, C.; Wattjes, M.P.; Beutel, G.; Eder, M.; David, S.; et al. Neurological management and work-up of neurotoxicity associated with CAR T cell therapy. Neurol. Res. Pract. 2022, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Sheth, V.S.; Gauthier, J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant. 2021, 56, 552–566. [Google Scholar] [CrossRef]
- Gust, J.; Taraseviciute, A.; Turtle, C.J. Neurotoxicity Associated with CD19-Targeted CAR-T Cell Therapies. CNS Drugs. 2018, 32, 1091–1101. [Google Scholar] [CrossRef]
- Shah, N.N.; Highfill, S.L.; Shalabi, H.; Yates, B.; Jin, J.; Wolters, P.L.; Ombrello, A.; Steinberg, S.M.; Martin, S.; Delbrook, C.; et al. CD4/CD8 T-cell selection affects chimeric antigen receptor (CAR) T-cell potency and toxicity: Updated results from a phase I anti-CD22 CAR T-cell trial. J. Clin. Oncol. 2020, 38, 1938–1950. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Phase of Treatment | Drugs | Toxicity-Related Gene | Mechanism of Neurotoxicity | Neurotoxicity | References |
|---|---|---|---|---|---|
| Induction | Vincristine | ABCC11 1, ABCC2 2, ABCC4 3, ABCC5 4, ABCB1 5, ABCC10 6, CEP72 7, SLC5A7 8, TUBB1 9, TUBB2A 10, TUBB2B 11, TUBB3 12, TUBB4A 13, MAP4 14, CYP3A4 15, CYP2C8 16, CYP3A5 17, CEP72 18 | Interferes with the assembly of microtubule structures leading to cell apoptosis. It affects the peripheral nerves but can also contribute to dysfunction of the cranial nerves and autonomic nervous system. | Peripheral neuropathy, sensory neuropathy: symmetry sensory/tactile impairment, numbness, and tingling in the hands and feet, paresthesia, decreased balance, tendon weakening, visual and hearing problems. | [8,9] |
| L-asparaginase | ZBTB1 19, GRIA1 20, HLA-DRB1 21 | L-asparaginase produces three neurotoxic agents: ammonia, L-aspartic acid, and glutamic acid. These two amino acids can induce cell death in CNS neurons by excessive stimulation through NMDA (N-methyl-D-aspartate) receptor, leading to a major intracellular calcium influx and apoptosis. | Myelosuppression, encephalopathy, hepatic toxicity. | [10,11,12] | |
| Consolidation | Methotrexate (intravenous infusion and intrathecally) | DHFR19bp 22, MTHFR 677C > T 23, MTHFR 677TT 24, SLC19A1 25, TYMS 26, ADORA2A 27 | Methotrexate is an antimetabolite that inhibits Dihydrofolate Reductase and thus tetrahydrofolate formation. This affects the synthesis of macromolecules such as myelin, and reversible leukoencephalopathy has been suggested to be secondary to impaired myelin turnover. Dihydrofolate Reductase inhibition leads to lack of folate and cobalamin, and increase in homocysteine, which is toxic to vascular endothelium may cause seizures and vascular disease. Dihydrofolate Reductase inhibition results in decreased levels of S-adenosylmethionine, which in turn plays a role in maintaining the myelin sheath, and this deficiency may lead to demyelination after intrathecal methotrexate administration. | Transverse myelopathy-symptoms include back pain with subsequent weakness, sensory loss and bladder or bowel incontinence, blurred vision, aphasia, anarthria, seizures, aphasia, mental status disorder, stroke-like episodes, delirium, leukoencephalopathy septic meningitis characterized by headache, neck stiffness, nausea, vomiting and potential fever and encephalopathy. | [13,14,15] |
| Cytarabine | DCK 28, NT5C2 29, CDA 30, RRM1 31, GIT1 32, NT5C 3 33, ENT1 34, SCL29A1 25 | Cytarabine exhibits preferential toxicity for CNS 35 progenitor cells and oligodendrocytes, compromises cell division in vitro, and causes cell death and reduced cell division in vivo. | Myelosuppression, neurotoxicity. | [16,17,18,19] | |
| Maintenance | Methotrexate (orally) | Genes have been described above. | Mechanism has been described above. | Seizures, aphasia, mental status disorder, stroke-like episodes, delirium, leukoencephalopathy, cognitive dysfunction, personality changes. | [13,14,15,20] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Śliwa-Tytko, P.; Kaczmarska, A.; Lejman, M.; Zawitkowska, J. Neurotoxicity Associated with Treatment of Acute Lymphoblastic Leukemia Chemotherapy and Immunotherapy. Int. J. Mol. Sci. 2022, 23, 5515. https://doi.org/10.3390/ijms23105515
Śliwa-Tytko P, Kaczmarska A, Lejman M, Zawitkowska J. Neurotoxicity Associated with Treatment of Acute Lymphoblastic Leukemia Chemotherapy and Immunotherapy. International Journal of Molecular Sciences. 2022; 23(10):5515. https://doi.org/10.3390/ijms23105515
Chicago/Turabian StyleŚliwa-Tytko, Patrycja, Agnieszka Kaczmarska, Monika Lejman, and Joanna Zawitkowska. 2022. "Neurotoxicity Associated with Treatment of Acute Lymphoblastic Leukemia Chemotherapy and Immunotherapy" International Journal of Molecular Sciences 23, no. 10: 5515. https://doi.org/10.3390/ijms23105515
APA StyleŚliwa-Tytko, P., Kaczmarska, A., Lejman, M., & Zawitkowska, J. (2022). Neurotoxicity Associated with Treatment of Acute Lymphoblastic Leukemia Chemotherapy and Immunotherapy. International Journal of Molecular Sciences, 23(10), 5515. https://doi.org/10.3390/ijms23105515