
PAI-1: A Major Player in the Vascular Dysfunction in Obstructive Sleep Apnea?


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. PAI-1 Sources, Structure, and Function
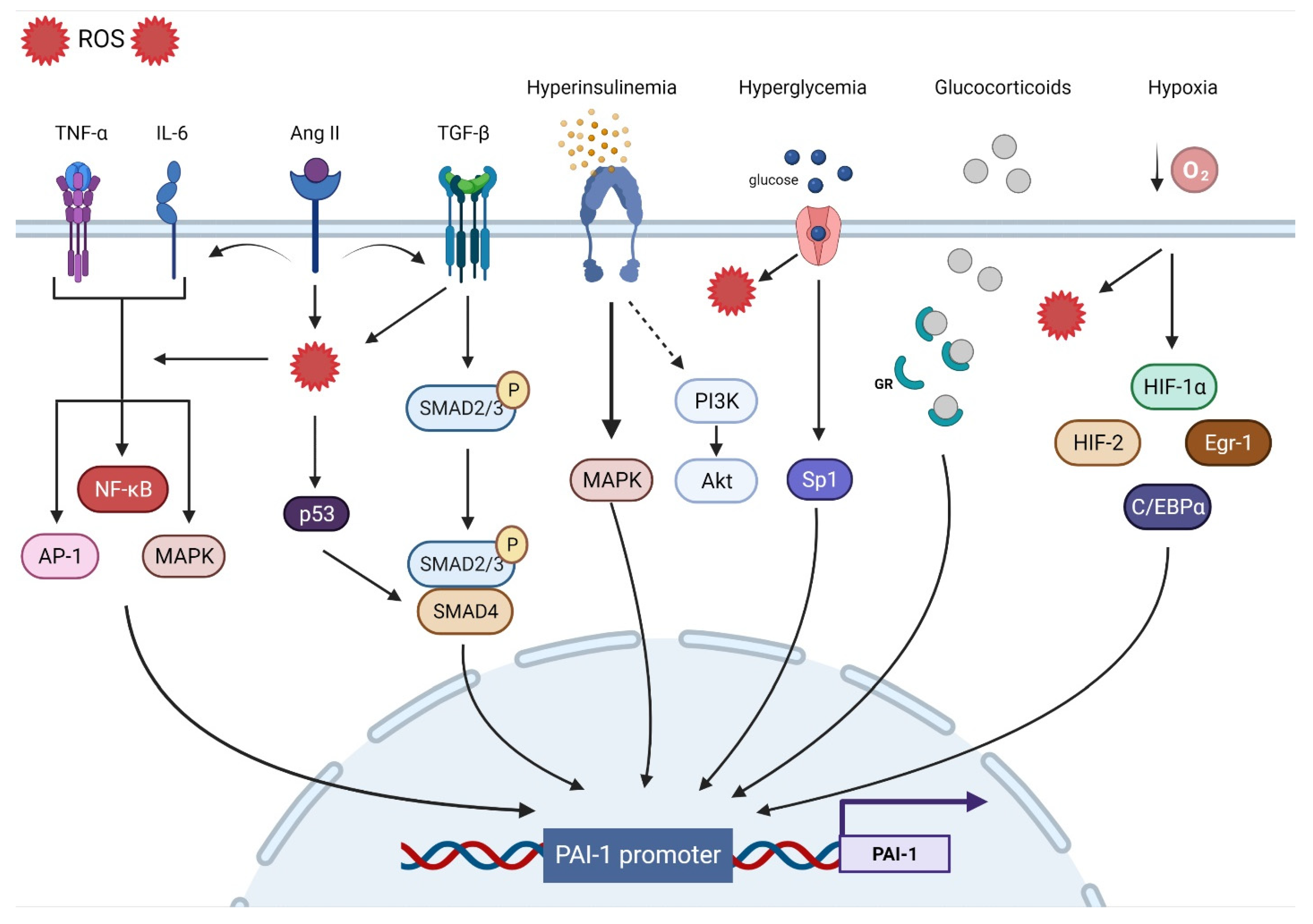
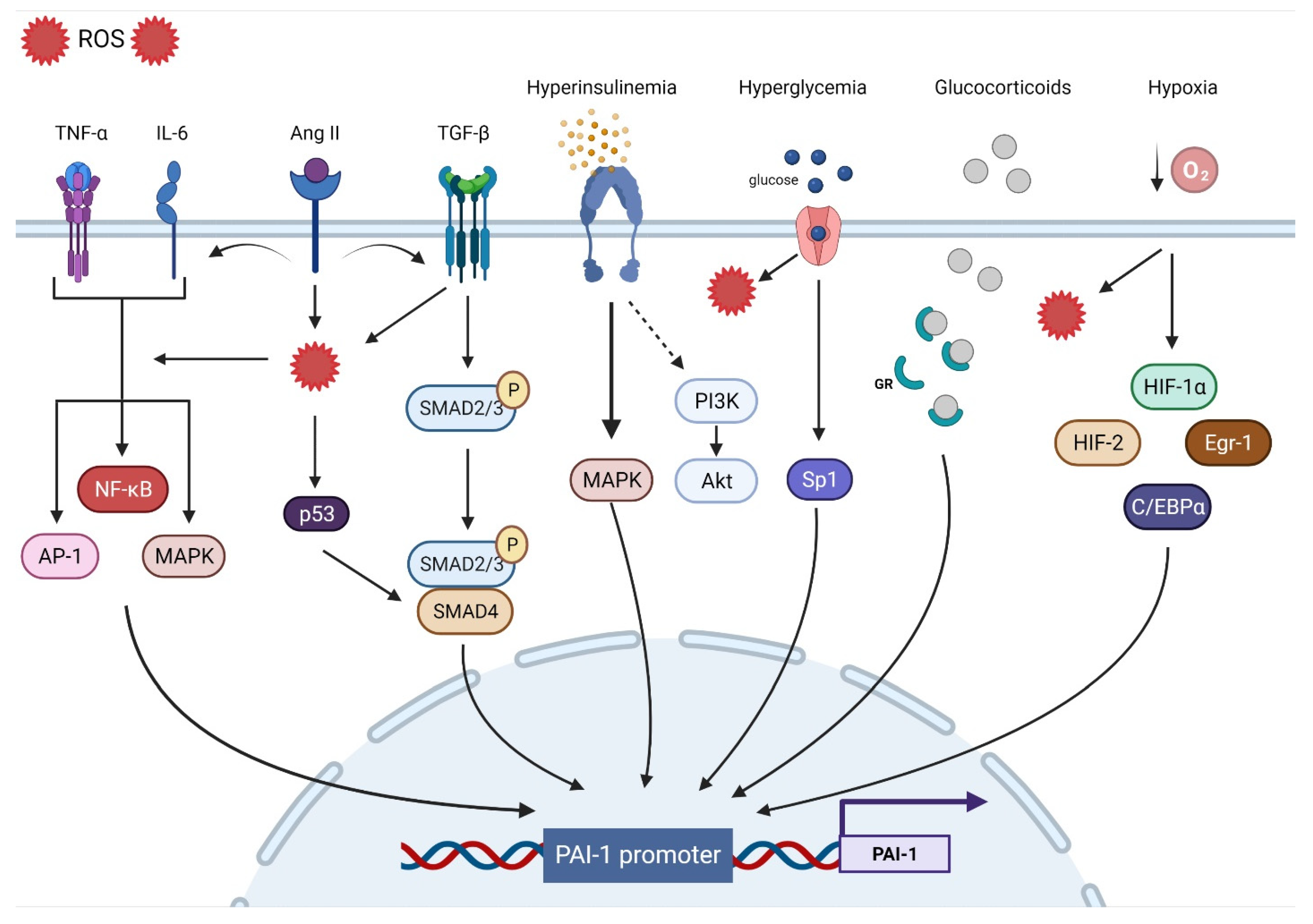
3. Mechanisms Involved in PAI-1 Upregulation
3.1. Oxidative Stress
3.2. Inflammation
3.3. Fibrosis
3.4. Hypoxia
3.5. Hormones
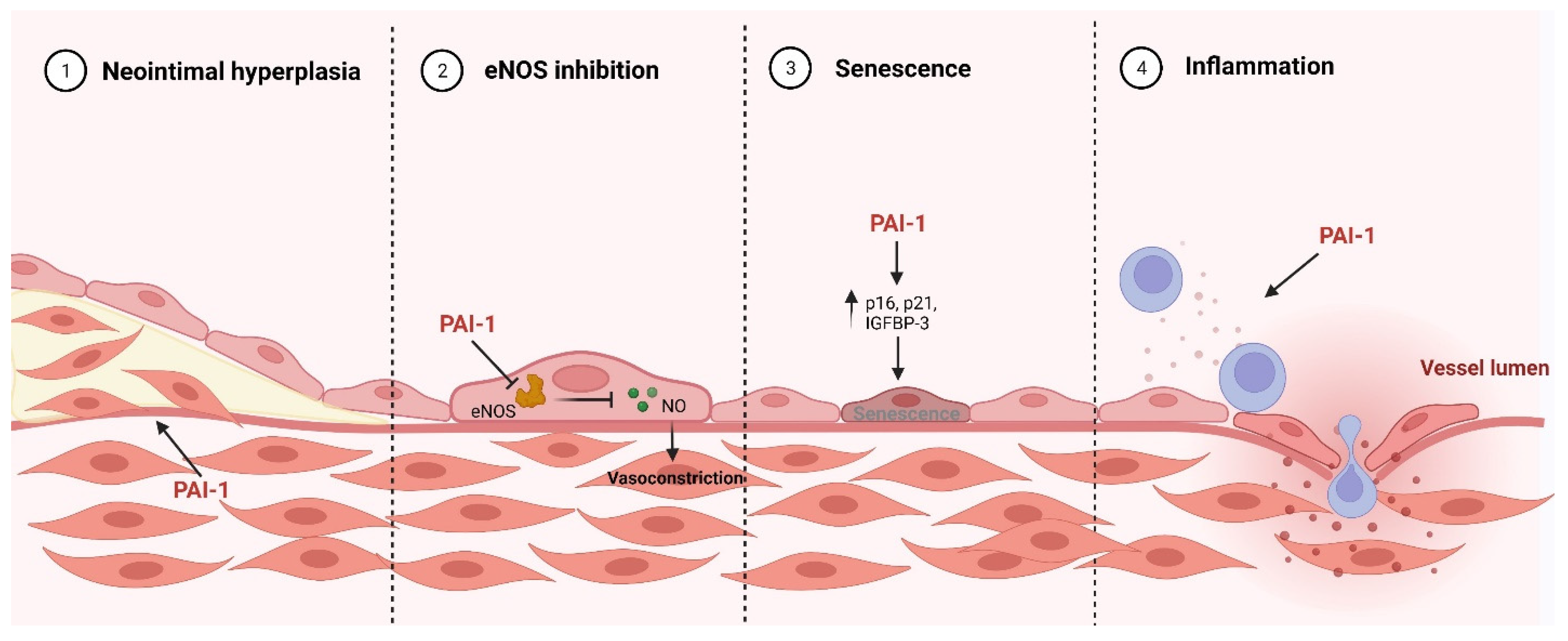
4. Pathological Role PAI-1 Role in Cardiovascular Disease
4.1. Pro-Inflammatory
4.2. eNOS Inhibition
4.3. Senescence
4.4. Neointimal Hyperplasia
5. Is PAI-1 a Mediator of OSA-Induced CVD?
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AP1 | Activation protein 1 |
| CAD | Coronary artery disease |
| CPAP | Continuous positive airway pressure |
| CRP | C-reactive protein |
| CVD | Cardiovascular disease |
| ECM | Extracellular matrix |
| eNOS | Endothelial nitric oxide synthase |
| GR | Glucocorticoid receptor |
| HIF-1α | Hypoxia-inducing factor-1α |
| IH | Intermittent hypoxia |
| IL-6 | Interleukin-6 |
| LRP1 | Low density lipoprotein receptor-related protein 1 |
| MACE | Major adverse cardiovascular events |
| MAPK | Mitogen-activated protein kinase |
| MI | Myocardial infarction |
| MMP | Matrix metalloproteinase |
| NF-κB | Nuclear factor kappa B |
| OSA | Obstructive sleep apnea |
| PAI-1 | Plasminogen activator inhibitor-1 |
| RCL | Reactive center loop |
| ROS | Reactive oxygen species |
| SMC | Smooth muscle cell |
| Sp1 | Specificity protein 1 |
| TGF-β | Transforming growth factor-β |
| TNF-α | Tumor necrosis factor |
| tPA | Tissue-type plasminogen activator |
| uPA | Urokinase-type plasminogen activator |
| uPAR | Urokinase-type plasminogen activator receptor |
References
- Benjafield, A.V.; Ayas, N.T.; Eastwood, P.R.; Heinzer, R.; Ip, M.S.M.; Morrell, M.J.; Nunez, C.M.; Patel, S.R.; Penzel, T.; Pépin, J.L.D.; et al. Estimation of the Global Prevalence and Burden of Obstructive Sleep Apnoea: A Literature-Based Analysis. Lancet Respir. Med. 2019, 7, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Kapur, V.K.; Auckley, D.H.; Chowdhuri, S.; Kuhlmann, D.C.; Mehra, R.; Ramar, K.; Harrod, C.G. Clinical Practice Guideline for Diagnostic Testing for Adult Obstructive Sleep Apnea: An American Academy of Sleep Medicine Clinical Practice Guideline. J. Clin. Sleep Med. 2017, 13, 479–504. [Google Scholar] [CrossRef] [PubMed]
- Badran, M.; Yassin, B.A.; Fox, N.; Laher, I.; Ayas, N. Epidemiology of Sleep Disturbances and Cardiovascular Consequences. Can. J. Cardiol. 2015, 31, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Golbidi, S.; Badran, M.; Ayas, N.; Laher, I. Cardiovascular Consequences of Sleep Apnea. Lung 2012, 190, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Badran, M.; Ayas, N.; Laher, I. Insights into Obstructive Sleep Apnea Research. Sleep Med. 2014, 15, 485–495. [Google Scholar] [CrossRef]
- Badran, M.; Yassin, B.A.; Lin, D.T.S.; Kobor, M.S.; Ayas, N.; Laher, I. Gestational Intermittent Hypoxia Induces Endothelial Dysfunction, Reduces Perivascular Adiponectin and Causes Epigenetic Changes in Adult Male Offspring. J. Physiol. 2019, 597, 5349–5364. [Google Scholar] [CrossRef]
- Badran, M.; Abuyassin, B.; Golbidi, S.; Ayas, N.; Laher, I. Uncoupling of Vascular Nitric Oxide Synthase Caused by Intermittent Hypoxia. Oxid. Med. Cell. Longev. 2016, 2354870. [Google Scholar] [CrossRef]
- Badran, M.; Golbidi, S.; Devlin, A.; Ayas, N.; Laher, I. Chronic Intermittent Hypoxia Causes Endothelial Dysfunction in a Mouse Model of Diet-Induced Obesity. Sleep Med. 2014, 15, 596–602. [Google Scholar] [CrossRef]
- Castro-Grattoni, A.L.; Alvarez-Buvé, R.; Torres, M.; Farré, R.; Montserrat, J.M.; Dalmases, M.; Almendros, I.; Barbé, F.; Sánchez-De-La-Torre, M. Intermittent Hypoxia-Induced Cardiovascular Remodeling Is Reversed by Normoxia in a Mouse Model of Sleep Apnea. Chest 2016, 149, 1400–1408. [Google Scholar] [CrossRef]
- Trzepizur, W.; Cortese, R.; Gozal, D. Murine Models of Sleep Apnea: Functional Implications of Altered Macrophage Polarity and Epigenetic Modifications in Adipose and Vascular Tissues. Metabolism 2018, 84, 44–55. [Google Scholar] [CrossRef]
- Carreras, A.; Kayali, F.; Zhang, J.; Hirotsu, C.; Wang, Y.; Gozal, D. Metabolic Effects of Intermittent Hypoxia in Mice: Steady versus High-Frequency Applied Hypoxia Daily during the Rest Period. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R700–R709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollicina, I.; Maniaci, A.; Lechien, J.R.; Iannella, G.; Vicini, C.; Cammaroto, G.; Cannavicci, A.; Magliulo, G.; Pace, A.; Cocuzza, S.; et al. Neurocognitive Performance Improvement after Obstructive Sleep Apnea Treatment: State of the Art. Behav. Sci. 2021, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Seda, G.; Han, T.S. Effect of Obstructive Sleep Apnea on Neurocognitive Performance. Sleep Med. Clin. 2020, 15, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Tietjens, J.R.; Claman, D.; Kezirian, E.J.; de Marco, T.; Mirzayan, A.; Sadroonri, B.; Goldberg, A.N.; Long, C.; Gerstenfeld, E.P.; Yeghiazarians, Y. Obstructive Sleep Apnea in Cardiovascular Disease: A Review of the Literature and Proposed Multidisciplinary Clinical Management Strategy. J. Am. Heart Assoc. 2019, 8, e010440. [Google Scholar] [CrossRef] [Green Version]
- Boehme, A.K.; Esenwa, C.; Elkind, M.S.V. Stroke Risk Factors, Genetics, and Prevention. Circ. Res. 2017, 120, 472. [Google Scholar] [CrossRef]
- Juhan-Vague, I.; Pyke, S.D.M.; Alessi, M.C.; Jespersen, J.; Haverkate, F.; Thompson, S.G. Fibrinolytic Factors and the Risk of Myocardial Infarction or Sudden Death in Patients with Angina Pectoris. Circulation 1996, 94, 2057–2063. [Google Scholar] [CrossRef]
- Rijken, D.C.; Lijnen, H.R. New Insights into the Molecular Mechanisms of the Fibrinolytic System. J. Thromb. Haemost. 2009, 7, 4–13. [Google Scholar] [CrossRef]
- Chapin, J.C.; Hajjar, K.A. Fibrinolysis and the Control of Blood Coagulation. Blood Rev. 2015, 29, 17. [Google Scholar] [CrossRef] [Green Version]
- Binder, B.R.; Christ, G.; Gruber, F.; Grubic, N.; Hufnagl, P.; Krebs, M.; Mihaly, J.; Prager, G.W. Plasminogen Activator Inhibitor 1: Physiological and Pathophysiological Roles. News Physiol. Sci. 2002, 17, 56–61. [Google Scholar] [CrossRef]
- Eun, A.L.; Ji, Y.S.; Jiang, Z.; Mi, R.Y.; Min, K.K.; Ha, H.; Hi, B.L. Reactive Oxygen Species Mediate High Glucose–Induced Plasminogen Activator Inhibitor-1 up-Regulation in Mesangial Cells and in Diabetic Kidney. Kidney Int. 2005, 67, 1762–1771. [Google Scholar] [CrossRef] [Green Version]
- Swiatkowska, M.; Szemraj, J.; Cierniewski, C.S. Induction of PAI-1 Expression by Tumor Necrosis Factor Alpha in Endothelial Cells Is Mediated by Its Responsive Element Located in the 4G/5G Site. FEBS J. 2005, 272, 5821–5831. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Inoki, K.; Isono, M.; Mori, H.; Kanasaki, K.; Sugimoto, T.; Akiba, S.; Sato, T.; Yang, B.; Kikkawa, R.; et al. MAPK/AP-1-Dependent Regulation of PAI-1 Gene Expression by TGF-Beta in Rat Mesangial Cells. Kidney Int. 2005, 68, 972–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Burgess, S.; Eicher, J.D.; O’Donnell, C.J.; Johnson, A.D.; Huang, J.; Sabater-Lleal, M.; Asselbergs, F.W.; Tregouet, D.; Shin, S.Y.; et al. Causal Effect of Plasminogen Activator Inhibitor Type 1 on Coronary Heart Disease. J. Am. Heart Assoc. 2017, 6, e004918. [Google Scholar] [CrossRef]
- Placencio, V.R.; DeClerck, Y.A. Plasminogen Activator Inhibitor-1 in Cancer: Rationale and Insight for Future Therapeutic Testing. Cancer Res. 2015, 75, 2969–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altalhi, R.; Pechlivani, N.; Ajjan, R.A. PAI-1 in Diabetes: Pathophysiology and Role as a Therapeutic Target. Int. J. Mol. Sci. 2021, 22, 3170. [Google Scholar] [CrossRef] [PubMed]
- MaŁgorzewicz, S.; Skrzypczak-Jankun, E.; Jankun, J. Plasminogen Activator Inhibitor-1 in Kidney Pathology (Review). Int. J. Mol. Med. 2013, 31, 503–510. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Li, X.; Chen, S.; Lu, N.; Yue, Y.; Liang, J.; Zhang, Z.; Yuan, Y. Plasminogen Activator Inhibitor-1 in Depression: Results from Animal and Clinical Studies. Sci. Rep. 2016, 6, 30464. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. PAI-1 Is a Marker and a Mediator of Senescence. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1446. [Google Scholar] [CrossRef] [Green Version]
- Praetner, M.; Zuchtriegel, G.; Holzer, M.; Uhl, B.; Schaubächer, J.; Mittmann, L.; Fabritius, M.; Fürst, R.; Zahler, S.; Funken, D.; et al. Plasminogen Activator Inhibitor-1 Promotes Neutrophil Infiltration and Tissue Injury on Ischemia-Reperfusion. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 829–842. [Google Scholar] [CrossRef] [Green Version]
- Garcia, V.; Park, E.J.; Siragusa, M.; Frohlich, F.; Haque, M.M.; Pascale, J.V.; Heberlein, K.R.; Isakson, B.E.; Stuehr, D.J.; Sessa, W.C. Unbiased Proteomics Identifies Plasminogen Activator Inhibitor-1 as a Negative Regulator of Endothelial Nitric Oxide Synthase. Proc. Natl. Acad. Sci. USA 2020, 117, 9497–9507. [Google Scholar] [CrossRef]
- Ji, Y.; Weng, Z.; Fish, P.; Goyal, N.; Luo, M.; Myears, S.P.; Strawn, T.L.; Chandrasekar, B.; Wu, J.; Fay, W.P. Pharmacological Targeting of Plasminogen Activator Inhibitor-1 Decreases Vascular Smooth Muscle Cell Migration and Neointima Formation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2167–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Känel, R.; Natarajan, L.; Ancoli-Israel, S.; Mills, P.J.; Loredo, J.S.; Dimsdale, J.E. Day/Night Rhythm of Hemostatic Factors in Obstructive Sleep Apnea. Sleep 2010, 33, 371–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gileles-Hillel, A.; Alonso-Álvarez, M.L.; Kheirandish-Gozal, L.; Peris, E.; Cordero-Guevara, J.A.; Terán-Santos, J.; Martinez, M.G.; Jurado-Luque, M.J.; Corral-Peñafiel, J.; Duran-Cantolla, J.; et al. Inflammatory Markers and Obstructive Sleep Apnea in Obese Children: The NANOS Study. Mediators Inflamm. 2014, e605280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakrzewski, M.; Zakrzewska, E.; Kiciński, P.; Przybylska-Kuć, S.; Dybała, A.; Myśliński, W.; Pastryk, J.; Tomaszewski, T.; Mosiewicz, J. Evaluation of Fibrinolytic Inhibitors: Alpha-2-Antiplasmin and Plasminogen Activator Inhibitor 1 in Patients with Obstructive Sleep Apnoea. PLoS ONE 2016, 11, 166725. [Google Scholar] [CrossRef]
- Nizankowska-Jȩdrzejczyk, A.; Almeida, F.R.; Lowe, A.A.; Kania, A.; Nastałek, P.; Mejza, F.; Foley, J.H.; Nizankowska-Mogilnicka, E.; Undas, A. Modulation of Inflammatory and Hemostatic Markers in Obstructive Sleep Apnea Patients Treated with Mandibular Advancement Splints: A Parallel, Controlled Trial. J. Clin. Sleep Med. 2014, 10, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Bagai, K.; Muldowney, J.A.S.; Song, Y.; Wang, L.; Bagai, J.; Artibee, K.J.; Vaughan, D.E.; Malow, B.A. Circadian Variability of Fibrinolytic Markers and Endothelial Function in Patients with Obstructive Sleep Apnea. Sleep 2014, 37, 359–367. [Google Scholar] [CrossRef]
- von Känel, R.; Loredo, J.S.; Ancoli-Israel, S.; Dimsdale, J.E. Association between Sleep Apnea Severity and Blood Coagulability: Treatment Effects of Nasal Continuous Positive Airway Pressure. Sleep Breath. 2006, 10, 139–146. [Google Scholar] [CrossRef]
- Phillips, C.L.; McEwen, B.J.; Morel-Kopp, M.C.; Yee, B.J.; Sullivan, D.R.; Ward, C.M.; Tofler, G.H.; Grunstein, R.R. Effects of Continuous Positive Airway Pressure on Coagulability in Obstructive Sleep Apnoea: A Randomised, Placebo-Controlled Crossover Study. Thorax 2012, 67, 639–644. [Google Scholar] [CrossRef] [Green Version]
- Rangemark, C.; Hedner, J.A.; Carlson, J.T.; Gleerup, G.; Winther, K. Platelet Function and Fibrinolytic Activity in Hypertensive and Normotensive Sleep Apnea Patients. Sleep 1995, 18, 188–194. [Google Scholar] [CrossRef] [Green Version]
- Kheirandish-Gozal, L.; Gileles-Hillel, A.; Alonso-Álvarez, M.L.; Peris, E.; Bhattacharjee, R.; Terán-Santos, J.; Duran-Cantolla, J.; Gozal, D. Effects of Adenotonsillectomy on Plasma Inflammatory Biomarkers in Obese Children with Obstructive Sleep Apnea: A Community-Based Study. Int. J. Obes. 2015, 39, 1094–1100. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.A.; Strosnider, C.; Giersch, G.; Womack, C.J.; Hargens, T.A. The Effect of Acute Aerobic Exercise on Hemostasis in Obstructive Sleep Apnea. Sleep Breath. 2017, 21, 623–629. [Google Scholar] [CrossRef] [PubMed]
- von Känel, R.; Loredo, J.S.; Ancoli-Israel, S.; Mills, P.J.; Dimsdale, J.E. Elevated Plasminogen Activator Inhibitor 1 in Sleep Apnea and Its Relation to the Metabolic Syndrome: An Investigation in 2 Different Study Samples. Metabolism 2007, 56, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-de-la-Torre, M.; Sánchez-de-la-Torre, A.; Bertran, S.; Abad, J.; Duran-Cantolla, J.; Cabriada, V.; Mediano, O.; Masdeu, M.J.; Alonso, M.L.; Masa, J.F.; et al. Effect of Obstructive Sleep Apnoea and Its Treatment with Continuous Positive Airway Pressure on the Prevalence of Cardiovascular Events in Patients with Acute Coronary Syndrome (ISAACC Study): A Randomised Controlled Trial. Lancet Respir. Med. 2020, 8, 359–367. [Google Scholar] [CrossRef]
- McEvoy, R.D.; Antic, N.A.; Heeley, E.; Luo, Y.; Ou, Q.; Zhang, X.; Mediano, O.; Chen, R.; Drager, L.F.; Liu, Z.; et al. CPAP for Prevention of Cardiovascular Events in Obstructive Sleep Apnea. N. Engl. J. Med. 2016, 375, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.J.; Booth, N.A.; Moore, N.R.; Bennett, B. Distribution of Plasminogen Activator Inhibitor (PAI-1) in Tissues. J. Clin. Pathol. 1991, 44, 139–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crandall, D.L.; Quinet, E.M.; Morgan, G.A.; Busler, D.E.; Mchendry-Rinde, B.; Kral, J.G. Synthesis and Secretion of Plasminogen Activator Inhibitor-1 by Human Preadipocytes. J. Clin. Endocrinol. Metab. 1999, 84, 3222–3227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Seiffert, D.; Fowler, B.J.; Jenkins, G.R.; Thinnes, T.C.; Loskutoff, D.J.; Parmer, R.J.; Miles, L.A. Plasminogen Has a Broad Extrahepatic Distribution. Thromb. Haemost. 2002, 87, 493–501. [Google Scholar] [CrossRef]
- Booth, N.A.; Simpson, A.J.; Croll, A.; Bennett, B.; MacGregor, I.R. Plasminogen Activator Inhibitor (PAI-1) in Plasma and Platelets. Br. J. Haematol. 1988, 70, 327–333. [Google Scholar] [CrossRef]
- Charlton, P. The Status of Plasminogen Activator Inhibitor-1 as a Therapeutic Target. Expert Opin. Investig. Drugs 1997, 6, 539–554. [Google Scholar] [CrossRef]
- Brogren, H.; Wallmark, K.; Deinum, J.; Karlsson, L.; Jern, S. Platelets Retain High Levels of Active Plasminogen Activator Inhibitor 1. PLoS ONE 2011, 6, e26762. [Google Scholar] [CrossRef]
- Torr-Brown, S.R.; Sobel, B.E. Attenuation of Thrombolysis by Release of Plasminogen Activator Inhibitor Type-1 from Platelets. Thromb. Res. 1993, 72, 413–421. [Google Scholar] [CrossRef]
- Morrow, G.B.; Whyte, C.S.; Mutch, N.J. Functional Plasminogen Activator Inhibitor 1 Is Retained on the Activated Platelet Membrane Following Platelet Activation. Haematologica 2020, 105, 2824–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sillen, M.; Declerck, P.J. A Narrative Review on Plasminogen Activator Inhibitor-1 and Its (Patho)Physiological Role: To Target or Not to Target? Int. J. Mol. Sci. 2021, 22, 2721. [Google Scholar] [CrossRef] [PubMed]
- Sillen, M.; Declerck, P.J. Targeting PAI-1 in Cardiovascular Disease: Structural Insights Into PAI-1 Functionality and Inhibition. Front. Cardiovasc. Med. 2020, 7, 364. [Google Scholar] [CrossRef] [PubMed]
- Rahman, F.A.; Krause, M.P. PAI-1, the Plasminogen System, and Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 7066. [Google Scholar] [CrossRef]
- Wind, T.; Hansen, M.; Jensen, J.K.; Andreasen, P.A. The Molecular Basis for Anti-Proteolytic and Non-Proteolytic Functions of Plasminogen Activator Inhibitor Type-1: Roles of the Reactive Centre Loop, the Shutter Region, the Flexible Joint Region and the Small Serpin Fragment. Biol. Chem. 2002, 383, 21–36. [Google Scholar] [CrossRef]
- Schroeck, F.; Arroyo de Prada, N.; Sperl, S.; Schmitt, M.; Magdolen, V. Interaction of Plasminogen Activator Inhibitor Type-1 (PAI-1) with Vitronectin (Vn): Mapping the Binding Sites on PAI-1 and Vn. Biol. Chem. 2002, 383, 1143–1149. [Google Scholar] [CrossRef]
- Wilczynska, M.; Fa, M.; Ohlsson, P.I.; Ny, T. The Inhibition Mechanism of Serpins. Evidence That the Mobile Reactive Center Loop Is Cleaved in the Native Protease-Inhibitor Complex. J. Biol. Chem. 1995, 270, 29652–29655. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, D.A.; Ginsburg, D.; Day, D.E.; Berkenpas, M.B.; Verhamme, I.M.; Kvassman, J.O.; Shore, J.D. Serpin-Protease Complexes Are Trapped as Stable Acyl-Enzyme Intermediates. J. Biol. Chem. 1995, 270, 25309–25312. [Google Scholar] [CrossRef] [Green Version]
- Boudier, C.; Gils, A.; Declerck, P.J.; Bieth, J.G. The Conversion of Active to Latent Plasminogen Activator Inhibitor-1 Is an Energetically Silent Event. Biophys. J. 2005, 88, 2848–2854. [Google Scholar] [CrossRef] [Green Version]
- Gettins, P.G.W.; Olson, S.T. Inhibitory Serpins. New Insights into Their Folding, Polymerization, Regulation and Clearance. Biochem. J. 2016, 473, 2273–2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simone, T.M.; Higgins, P.J. Low Molecular Weight Antagonists of Plasminogen Activator Inhibitor-1: Therapeutic Potential in Cardiovascular Disease. Mol. Med. Ther. 2012, 1, 101. [Google Scholar] [CrossRef] [PubMed]
- Dupont, D.M.; Madsen, J.B.; Kristensen, T.; Bodker, J.S.; Blouse, G.E.; Wind, T.; Andreasen, P.A. Biochemical Properties of Plasminogen Activator Inhibitor-1. Front. Biosci. 2009, 14, 1337–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fjellström, O.; Deinum, J.; Sjögren, T.; Johansson, C.; Geschwindner, S.; Nerme, V.; Legnehed, A.; McPheat, J.; Olsson, K.; Bodin, C.; et al. Characterization of a Small Molecule Inhibitor of Plasminogen Activator Inhibitor Type 1 That Accelerates the Transition into the Latent Conformation. J. Biol. Chem. 2013, 288, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, D.A.; Ginsburg, D.; Olson, S.T.; Palaniappan, S. Engineering Plasminogen Activator Inhibitor 1 Mutants with Increased Functional Stability. Biochemistry 1994, 33, 3643–3648. [Google Scholar] [CrossRef]
- Van De Craen, B.; Declerck, P.J.; Gils, A. The Biochemistry, Physiology and Pathological Roles of PAI-1 and the Requirements for PAI-1 Inhibition in Vivo. Thromb. Res. 2012, 130, 576–585. [Google Scholar] [CrossRef]
- Thorsen, S.; Philips, M.; Selmer, J.; Lecander, I.; Åstedt, B. Kinetics of Inhibition of Tissue-Type and Urokinase-Type Plasminogen Activator by Plasminogen-Activator Inhibitor Type 1 and Type 2. Eur. J. Biochem. 1988, 175, 33–39. [Google Scholar] [CrossRef]
- Lee, E.; Vaughan, D.E.; Parikh, S.H.; Grodzinsky, A.J.; Libby, P.; Lark, M.W.; Lee, R.T. Regulation of Matrix Metalloproteinases and Plasminogen Activator Inhibitor-1 Synthesis by Plasminogen in Cultured Human Vascular Smooth Muscle Cells. Circ. Res. 1996, 78, 44–49. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [Green Version]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef] [PubMed]
- Oszajca, K.; Bieniasz, M.; Brown, G.; Swiatkowska, M.; Bartkowiak, J.; Szemraj, J. Effect of Oxidative Stress on the Expression of T-PA, u-PA, u-PAR, and PAI-1 in Endothelial Cells. Biochem. Cell Biol. 2008, 86, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Swiatkowska, M.; Szemraj, J.; Al-Nedawi, K.; Pawlowska, Z. Reactive Oxygen Species Upregulate Expression of PAI-1 in Endothelial Cells. Cell. Mol. Biol. Lett. 2002, 7, 1065–1071. [Google Scholar] [PubMed]
- Jaulmes, A.; Sansilvestri-Morel, P.; Rolland-Valognes, G.; Bernhardt, F.; Gaertner, R.; Lockhart, B.P.; Cordi, A.; Wierzbicki, M.; Rupin, A.; Verbeuren, T.J. Nox4 Mediates the Expression of Plasminogen Activator Inhibitor-1 via P38 MAPK Pathway in Cultured Human Endothelial Cells. Thromb. Res. 2009, 124, 439–446. [Google Scholar] [CrossRef]
- Orbe, J.; Rodriguez, J.A.; Calvo, A.; Grau, A.; Belzunce, M.S.; Martinez-Caro, D.; Páramo, J.A. Vitamins C and E Attenuate Plasminogen Activator Inhibitor-1 (PAI-1) Expression in a Hypercholesterolemic Porcine Model of Angioplasty. Cardiovasc. Res. 2001, 49, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Gomaa, A.M.S.; Abd El-Mottaleb, N.A.; Aamer, H.A. Antioxidant and Anti-Inflammatory Activities of Alpha Lipoic Acid Protect against Indomethacin-Induced Gastric Ulcer in Rats. Biomed. Pharmacother. 2018, 101, 188–194. [Google Scholar] [CrossRef]
- Martina, V.; Bruno, G.A.; Pannocchia, A.; Zumpano, E.; Tagliabue, M.; Trucco, F.; Giorgianni, A.; Stella, S.; Pescarmona, G.P. PAI-1 Reduction after Treatment with Glutathione in NIDDM. Fibrinolysis 1996, 10, 63–65. [Google Scholar] [CrossRef]
- Bonfigli, A.R.; Pieri, C.; Manfrini, S.; Testa, I.; Sirolla, C.; Ricciotti, R.; Marra, M.; Compagnucci, P.; Testa, R. Vitamin E Intake Reduces Plasminogen Activator Inhibitor Type 1 in T2DM Patients. Diabetes Nutr. Metab. 2001, 14, 71–77. [Google Scholar]
- Antoniades, C.; Tousoulis, D.; Tentolouris, C.; Toutouza, M.; Marinou, K.; Goumas, G.; Tsioufis, C.; Toutouzas, P.; Stefanadis, C. Effects of Antioxidant Vitamins C and E on Endothelial Function and Thrombosis/Fibrinolysis System in Smokers. Thromb. Haemost. 2003, 89, 990–995. [Google Scholar] [CrossRef]
- Zhao, R.; Ma, X.; Xie, X.; Shen, G.X. Involvement of NADPH Oxidase in Oxidized LDL-Induced Upregulation of Heat Shock Factor-1 and Plasminogen Activator Inhibitor-1 in Vascular Endothelial Cells. Am. J. Physiol. Endocrinol. Metab. 2009, 297, 104–111. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, H.; Seki, T.; Ariga, T. The Effect of Pre-Germinated Brown Rice Intake on Blood Glucose and PAI-1 Levels in Streptozotocin-Induced Diabetic Rats. Biosci. Biotechnol. Biochem. 2004, 68, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Diebold, I.; Schini-Kerth, V.B.; Berchner-Pfannschmidt, U.; Roth, U.; Brandes, R.P.; Kietzmann, T.; Busse, R. Thrombin Activates the Hypoxia-Inducible Factor-1 Signaling Pathway in Vascular Smooth Muscle Cells. Circ. Res. 2001, 89, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, S.; Shen, G.X. Impact of Antioxidants and HDL on Glycated LDL–Induced Generation of Fibrinolytic Regulators from Vascular Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1688–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, I.S.; Kim, J.; Rhee, D.K.; Kim, B.O.; Pyo, S. Pneumolysin Induces Cellular Senescence by Increasing ROS Production and Activation of MAPK/NF-ΚB Signal Pathway in Glial Cells. Toxicon 2017, 129, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Vayalil, P.K.; Iles, K.E.; Choi, J.; Yi, A.K.; Postlethwait, E.M.; Liu, R.M. Glutathione Suppresses TGF-Beta-Induced PAI-1 Expression by Inhibiting P38 and JNK MAPK and the Binding of AP-1, SP-1, and Smad to the PAI-1 Promoter. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1281–L1292. [Google Scholar] [CrossRef] [Green Version]
- Dimova, E.Y.; Kietzmann, T. Metabolic, Hormonal and Environmental Regulation of Plasminogen Activator Inhibitor-1 (PAI-1) Expression: Lessons from the Liver. Thromb. Haemost. 2008, 100, 992–1006. [Google Scholar] [CrossRef]
- Libby, P. Inflammation and Cardiovascular Disease Mechanisms. Am. J. Clin. Nutr. 2006, 83, 456S–460S. [Google Scholar] [CrossRef] [Green Version]
- Ruparelia, N.; Chai, J.T.; Fisher, E.A.; Choudhury, R.P. Inflammatory Processes in Cardiovascular Disease: A Route to Targeted Therapies. Nat. Rev. Cardiol. 2016, 14, 133–144. [Google Scholar] [CrossRef]
- Alfaddagh, A.; Martin, S.S.; Leucker, T.M.; Michos, E.D.; Blaha, M.J.; Lowenstein, C.J.; Jones, S.R.; Toth, P.P. Inflammation and Cardiovascular Disease: From Mechanisms to Therapeutics. Am. J. Prev. Cardiol. 2020, 4, 100130. [Google Scholar] [CrossRef]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Jimenez, M.T.B.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxid. Med. Cell. Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef] [Green Version]
- Cesari, M.; Pahor, M.; Incalzi, R.A. REVIEW: Plasminogen Activator Inhibitor-1 (PAI-1): A Key Factor Linking Fibrinolysis and Age-Related Subclinical and Clinical Conditions. Cardiovasc. Ther. 2010, 28, e72–e91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hube, F.; Hauner, H. The Role of TNF-Alpha in Human Adipose Tissue: Prevention of Weight Gain at the Expense of Insulin Resistance? Horm. Metab. Res. 1999, 31, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, Y.; Takamura, T.; Hamaguchi, E.; Shimizu, A.; Ota, T.; Sakurai, M.; Kaneko, S. Tumor Necrosis Factor-Alpha-Induced Production of Plasminogen Activator Inhibitor 1 and Its Regulation by Pioglitazone and Cerivastatin in a Nonmalignant Human Hepatocyte Cell Line. Metabolism 2006, 55, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, M.; Loskutoff, D.J.; Samad, F. Molecular Mechanisms of Tumor Necrosis Factor-Alpha-Mediated Plasminogen Activator Inhibitor-1 Expression in Adipocytes. FASEB J. 2005, 19, 1317–1319. [Google Scholar] [CrossRef] [Green Version]
- Macfelda, K.; Weiss, T.W.; Kaun, C.; Breuss, J.M.; Kapeller, B.; Zorn, G.; Oberndorfer, U.; Voegele-Kadletz, M.; Huber-Beckmann, R.; Ullrich, R.; et al. Plasminogen Activator Inhibitor 1 Expression Is Regulated by the Inflammatory Mediators Interleukin-1alpha, Tumor Necrosis Factor-Alpha, Transforming Growth Factor-Beta and Oncostatin M in Human Cardiac Myocytes. J. Mol. Cell. Cardiol. 2002, 34, 1681–1691. [Google Scholar] [CrossRef]
- Hou, B.; Eren, M.; Painter, C.A.; Covington, J.W.; Dixon, J.D.; Schoenhard, J.A.; Vaughan, D.E. Tumor Necrosis Factor Alpha Activates the Human Plasminogen Activator Inhibitor-1 Gene through a Distal Nuclear Factor KappaB Site. J. Biol. Chem. 2004, 279, 18127–18136. [Google Scholar] [CrossRef] [Green Version]
- Samad, F.; Yamamoto, K.; Loskutoff, D.J. Distribution and Regulation of Plasminogen Activator Inhibitor-1 in Murine Adipose Tissue In Vivo. Induction by Tumor Necrosis Factor-Alpha and Lipopolysaccharide. J. Clin. Investig. 1996, 97, 37–46. [Google Scholar] [CrossRef]
- Cigolini, M.; Tonoli, M.; Borgato, L.; Frigotto, L.; Manzato, F.; Zeminian, S.; Cardinale, C.; Camin, M.; Chiaramonte, E.; De Sandre, G.; et al. Expression of Plasminogen Activator Inhibitor-1 in Human Adipose Tissue: A Role for TNF-Alpha? Atherosclerosis 1999, 143, 81–90. [Google Scholar] [CrossRef]
- Papanicolaou, D.A.; Wilder, R.L.; Manolagas, S.C.; Chrousos, G.P. The Pathophysiologic Roles of Interleukin-6 in Human Disease. Ann. Intern. Med. 1998, 128, 127–137. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Inoue, H.; Ono, C.; Hashimoto, S.; Kioi, Y.; Matsumoto, H.; Matsuura, H.; Matsubara, T.; Shimizu, K.; et al. IL-6 Trans-Signaling Induces Plasminogen Activator Inhibitor-1 from Vascular Endothelial Cells in Cytokine Release Syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 22351–22356. [Google Scholar] [CrossRef]
- Mestries, J.C.; Kruithof, E.K.O.; Gascon, M.P.; Herodin, F.; Agay, D.; Ythier, A. In Vivo Modulation of Coagulation and Fibrinolysis by Recombinant Glycosylated Human Interleukin-6 in Baboons. Eur. Cytokine Netw. 1994, 5, 275–281. [Google Scholar] [PubMed]
- Kruithof, E.K.O. Regulation of Plasminogen Activator Inhibitor Type 1 Gene Expression by Inflammatory Mediators and Statins. Thromb. Haemost. 2008, 100, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Toma, I.; McCaffrey, T.A. Transforming Growth Factor-β and Atherosclerosis: Interwoven Atherogenic and Atheroprotective Aspects. Cell Tissue Res. 2012, 347, 155. [Google Scholar] [CrossRef] [Green Version]
- Verrecchia, F.; Mauviel, A. Transforming Growth Factor-β and Fibrosis. World J. Gastroenterol. 2007, 13, 3056. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Li, G.; Wang, Z.; Dahlman, J.E.; Malagon-Lopez, J.; Gujja, S.; Cilfone, N.A.; Kauffman, K.J.; Sun, L.; et al. Endothelial TGF-β Signalling Drives Vascular Inflammation and Atherosclerosis. Nat. Metab. 2019, 1, 912–926. [Google Scholar] [CrossRef] [PubMed]
- Seeland, U.; Haeuseler, C.; Hinrichs, R.; Rosenkranz, S.; Pfitzner, T.; Scharffetter-Kochanek, K.; Böhm, M. Myocardial Fibrosis in Transforming Growth Factor-Beta(1) (TGF-Beta(1)) Transgenic Mice Is Associated with Inhibition of Interstitial Collagenase. Eur. J. Clin. Investig. 2002, 32, 295–303. [Google Scholar] [CrossRef]
- Grandaliano, G.; Di Paolo, S.; Monno, R.; Stallone, G.; Ranieri, E.; Pontrelli, P.; Gesualdo, L.; Schena, F.P. Protease-Activated Receptor 1 and Plasminogen Activator Inhibitor 1 Expression in Chronic Allograft Nephropathy: The Role of Coagulation and Fibrinolysis in Renal Graft Fibrosis. Transplantation 2001, 72, 1437–1443. [Google Scholar] [CrossRef]
- De Gouville, A.C.; Boullay, V.; Krysa, G.; Pilot, J.; Brusq, J.M.; Loriolle, F.; Gauthier, J.M.; Papworth, S.A.; Laroze, A.; Gellibert, F.; et al. Inhibition of TGF-Beta Signaling by an ALK5 Inhibitor Protects Rats from Dimethylnitrosamine-Induced Liver Fibrosis. Br. J. Pharmacol. 2005, 145, 166–177. [Google Scholar] [CrossRef] [Green Version]
- Kutz, S.M.; Hordines, J.; McKeown-Longo, P.J.; Higgins, P.J. TGF-Beta1-Induced PAI-1 Gene Expression Requires MEK Activity and Cell-to-Substrate Adhesion. J. Cell Sci. 2001, 114, 3905–3914. [Google Scholar] [CrossRef]
- Hirashima, Y.; Kobayashi, H.; Suzuki, M.; Tanaka, Y.; Kanayama, N.; Terao, T. Transforming Growth Factor-Beta1 Produced by Ovarian Cancer Cell Line HRA Stimulates Attachment and Invasion through an up-Regulation of Plasminogen Activator Inhibitor Type-1 in Human Peritoneal Mesothelial Cells. J. Biol. Chem. 2003, 278, 26793–26802. [Google Scholar] [CrossRef] [Green Version]
- Datta, P.K.; Blake, M.C.; Moses, H.L. Regulation of Plasminogen Activator Inhibitor-1 Expression by Transforming Growth Factor-Beta -Induced Physical and Functional Interactions between Smads and Sp1. J. Biol. Chem. 2000, 275, 40014–40019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, L.R.; Riccio, A.; Andreasen, P.A.; Nielsen, L.S.; Kristensen, P.; Laiho, M.; Saksela, O.; Blasi, F.; Danø, K. Transforming Growth Factor-Beta Is a Strong and Fast Acting Positive Regulator of the Level of Type-1 Plasminogen Activator Inhibitor MRNA in WI-38 Human Lung Fibroblasts. EMBO J. 1987, 6, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Jaffer, O.A.; Carter, A.B.; Sanders, P.N.; Dibbern, M.E.; Winters, C.J.; Murthy, S.; Ryan, A.J.; Rokita, A.G.; Prasad, A.M.; Zabner, J.; et al. Mitochondrial-Targeted Antioxidant Therapy Decreases Transforming Growth Factor-β-Mediated Collagen Production in a Murine Asthma Model. Am. J. Respir. Cell Mol. Biol. 2015, 52, 106–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, W.; Hong, Y.; He, H.; Huang, X.; Tao, W.; Liang, X.; Zhang, Y.; Li, X. TGF-β Mediates Aortic Smooth Muscle Cell Senescence in Marfan Syndrome. Aging 2019, 11, 3574–3584. [Google Scholar] [CrossRef]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial Reactive Oxygen Species Regulate Transforming Growth Factor-β Signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [Green Version]
- García-Trevijano, E.R.; Iraburu, M.J.; Fontana, L.; Domínguez-Rosales, J.A.; Auster, A.; Covarrubias-Pinedo, A.; Rojkind, M. Transforming Growth Factor Beta1 Induces the Expression of Alpha1(I) Procollagen MRNA by a Hydrogen Peroxide-C/EBPbeta-Dependent Mechanism in Rat Hepatic Stellate Cells. Hepatology 1999, 29, 960–970. [Google Scholar] [CrossRef]
- Herrera, B.; Murillo, M.M.; Álvarez-Barrientos, A.; Beltrán, J.; Fernández, M.; Fabregat, I. Source of Early Reactive Oxygen Species in the Apoptosis Induced by Transforming Growth Factor-Beta in Fetal Rat Hepatocytes. Free Radic. Biol. Med. 2004, 36, 16–26. [Google Scholar] [CrossRef]
- Franklin, C.C.; Rosenfeld-Franklin, M.E.; White, C.; Kavanagh, T.J.; Fausto, N. TGFbeta1-Induced Suppression of Glutathione Antioxidant Defenses in Hepatocytes: Caspase-Dependent Post-Translational and Caspase-Independent Transcriptional Regulatory Mechanisms. FASEB J. 2003, 17, 1535–1537. [Google Scholar] [CrossRef]
- Samarakoon, R.; Chitnis, S.S.; Higgins, S.P.; Higgins, C.E.; Krepinsky, J.C.; Higgins, P.J. Redox-Induced Src Kinase and Caveolin-1 Signaling in TGF-Β1-Initiated SMAD2/3 Activation and PAI-1 Expression. PLoS ONE 2011, 6, e22896. [Google Scholar] [CrossRef]
- Furukawa, F.; Matsuzaki, K.; Mori, S.; Tahashi, Y.; Yoshida, K.; Sugano, Y.; Yamagata, H.; Matsushita, M.; Seki, T.; Inagaki, Y.; et al. P38 MAPK Mediates Fibrogenic Signal through Smad3 Phosphorylation in Rat Myofibroblasts. Hepatology 2003, 38, 879–889. [Google Scholar] [CrossRef]
- Woodward, R.N.; Finn, A.V.; Dichek, D.A. Identification of Intracellular Pathways through Which TGF-Beta1 Upregulates PAI-1 Expression in Endothelial Cells. Atherosclerosis 2006, 186, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Q.; Su, M.; Walia, R.R.; Hao, Q.; Covington, J.W.; Vaughan, D.E. Sp1 Sites Mediate Activation of the Plasminogen Activator Inhibitor-1 Promoter by Glucose in Vascular Smooth Muscle Cells. J. Biol. Chem. 1998, 273, 8225–8231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsch, E.; Sluimer, J.C.; Daemen, M.J.A.P. Hypoxia in Atherosclerosis and Inflammation. Curr. Opin. Lipidol. 2013, 24, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Rey, S.; Semenza, G.L. Hypoxia-Inducible Factor-1-Dependent Mechanisms of Vascularization and Vascular Remodelling. Cardiovasc. Res. 2010, 86, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Chen, Q.; Zhou, X.; Fan, L. The Role of Hypoxia-Inducible Factor 1 in Atherosclerosis. J. Clin. Pathol. 2012, 65, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-Inducible Factor 1 Is a Basic-Helix-Loop-Helix-PAS Heterodimer Regulated by Cellular O2 Tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Kuo, I.H.; Chang, C.C.; Chu, C.Y.; Chen, H.Y.; Lin, B.R.; Sureshbabu, M.; Shih, H.J.; Kuo, M.L. Involvement of Hypoxia-Inducing Factor-1α-Dependent Plasminogen Activator Inhibitor-1 up-Regulation in Cyr61/CCN1-Induced Gastric Cancer Cell Invasion. J. Biol. Chem. 2016, 291, 27433. [Google Scholar] [CrossRef] [Green Version]
- Sanagawa, A.; Iwaki, S.; Asai, M.; Sakakibara, D.; Norimoto, H.; Sobel, B.E.; Fujii, S. Sphingosine 1-phosphate Induced by Hypoxia Increases the Expression of PAI-1 in HepG2 Cells via HIF-1α. Mol. Med. Rep. 2016, 14, 1841–1848. [Google Scholar] [CrossRef]
- Kabei, K.; Tateishi, Y.; Nozaki, M.; Tanaka, M.; Shiota, M.; Osada-Oka, M.; Nishide, S.; Uchida, J.; Nakatani, T.; Tomita, S.; et al. Role of Hypoxia-Inducible Factor-1 in the Development of Renal Fibrosis in Mouse Obstructed Kidney: Special References to HIF-1 Dependent Gene Expression of Profibrogenic Molecules. J. Pharmacol. Sci. 2018, 136, 31–38. [Google Scholar] [CrossRef]
- Uchiyama, T.; Kurabayashi, M.; Ohyama, Y.; Utsugi, T.; Akuzawa, N.; Sato, M.; Tomono, S.; Kawazu, S.; Nagai, R. Hypoxia Induces Transcription of the Plasminogen Activator Inhibitor-1 Gene through Genistein-Sensitive Tyrosine Kinase Pathways in Vascular Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1155–1161. [Google Scholar] [CrossRef] [Green Version]
- Kimura, D.; Imaizumi, T.; Tamo, W.; Sakai, T.; Ito, K.; Hatanaka, R.; Yoshida, H.; Tsushima, T.; Satoh, K.; Fukuda, I. Hypoxia Enhances the Expression of Plasminogen Activator Inhibitor-1 in Human Lung Cancer Cells, EBC-1. Tohoku J. Exp. Med. 2002, 196, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toullec, A.; Buard, V.; Rannou, E.; Tarlet, G.; Guipaud, O.; Robine, S.; Iruela-Arispe, M.L.; François, A.; Milliat, F. HIF-1α Deletion in the Endothelium, but Not in the Epithelium, Protects from Radiation-Induced Enteritis. Cell. Mol. Gastroenterol. Hepatol. 2017, 5, 15–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petry, A.; Belaiba, R.S.; Weitnauer, M.; Görlach, A. Inhibition of Endothelial Nitric Oxyde Synthase Increases Capillary Formation via Rac1-Dependent Induction of Hypoxia-Inducible Factor-1α and Plasminogen Activator Inhibitor-1. Thromb. Haemost. 2012, 108, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Berchner-Pfannschmidt, U.; Wotzlaw, C.; Cool, R.H.; Fandrey, J.; Acker, H.; Jungermann, K.; Kietzmann, T. Reactive Oxygen Species Modulate HIF-I Mediated PAI-I Expression: Involvement of the GTPase RacI. Thromb. Haemost. 2003, 89, 926–935. [Google Scholar] [CrossRef]
- Bonello, S.; Zähringer, C.; BelAiba, R.S.; Djordjevic, T.; Hess, J.; Michiels, C.; Kietzmann, T.; Görlach, A. Reactive Oxygen Species Activate the HIF-1α Promoter via a Functional NFκB Site. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 755–761. [Google Scholar] [CrossRef] [Green Version]
- Diebold, I.; Djordjevic, T.; Hess, J.; Görlach, A. Rac-1 Promotes Pulmonary Artery Smooth Muscle Cell Proliferation by Upregulation of Plasminogen Activator Inhibitor-1: Role of NFkappaB-Dependent Hypoxia-Inducible Factor-1alpha Transcription. Thromb. Haemost. 2008, 100, 1021–1028. [Google Scholar] [CrossRef]
- An, W.G.; Ahn, Y.-T.; Chua, M.-S.; Whitlock, J.P.; Shin, Y.-C.; Song, W.-H.; Kim, Y.; Eom, C.-Y. Rodent-Specific Hypoxia Response Elements Enhance PAI-1 Expression through HIF-1 or HIF-2 in Mouse Hepatoma Cells. Int. J. Oncol. 2010, 37, 1627–1638. [Google Scholar] [CrossRef]
- Liao, H.; Hyman, M.C.; Lawrence, D.A.; Pinsky, D.J. Molecular Regulation of the PAI-1 Gene by Hypoxia: Contributions of Egr-1, HIF-1alpha, and C/EBPalpha. FASEB J. 2007, 21, 935–949. [Google Scholar] [CrossRef]
- Anfosso, F.; Chomiki, N.; Alessi, M.C.; Vague, P.; Juhan-Vague, I. Plasminogen Activator Inhibitor-1 Synthesis in the Human Hepatoma Cell Line Hep G2 Metformin Inhibits the Stimulating Effect of Insulin. J. Clin. Investig. 1993, 91, 2185–2193. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.J.; Sobel, B.E. Augmentation of Synthesis of Plasminogen Activator Inhibitor Type 1 by Insulin and Insulin-like Growth Factor Type I: Implications for Vascular Disease in Hyperinsulinemic States. Proc. Natl. Acad. Sci. USA 1991, 88, 9959–9963. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.J.; Absher, P.M.; Ricci, M.A. Dependence of Augmentation of Arterial Endothelial Cell Expression of Plasminogen Activator Inhibitor Type 1 by Insulin on Soluble Factors Released from Vascular Smooth Muscle Cells. Circulation 1997, 96, 2868–2876. [Google Scholar] [CrossRef] [PubMed]
- Montagnani, M.; Golovchenko, I.; Kim, I.; Koh, G.Y.; Goalstone, M.L.; Mundhekar, A.N.; Johansen, M.; Kucik, D.F.; Quon, M.J.; Draznin, B. Inhibition of Phosphatidylinositol 3-Kinase Enhances Mitogenic Actions of Insulin in Endothelial Cells. J. Biol. Chem. 2002, 277, 1794–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusi, K.; Maezono, K.; Osman, A.; Pendergrass, M.; Patti, M.E.; Pratipanawatr, T.; DeFronzo, R.A.; Kahn, C.R.; Mandarino, L.J. Insulin Resistance Differentially Affects the PI 3-Kinase- and MAP Kinase-Mediated Signaling in Human Muscle. J. Clin. Investig. 2000, 105, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessi, M.C.; Juhan-Vague, I. PAI-1 and the Metabolic Syndrome: Links, Causes, and Consequences. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2200–2207. [Google Scholar] [CrossRef]
- Shimomura, I.; Funahashi, T.; Takahashi, M.; Maeda, K.; Kotani, K.; Nakamura, T.; Yamashita, S.; Miura, M.; Fukuda, Y.; Takemura, K.; et al. Enhanced Expression of PAI-1 in Visceral Fat: Possible Contributor to Vascular Disease in Obesity. Nat. Med. 1996, 2, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Bouarab, C.; Roullot-Lacarrière, V.; Vallée, M.; Le Roux, A.; Guette, C.; Mennesson, M.; Marighetto, A.; Desmedt, A.; Piazza, P.V.; Revest, J.M. PAI-1 Protein Is a Key Molecular Effector in the Transition from Normal to PTSD-like Fear Memory. Mol. Psychiatry 2021, 26, 4968–4981. [Google Scholar] [CrossRef]
- Vaughan, D.E.; Lazos, S.A.; Tong, K. Angiotensin II Regulates the Expression of Plasminogen Activator Inhibitor-1 in Cultured Endothelial Cells. A Potential Link between the Renin-Angiotensin System and Thrombosis. J. Clin. Investig. 1995, 95, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Rüster, C.; Wolf, G. Angiotensin II as a Morphogenic Cytokine Stimulating Renal Fibrogenesis. J. Am. Soc. Nephrol. 2011, 22, 1189–1199. [Google Scholar] [CrossRef] [Green Version]
- Fogari, R.; Zoppi, A.; Mugellini, A.; Maffioli, P.; Lazzari, P.; Derosa, G. Role of Angiotensin II in Plasma PAI-1 Changes Induced by Imidapril or Candesartan in Hypertensive Patients with Metabolic Syndrome. Hypertens. Res. 2011, 34, 1321–1326. [Google Scholar] [CrossRef]
- Skurk, T.; Lee, Y.M.; Hauner, H. Angiotensin II and Its Metabolites Stimulate PAI-1 Protein Release from Human Adipocytes in Primary Culture. Hypertension 2001, 37, 1336–1340. [Google Scholar] [CrossRef] [Green Version]
- Fay, W.P.; Parker, A.C.; Condrey, L.R.; Shapiro, A.D. Human Plasminogen Activator Inhibitor-1 (PAI-1) Deficiency: Characterization of a Large Kindred with a Null Mutation in the PAI-1 Gene. Blood 1997, 90, 204–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.H.; Vosburgh, E.; Anderson, K.; McDonagh, J. Deficiency of Plasma Plasminogen Activator Inhibitor 1 Results in Hyperfibrinolytic Bleeding. Blood 1993, 81, 2357–2362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleef, R.; Higgins, D.L.; Pillemer, E.; Levitt, L.J. Bleeding Diathesis Due to Decreased Functional Activity of Type 1 Plasminogen Activator Inhibitor. J. Clin. Investig. 1989, 83, 1747–1752. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, T.; Nagahashi, K.; Kobayashi, T.; Umemura, K.; Terao, T.; Kanayama, N. The First Report of Uncontrollable Subchorionic and Retroplacental Haemorrhage Inducing Preterm Labour in Complete PAI-1 Deficiency in a Human. Thromb. Res. 2012, 129, e161–e163. [Google Scholar] [CrossRef]
- Mehta, R.; Shapiro, A.D. Plasminogen Activator Inhibitor Type 1 Deficiency. Haemophilia 2008, 14, 1255–1260. [Google Scholar] [CrossRef]
- Kathiresan, S.; Gabriel, S.B.; Yang, Q.; Lochner, A.L.; Larson, M.G.; Levy, D.; Tofler, G.H.; Hirschhorn, J.N.; O’Donnell, C.J. Comprehensive Survey of Common Genetic Variation at the Plasminogen Activator Inhibitor-1 Locus and Relations to Circulating Plasminogen Activator Inhibitor-1 Levels. Circulation 2005, 112, 1728–1735. [Google Scholar] [CrossRef] [Green Version]
- Dawson, S.; Hamsten, A.; Wiman, B.; Henney, A.; Humphries, S. Genetic Variation at the Plasminogen Activator Inhibitor-1 Locus Is Associated with Altered Levels of Plasma Plasminogen Activator Inhibitor-1 Activity. Arterioscler. Thromb. A J. Vasc. Biol. 1991, 11, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Grubic, N.; Stegnar, M.; Peternel, P.; Kaider, A.; Binder, B.R. A Novel G/A and the 4G/5G Polymorphism within the Promoter of the Plasminogen Activator Inhibitor-1 Gene in Patients with Deep Vein Thrombosis. Thromb. Res. 1996, 84, 431–443. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, J.; Guo, X.; Mo, J.; Gao, B.; Zhou, H.; Wu, Y.; Li, Z. The Roles of PAI-1 Gene Polymorphisms in Atherosclerotic Diseases: A Systematic Review and Meta-Analysis Involving 149,908 Subjects. Gene 2018, 673, 167–173. [Google Scholar] [CrossRef]
- Huang, G.; Wang, P.; Li, T.; Deng, X. Genetic Association between Plasminogen Activator Inhibitor-1 Rs1799889 Polymorphism and Venous Thromboembolism: Evidence from a Comprehensive Meta-Analysis. Clin. Cardiol. 2019, 42, 1232–1238. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Jiang, W.; Ouyang, M.; Yang, K. PAI-1 4G/5G Polymorphism and Coronary Artery Disease Risk: A Meta-Analysis. Int. J. Clin. Exp. Med. 2015, 8, 2097. [Google Scholar] [PubMed]
- Tsantes, A.E.; Nikolopoulos, G.K.; Bagos, P.G.; Tsiara, C.G.; Kapsimali, V.; Travlou, A.; Vaiopoulos, G. Plasminogen Activator Inhibitor-1 4G/5G Polymorphism and Risk of Ischemic Stroke: A Meta-Analysis. Blood Coagul. Fibrinolysis 2007, 18, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Garciá-González, I.J.; Valle, Y.; Sandoval-Pinto, E.; Valdés-Alvarado, E.; Valdez-Haro, A.; Francisco Munõz-Valle, J.; Flores-Salinas, H.E.; Figuera-Villanueva, L.E.; Dávalos-Rodríguez, N.O.; Padilla-Gutiérrez, J.R. The -844 G>A PAI-1 Polymorphism Is Associated with Acute Coronary Syndrome in Mexican Population. Dis. Markers 2015, 460974. [Google Scholar] [CrossRef] [PubMed]
- Saidi, S.; Slamia, L.B.; Mahjoub, T.; Ammou, S.B.; Almawi, W.Y. Association of PAI-1 4G/5G and -844G/A Gene Polymorphism and Changes in PAI-1/TPA Levels in Stroke: A Case-Control Study. J. Stroke Cerebrovasc. Dis. 2007, 16, 153–159. [Google Scholar] [CrossRef]
- Abboud, N.; Ghazouani, L.; Saidi, S.; Ben-Hadj-Khalifa, S.; Addad, F.; Almawi, W.Y.; Mahjoub, T. Association of PAI-1 4G/5G and -844G/A Gene Polymorphisms and Changes in PAI-1/Tissue Plasminogen Activator Levels in Myocardial Infarction: A Case-Control Study. Genet. Test. Mol. Biomarkers 2010, 14, 23–27. [Google Scholar] [CrossRef]
- Kollabathula, A.; Sharma, S.; Kumar, N.; Ahluwalia, J.; Das, R.; Varma, N.; Rana, S.S. Plasminogen Activator Inhibitor-1 4G/5G Promoter Polymorphism in Adults with Splanchnic Vein Thrombosis: A Case-Control Study. Indian J. Hematol. Blood Transfus. 2022, 38, 169–172. [Google Scholar] [CrossRef]
- Frischmuth, T.; Hindberg, K.; Aukrust, P.; Ueland, T.; Brækkan, S.K.; Hansen, J.; Morelli, V.M. Elevated Plasma Levels of Plasminogen Activator Inhibitor-1 Are Associated with Risk of Future Incident Venous Thromboembolism. J. Thromb. Haemost. 2022. [Google Scholar] [CrossRef]
- Tofler, G.H.; Massaro, J.; O’Donnell, C.J.; Wilson, P.W.F.; Vasan, R.S.; Sutherland, P.A.; Meigs, J.B.; Levy, D.; D’Agostino, R.B. Plasminogen Activator Inhibitor and the Risk of Cardiovascular Disease: The Framingham Heart Study. Thromb. Res. 2016, 140, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Meltzer, M.E.; Lisman, T.; De Groot, P.G.; Meijers, J.C.M.; Le Cessie, S.; Doggen, C.J.M.; Rosendaal, F.R. Venous Thrombosis Risk Associated with Plasma Hypofibrinolysis Is Explained by Elevated Plasma Levels of TAFI and PAI-1. Blood 2010, 116, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Schneiderman, J.; Sawdey, M.S.; Keeton, M.R.; Bordin, G.M.; Bernstein, E.F.; Dilley, R.B.; Loskutoff, D.J. Increased Type 1 Plasminogen Activator Inhibitor Gene Expression in Atherosclerotic Human Arteries. Proc. Natl. Acad. Sci. USA 1992, 89, 6998–7002. [Google Scholar] [CrossRef] [Green Version]
- Jung, R.G.; Motazedian, P.; Ramirez, F.D.; Simard, T.; Di Santo, P.; Visintini, S.; Faraz, M.A.; Labinaz, A.; Jung, Y.; Hibbert, B. Association between Plasminogen Activator Inhibitor-1 and Cardiovascular Events: A Systematic Review and Meta-Analysis. Thromb. J. 2018, 16, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugano, T.; Tsuji, H.; Masuda, H.; Nakagawa, K.; Nishimura, H.; Kasahara, T.; Yoshizumi, M.; Nakahara, Y.; Kitamura, H.; Yamada, K.; et al. Plasminogen Activator Inhibitor-1 Promoter 4G/5G Genotype Is Not a Risk Factor for Myocardial Infarction in a Japanese Population. Blood Coagul. Fibrinolysis 1998, 9, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Crainich, P.; Jenny, N.S.; Tang, Z.; Arnold, A.M.; Kuller, L.H.; Manolio, T.; Sharrettu, A.R.; Tracy, R.P. Lack of Association of the Plasminogen Activator Inhibitor-1 4G/5G Promoter Polymorphism with Cardiovascular Disease in the Elderly. J. Thromb. Haemost. 2003, 1, 1799–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, L.; Jansson, J.H.; Boman, K.; Nilsson, T.K.; Stegmayr, B.; Hallmans, G. Tissue Plasminogen Activator, Plasminogen Activator Inhibitor-1, and Tissue Plasminogen Activator/Plasminogen Activator Inhibitor-1 Complex as Risk Factors for the Development of a First Stroke. Stroke 2000, 31, 26–32. [Google Scholar] [CrossRef]
- Folsom, A.R.; Cushman, M.; Heckbert, S.R.; Rosamond, W.D.; Aleksic, N. Prospective Study of Fibrinolytic Markers and Venous Thromboembolism. J. Clin. Epidemiol. 2003, 56, 598–603. [Google Scholar] [CrossRef]
- Chen, R.; Yan, J.; Liu, P.; Wang, Z.; Wang, C. Plasminogen Activator Inhibitor Links Obesity and Thrombotic Cerebrovascular Diseases: The Roles of PAI-1 and Obesity on Stroke. Metab. Brain Dis. 2017, 32, 667–673. [Google Scholar] [CrossRef]
- van der Weerd, N.; van Os, H.J.A.; Ali, M.; Schoones, J.W.; van den Maagdenberg, A.M.J.M.; Kruyt, N.D.; Siegerink, B.; Wermer, M.J.H. Sex Differences in Hemostatic Factors in Patients with Ischemic Stroke and the Relation with Migraine—A Systematic Review. Front. Cell. Neurosci. 2021, 15, 7116. [Google Scholar] [CrossRef]
- Yamamoto, K.; Takeshita, K.; Kojima, T.; Takamatsu, J.; Saito, H. Aging and Plasminogen Activator Inhibitor-1 (PAI-1) Regulation: Implication in the Pathogenesis of Thrombotic Disorders in the Elderly. Cardiovasc. Res. 2005, 66, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Carmeliet, P.; Kieckens, L.; Schoonjans, L.; Ream, B.; Van Nuffelen, A.; Prendergast, G.; Cole, M.; Bronson, R.; Collen, D.; Mulligan, R.C. Plasminogen Activator Inhibitor-1 Gene-Deficient Mice. I. Generation by Homologous Recombination and Characterization. J. Clin. Investig. 1993, 92, 2746–2755. [Google Scholar] [CrossRef] [Green Version]
- Eitzman, D.T.; Westrick, R.J.; Xu, Z.; Tyson, J.; Ginsburg, D. Plasminogen Activator Inhibitor-1 Deficiency Protects against Atherosclerosis Progression in the Mouse Carotid Artery. Blood 2000, 96, 4212–4215. [Google Scholar] [CrossRef]
- Zhu, Y.; Farrehi, P.M.; Fay, W.P. Plasminogen Activator Inhibitor Type 1 Enhances Neointima Formation after Oxidative Vascular Injury in Atherosclerosis-Prone Mice. Circulation 2001, 103, 3105–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Stassen, J.M.; Schoonjans, L.; Ream, B.; Van Den Oord, J.J.; De Mol, M.; Mulligan, R.C.; Collen, D. Plasminogen Activator Inhibitor-1 Gene-Deficient Mice. II. Effects on Hemostasis, Thrombosis, and Thrombolysis. J. Clin. Investig. 1993, 92, 2756–2760. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.K.; Donahue, D.L.; Sandoval-Cooper, M.J.; Castellino, F.J.; Ploplis, V.A. Plasminogen Activator Inhibitor-1 Protects Mice against Cardiac Fibrosis by Inhibiting Urokinase-Type Plasminogen Activator-Mediated Plasminogen Activation. Sci. Rep. 2017, 7, 365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luttun, A.; Lupu, F.; Storkebaum, E.; Hoylaerts, M.F.; Moons, L.; Crawley, J.; Bono, F.; Poole, A.R.; Tipping, P.; Herbert, J.M.; et al. Lack of Plasminogen Activator Inhibitor-1 Promotes Growth and Abnormal Matrix Remodeling of Advanced Atherosclerotic Plaques in Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 499–505. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Castellino, F.J.; Ploplis, V.A. Plasminogen Activator Inhibitor-1 (PAI-1) Is Cardioprotective in Mice by Maintaining Microvascular Integrity and Cardiac Architecture. Blood 2010, 115, 2038–2047. [Google Scholar] [CrossRef] [Green Version]
- Kremen, M.; Krishnan, R.; Emery, I.; Jie, H.H.; Slezicki, K.I.; Wu, A.; Qian, K.; Du, L.; Plawman, A.; Stempien-Otero, A.; et al. Plasminogen Mediates the Atherogenic Effects of Macrophage-Expressed Urokinase and Accelerates Atherosclerosis in ApoE-Knockout Mice. Proc. Natl. Acad. Sci. USA 2008, 105, 17109–17114. [Google Scholar] [CrossRef] [Green Version]
- Stempien-Otero, A.; Plawman, A.; Meznarich, J.; Dyamenahalli, T.; Otsuka, G.; Dichek, D.A. Mechanisms of Cardiac Fibrosis Induced by Urokinase Plasminogen Activator. J. Biol. Chem. 2006, 281, 15345–15351. [Google Scholar] [CrossRef] [Green Version]
- Haka, A.S.; Grosheva, I.; Singh, R.K.; Maxfield, F.R. Plasmin Promotes Foam Cell Formation by Increasing Macrophage Catabolism of Aggregated Low-Density Lipoprotein. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1768–1778. [Google Scholar] [CrossRef] [Green Version]
- Eren, M.; Gleaves, L.A.; Atkinson, J.B.; King, L.E.; Declerck, P.J.; Vaughan, D.E. Reactive Site-Dependent Phenotypic Alterations in Plasminogen Activator Inhibitor-1 Transgenic Mice. J. Thromb. Haemost. 2007, 5, 1500–1508. [Google Scholar] [CrossRef]
- Eren, M.; Painter, C.A.; Atkinson, J.B.; Declerck, P.J.; Vaughan, D.E. Age-Dependent Spontaneous Coronary Arterial Thrombosis in Transgenic Mice That Express a Stable Form of Human Plasminogen Activator Inhibitor-1. Circulation 2002, 106, 491–496. [Google Scholar] [CrossRef]
- Erickson, L.A.; Fici, G.J.; Lund, J.E.; Boyle, T.P.; Polites, H.G.; Marotti, K.R. Development of Venous Occlusions in Mice Transgenic for the Plasminogen Activator Inhibitor-1 Gene. Nature 1990, 346, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Fay, W.P.; Garg, N.; Sunkar, M. Vascular Functions of the Plasminogen Activation System. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, H.; Wang, Z.; Xiao, W. Plasminogen Activator Inhibitor-1 Promotes Inflammatory Process Induced by Cigarette Smoke Extraction or Lipopolysaccharides in Alveolar Epithelial Cells. Exp. Lung Res. 2009, 35, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.K.; Xu, Z.; Castellino, F.J.; Ploplis, V.A. Plasminogen Activator Inhibitor-1 Stimulates Macrophage Activation through Toll-like Receptor-4. Biochem. Biophys. Res. Commun. 2016, 477, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkaliagkousi, E.; Ferro, A. Nitric Oxide Signalling in the Regulation of Cardiovascular and Platelet Function. Front. Biosci. 2011, 16, 1873–1897. [Google Scholar] [CrossRef] [Green Version]
- Farah, C.; Michel, L.Y.M.; Balligand, J.L. Nitric Oxide Signalling in Cardiovascular Health and Disease. Nat. Rev. Cardiol. 2018, 15, 292–316. [Google Scholar] [CrossRef]
- Walford, G.; Loscalzo, J. Nitric Oxide in Vascular Biology. J. Thromb. Haemost. 2003, 1, 2112–2118. [Google Scholar] [CrossRef]
- Garcia, V.; Sessa, W.C. Endothelial NOS: Perspective and Recent Developments. Br. J. Pharmacol. 2019, 176, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Shu, X.; Ruddiman, C.A.; Keller, T.C.S.; Keller, A.S.; Yang, Y.Y.; Good, M.E.; Best, A.K.; Columbus, L.; Isakson, B.E. Heterocellular Contact Can Dictate Arterial Function. Circ. Res. 2019, 124, 1473. [Google Scholar] [CrossRef]
- Childs, B.G.; Li, H.; Van Deursen, J.M. Senescent Cells: A Therapeutic Target for Cardiovascular Disease. J. Clin. Investig. 2018, 128, 1217. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular Senescence in Aging and Age-Related Disease: From Mechanisms to Therapy. Nat. Med. 2015, 21, 1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermolaeva, M.; Neri, F.; Ori, A.; Rudolph, K.L. Cellular and Epigenetic Drivers of Stem Cell Ageing. Nat. Rev. Mol. Cell Biol. 2018, 19, 594–610. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Samarakoon, R.; Higgins, S.P.; Higgins, C.E.; Higgins, P.J. The TGF-Β1/P53/PAI-1 Signaling Axis in Vascular Senescence: Role of Caveolin-1. Biomolecules 2019, 9, 341. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Takeshita, K.; Shimokawa, T.; Yi, H.; Isobe, K.I.; Loskutoff, D.J.; Saito, H. Plasminogen Activator Inhibitor-1 Is a Major Stress-Regulated Gene: Implications for Stress-Induced Thrombosis in Aged Individuals. Proc. Natl. Acad. Sci. USA 2002, 99, 890. [Google Scholar] [CrossRef] [Green Version]
- McDonald, A.P.; Meier, T.R.; Hawley, A.E.; Thibert, J.N.; Farris, D.M.; Wrobleski, S.K.; Henke, P.K.; Wakefield, T.W.; Myers, D.D. Aging Is Associated with Impaired Thrombus Resolution in a Mouse Model of Stasis Induced Thrombosis. Thromb. Res. 2010, 125, 72–78. [Google Scholar] [CrossRef]
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. Sirt1 Modulates Premature Senescence-like Phenotype in Human Endothelial Cells. J. Mol. Cell. Cardiol. 2007, 43, 571–579. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Higgins, P.J.; Bernards, R. Plasminogen Activator Inhibitor-1 Is a Critical Downstream Target of P53 in the Induction of Replicative Senescence. Nat. Cell Biol. 2006, 8, 878–884. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Nijwening, J.H.; Bernards, R. Transforming Growth Factor-Beta Requires Its Target Plasminogen Activator Inhibitor-1 for Cytostatic Activity. J. Biol. Chem. 2008, 283, 24308–24313. [Google Scholar] [CrossRef] [Green Version]
- Boe, A.E.; Eren, M.; Murphy, S.B.; Kamide, C.E.; Ichimura, A.; Terry, D.; McAnally, D.; Smith, L.H.; Miyata, T.; Vaughan, D.E. The PAI-1 Antagonist TM5441 Attenuates L-NAME-Induced Hypertension and Vascular Senescence. Circulation 2013, 128, 2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eren, M.; Boe, A.E.; Murphy, S.B.; Place, A.T.; Nagpal, V.; Morales-Nebreda, L.; Urich, D.; Quaggin, S.E.; Scott Budinger, G.R.; Mutlu, G.M.; et al. PAI-1-Regulated Extracellular Proteolysis Governs Senescence and Survival in Klotho Mice. Proc. Natl. Acad. Sci. USA 2014, 111, 7090–7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elzi, D.J.; Lai, Y.; Song, M.; Hakala, K.; Weintraub, S.T.; Shiio, Y. Plasminogen Activator Inhibitor 1--Insulin-like Growth Factor Binding Protein 3 Cascade Regulates Stress-Induced Senescence. Proc. Natl. Acad. Sci. USA 2012, 109, 12052–12057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, A.K.; Rai, R.; Park, K.E.; Eren, M.; Miyata, T.; Wilsbacher, L.D.; Vaughan, D.E. A Small Molecule Inhibitor of PAI-1 Protects against Doxorubicin-Induced Cellular Senescence. Oncotarget 2016, 7, 72443–72457. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.-N.; Parkinson, J.; Haskell, C.; Wang, Y.-X. Mechanisms of Intimal Hyperplasia Learned from a Murine Carotid Artery Ligation Model. Curr. Vasc. Pharmacol. 2008, 6, 37–43. [Google Scholar] [CrossRef]
- Pandolfi, A.; Cetrullo, D.; Polishuck, R.; Alberta, M.M.; Calafiore, A.; Pellegrini, G.; Vitacolonna, E.; Capani, F.; Consoli, A. Plasminogen Activator Inhibitor Type 1 Is Increased in the Arterial Wall of Type II Diabetic Subjects. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1378–1382. [Google Scholar] [CrossRef] [Green Version]
- Stefansson, S.; Lawrence, D.A. The Serpin PAI-1 Inhibits Cell Migration by Blocking Integrin Alpha V Beta 3 Binding to Vitronectin. Nature 1996, 383, 441–443. [Google Scholar] [CrossRef]
- Brown, S.L.; Lundgren, C.H.; Nordt, T.; Fujii, S. Stimulation of Migration of Human Aortic Smooth Muscle Cells by Vitronectin: Implications for Atherosclerosis. Cardiovasc. Res. 1994, 28, 1815–1820. [Google Scholar] [CrossRef]
- Degryse, B.; Neels, J.G.; Czekay, R.P.; Aertgeerts, K.; Kamikubo, Y.I.; Loskutoff, D.J. The Low Density Lipoprotein Receptor-Related Protein Is a Motogenic Receptor for Plasminogen Activator Inhibitor-1. J. Biol. Chem. 2004, 279, 22595–22604. [Google Scholar] [CrossRef] [Green Version]
- Simone, T.M.; Higgins, S.P.; Archambeault, J.; Higgins, C.E.; Ginnan, R.G.; Singer, H.; Higgins, P.J. A Small Molecule PAI-1 Functional Inhibitor Attenuates Neointimal Hyperplasia and Vascular Smooth Muscle Cell Survival by Promoting PAI-1 Cleavage. Cell. Signal. 2015, 27, 923. [Google Scholar] [CrossRef] [Green Version]
- Hermann, D.M.; Bassetti, C.L. Sleep-Related Breathing and Sleep-Wake Disturbances in Ischemic Stroke. Neurology 2009, 73, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rio, F.; Alonso-Fernández, A.; Armada, E.; Mediano, O.; Lores, V.; Rojo, B.; Fernández-Lahera, J.; Fernández-Navarro, I.; Carpio, C.; Ramírez, T. CPAP Effect on Recurrent Episodes in Patients with Sleep Apnea and Myocardial Infarction. Int. J. Cardiol. 2013, 168, 1328–1335. [Google Scholar] [CrossRef] [PubMed]
- Badran, M.; Ayas, N.; Laher, I. Cardiovascular Complications of Sleep Apnea: Role of Oxidative Stress. Oxid. Med. Cell. Longev. 2014, 985258. [Google Scholar] [CrossRef] [PubMed]
- Zapater, A.; Sánchez-De-La-Torre, M.; Benítez, I.D.; Targa, A.; Bertran, S.; Torres, G.; Aldomà, A.; de Batlle, J.; Abad, J.; Duran-Cantolla, J.; et al. The Effect of Sleep Apnea on Cardiovascular Events in Different Acute Coronary Syndrome Phenotypes. Am. J. Respir. Crit. Care Med. 2020, 202, 1698–1706. [Google Scholar] [CrossRef]
- Cortese, R.; Gileles-Hillel, A.; Khalyfa, A.; Almendros, I.; Akbarpour, M.; Khalyfa, A.A.; Qiao, Z.; Garcia, T.; Andrade, J.; Gozal, D. Aorta Macrophage Inflammatory and Epigenetic Changes in a Murine Model of Obstructive Sleep Apnea: Potential Role of CD36. Sci. Rep. 2017, 7, 43648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, A.M.; Wang, L.; Strohl, K.P.; Walia, H.; Hazen, S.L.; Mehra, R. Sex-Specific Differential Responses of Circulating Biomarkers in Obstructive Sleep Apnea Treatment A Post Hoc Analysis of a Randomized Controlled Trial. Ann. Am. Thorac. Soc. 2020, 17, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Bikov, A.; Meszaros, M.; Schwarz, E.I. Coagulation and Fibrinolysis in Obstructive Sleep Apnoea. Int. J. Mol. Sci. 2021, 22, 2834. [Google Scholar] [CrossRef]
- Seckin, Z.I.; Helmi, H.; Weister, T.J.; Lee, A.; Festic, E. Acute Pulmonary Embolism in Patientswith Obstructive Sleep Apnea: Frequency, Hospital Outcomes, and Recurrence. J. Clin. Sleep Med. 2020, 16, 1029–1036. [Google Scholar] [CrossRef]
- Lippi, G.; Mattiuzzi, C.; Franchini, M. Sleep Apnea and Venous Thromboembolism. A Systematic Review. Thromb. Haemost. 2015, 114, 958–963. [Google Scholar] [CrossRef]
- García-Ortega, A.; Mañas, E.; López-Reyes, R.; Selma, M.J.; García-Sánchez, A.; Oscullo, G.; Jiménez, D.; Martínez-García, M.Á. Obstructive Sleep Apnoea and Venous Thromboembolism: Pathophysiological Links and Clinical Implications. Eur. Respir. J. 2019, 53, 1800893. [Google Scholar] [CrossRef]
- Park, A.M.; Suzuki, Y.J. Effects of Intermittent Hypoxia on Oxidative Stress-Induced Myocardial Damage in Mice. J. Appl. Physiol. 2007, 102, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Rosa, D.P.; Martinez, D.; Picada, J.N.; Semedo, J.G.; Marroni, N.P. Hepatic Oxidative Stress in an Animal Model of Sleep Apnoea: Effects of Different Duration of Exposure. Comp. Hepatol. 2011, 10, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, J.; Reinke, C.; Bedja, D.; Berkowitz, D.; Bevans-Fonti, S.; Li, J.; Barouch, L.A.; Gabrielson, K.; Polotsky, V.Y. Effect of Intermittent Hypoxia on Atherosclerosis in Apolipoprotein E-Deficient Mice. Atherosclerosis 2010, 209, 381–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, M.; Nakano, H.; Maekawa, J.; Okamoto, J.; Ohnishi, Y.; Suzuki, T.; Kimura, H. Oxidative Stress in Obstructive Sleep Apnea. Chest 2005, 127, 1674–1679. [Google Scholar] [CrossRef] [Green Version]
- Vatansever, E.; Surmen-Gur, E.; Ursavas, A.; Karadag, M. Obstructive Sleep Apnea Causes Oxidative Damage to Plasma Lipids and Proteins and Decreases Adiponectin Levels. Sleep Breath. 2011, 15, 275–282. [Google Scholar] [CrossRef]
- Kizawa, T.; Nakamura, Y.; Takahashi, S.; Sakurai, S.; Yamauchi, K.; Inoue, H. Pathogenic Role of Angiotensin II and Oxidised LDL in Obstructive Sleep Apnoea. Eur. Respir. J. 2009, 34, 1390–1398. [Google Scholar] [CrossRef]
- Maniaci, A.; Iannella, G.; Cocuzza, S.; Vicini, C.; Magliulo, G.; Ferlito, S.; Cammaroto, G.; Meccariello, G.; De Vito, A.; Nicolai, A.; et al. Oxidative Stress and Inflammation Biomarker Expression in Obstructive Sleep Apnea Patients. J. Clin. Med. 2021, 10, 277. [Google Scholar] [CrossRef]
- Li, Q.; Zheng, X.; Li, Q.; Zheng, X. Tumor Necrosis Factor Alpha Is a Promising Circulating Biomarker for the Development of Obstructive Sleep Apnea Syndrome: A Meta-Analysis. Oncotarget 2017, 8, 27616–27626. [Google Scholar] [CrossRef] [Green Version]
- Schulz, R.; Mahmoudi, S.; Hattar, K.; Sibelius, U.L.F.; Olschewski, H.; Mayer, K.; Seeger, W.; Grimminger, F. Enhanced Release of Superoxide from Polymorphonuclear Neutrophils in Obstructive Sleep Apnea. Impact of Continuous Positive Airway Pressure Therapy. Am. J. Respir. Crit. Care Med. 2000, 162, 566–570. [Google Scholar] [CrossRef]
- Dyugovskaya, L.; Lavie, P.; Lavie, L. Increased Adhesion Molecules Expression and Production of Reactive Oxygen Species in Leukocytes of Sleep Apnea Patients. Am. J. Respir. Crit. Care Med. 2002, 165, 934–939. [Google Scholar] [CrossRef]
- Imani, M.M.; Sadeghi, M.; Khazaie, H.; Emami, M.; Sadeghi Bahmani, D.; Brand, S. Evaluation of Serum and Plasma Interleukin-6 Levels in Obstructive Sleep Apnea Syndrome: A Meta-Analysis and Meta-Regression. Front. Immunol. 2020, 11, 1343. [Google Scholar] [CrossRef] [PubMed]
- Unnikrishnan, D.; Jun, J.; Polotsky, V. Inflammation in Sleep Apnea: An Update. Rev. Endocr. Metab. Disord. 2015, 16, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozal, D.; Kheirandish-Gozal, L. Cardiovascular Morbidity in Obstructive Sleep Apnea: Oxidative Stress, Inflammation, and Much More. Am. J. Respir. Crit. Care Med. 2008, 177, 369–375. [Google Scholar] [CrossRef]
- Kheirandish-Gozal, L.; Gozal, D. Obstructive Sleep Apnea and Inflammation: Proof of Concept Based on Two Illustrative Cytokines. Int. J. Mol. Sci. 2019, 20, 459. [Google Scholar] [CrossRef] [Green Version]
- Htoo, A.K.; Greenberg, H.; Tongia, S.; Chen, G.; Henderson, T.; Wilson, D.; Liu, S.F. Activation of Nuclear Factor KappaB in Obstructive Sleep Apnea: A Pathway Leading to Systemic Inflammation. Sleep Breath. 2006, 10, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Song, Y.; Ning, P.; Zhang, L.; Wu, S.; Quan, J.; Li, Q. Association between Tumor Necrosis Factor Alpha and Obstructive Sleep Apnea in Adults: A Meta-Analysis Update. BMC Pulm. Med. 2020, 20, 215. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, N.R.; Peng, Y.J.; Nanduri, J. Hypoxia-Inducible Factors and Obstructive Sleep Apnea. J. Clin. Investig. 2020, 130, 5042–5051. [Google Scholar] [CrossRef]
- Steffanina, A.; Proietti, L.; Antonaglia, C.; Palange, P.; Angelici, E.; Canipari, R. The Plasminogen System and Transforming Growth Factor-β in Subjects with Obstructive Sleep Apnea Syndrome: Effects of CPAP Treatment. Respir. Care 2015, 60, 1643–1651. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.C.; Liaw, S.F.; Chiu, C.H.; Lin, M.W. Effects of Continuous Positive Airway Pressure on Exhaled Transforming Growth Factor-β and Vascular Endothelial Growth Factor in Patients with Obstructive Sleep Apnea. J. Thorac. Dis. 2020, 12, 932–941. [Google Scholar] [CrossRef]
- Zhou, J.P.; Lin, Y.N.; Li, N.; Sun, X.W.; Ding, Y.J.; Yan, Y.R.; Zhang, L.; Li, Q.Y. Angiotensin-(1-7) Rescues Chronic Intermittent Hypoxia-Aggravated TGF-β-Mediated Airway Remodeling in Murine and Cellular Models of Asthma. J. Pharmacol. Exp. Ther. 2020, 375, 268–275. [Google Scholar] [CrossRef]
- Ding, W.X.; Dong, Y.B.; Ding, N.; Zhang, X.F.; Zhang, S.J.; Zhang, X.L.; Liu, J.N.; Lu, G. Adiponectin Protects Rat Heart from Left Ventricular Remodeling Induced by Chronic Intermittent Hypoxia via Inhibition of TGF-β/Smad2/3 Pathway. J. Thorac. Dis. 2014, 6, 1278. [Google Scholar] [CrossRef] [PubMed]
- Abuyassin, B.; Badran, M.; Ayas, N.T.; Laher, I. Intermittent Hypoxia Causes Histological Kidney Damage and Increases Growth Factor Expression in a Mouse Model of Obstructive Sleep Apnea. PLoS ONE 2018, 13, e0192084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resta, O.; Foschino-Barbaro, M.P.; Legari, G.; Talamo, S.; Bonfitto, P.; Palumbo, A.; Minenna, A.; Giorgino, R.; De Pergola, G. Sleep-Related Breathing Disorders, Loud Snoring and Excessive Daytime Sleepiness in Obese Subjects. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 669–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaneda, A.; Jauregui-Maldonado, E.; Ratnani, I.; Varon, J.; Surani, S. Correlation between Metabolic Syndrome and Sleep Apnea. World J. Diabetes 2018, 9, 66. [Google Scholar] [CrossRef]
- Fallahi, A.; Jamil, D.I.; Karimi, E.B.; Baghi, V.; Gheshlagh, R.G. Prevalence of Obstructive Sleep Apnea in Patients with Type 2 Diabetes: A Systematic Review and Meta-Analysis. Diabetes Metab. Syndr. 2019, 13, 2463–2468. [Google Scholar] [CrossRef]
- Patel, A.R.; Patel, A.R.; Singh, S.; Singh, S.; Khawaja, I. The Association of Obstructive Sleep Apnea and Hypertension. Cureus 2019, 11, e4858. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Roberts-Thomson, P.; Phillips, B.G.; Haynes, W.G.; Winnicki, M.; Accurso, V.; Somers, V.K. Impairment of Endothelium-Dependent Vasodilation of Resistance Vessels in Patients with Obstructive Sleep Apnea. Circulation 2000, 102, 2607–2610. [Google Scholar] [CrossRef] [Green Version]
- Gozal, D.; Kheirandish-Gozal, L.; Serpero, L.D.; Capdevila, O.S.; Dayyat, E. Obstructive Sleep Apnea and Endothelial Function in School-Aged Nonobese Children: Effect of Adenotonsillectomy. Circulation 2007, 116, 2307–2314. [Google Scholar] [CrossRef] [Green Version]
- Harki, O.; Boete, Q.; Pépin, J.-L.; Arnaud, C.; Belaidi, E.; Faury, G.; Khouri, C.; Briançon-Marjollet, A. Intermittent Hypoxia-Related Alterations in Vascular Structure and Function: A Systematic Review and Meta-Analysis of Rodent Data. Eur. Respir. J. 2021, 59, 2100866. [Google Scholar] [CrossRef]
- Varadharaj, S.; Porter, K.; Pleister, A.; Wannemacher, J.; Sow, A.; Jarjoura, D.; Zweier, J.L.; Khayat, R.N. Endothelial Nitric Oxide Synthase Uncoupling: A Novel Pathway in OSA Induced Vascular Endothelial Dysfunction. Respir. Physiol. Neurobiol. 2014, 207, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Badran, M.; Abuyassin, B.; Ayas, N.; Laher, I. Intermittent Hypoxia Impairs Uterine Artery Function in Pregnant Mice. J. Physiol. 2019, 597, 2639–2650. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Dong, Y.; Somers, V.K.; Peterson, T.E.; Zhang, Y.; Wang, S.; Li, G.; Singh, P. Intermittent Hypoxia Regulates Vasoactive Molecules and Alters Insulin-Signaling in Vascular Endothelial Cells. Sci. Rep. 2018, 8, 14110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Guo, B.; Wang, Y.; Yan, D.; Lin, C.; Shi, Z. The Association between Obstructive Sleep Apnea and Carotid Intima-Media Thickness: A Systematic Review and Meta-Analysis. Angiology 2017, 68, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Turkiewicz, S.; Ditmer, M.; Sochal, M.; Białasiewicz, P.; Strzelecki, D.; Gabryelska, A. Obstructive Sleep Apnea as an Acceleration Trigger of Cellular Senescence Processes through Telomere Shortening. Int. J. Mol. Sci. 2021, 22, 12536. [Google Scholar] [CrossRef]
- Khalyfa, A.; Marin, J.M.; Qiao, Z.; Rubio, D.S.; Kheirandish-Gozal, L.; Gozal, D. Plasma Exosomes in OSA Patients Promote Endothelial Senescence: Effect of Long-Term Adherent Continuous Positive Airway Pressure. Sleep 2020, 43, 217. [Google Scholar] [CrossRef]
- Cortese, R.; Sanz-Rubio, D.; Kheirandish-Gozal, L.; Marin, J.M.; Gozal, D. Epigenetic Age Acceleration in Obstructive Sleep Apnea Is Reversible with Adherent Treatment. Eur. Respir. J. 2022, 59, 2103042. [Google Scholar] [CrossRef]
- Wang, L.; Chen, L.; Liu, Z.; Liu, Y.; Luo, M.; Chen, N.; Deng, X.; Luo, Y.; He, J.; Zhang, L.; et al. PAI-1 Exacerbates White Adipose Tissue Dysfunction and Metabolic Dysregulation in High Fat Diet-Induced Obesity. Front. Pharmacol. 2018, 9, 1087. [Google Scholar] [CrossRef]
- Ichimura, A.; Matsumoto, S.; Suzuki, S.; Dan, T.; Yamaki, S.; Sato, Y.; Kiyomoto, H.; Ishii, N.; Okada, K.; Matsuo, O.; et al. A Small Molecule Inhibitor to Plasminogen Activator Inhibitor 1 Inhibits Macrophage Migration. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 935–942. [Google Scholar] [CrossRef] [Green Version]
- Khoukaz, H.B.; Ji, Y.; Braet, D.J.; Vadali, M.; Abdelhamid, A.A.; Emal, C.D.; Lawrence, D.A.; Fay, W.P. Drug Targeting of Plasminogen Activator Inhibitor-1 Inhibits Metabolic Dysfunction and Atherosclerosis in a Murine Model of Metabolic Syndrome. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1479–1490. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Badran, M.; Gozal, D. PAI-1: A Major Player in the Vascular Dysfunction in Obstructive Sleep Apnea? Int. J. Mol. Sci. 2022, 23, 5516. https://doi.org/10.3390/ijms23105516
Badran M, Gozal D. PAI-1: A Major Player in the Vascular Dysfunction in Obstructive Sleep Apnea? International Journal of Molecular Sciences. 2022; 23(10):5516. https://doi.org/10.3390/ijms23105516
Chicago/Turabian StyleBadran, Mohammad, and David Gozal. 2022. "PAI-1: A Major Player in the Vascular Dysfunction in Obstructive Sleep Apnea?" International Journal of Molecular Sciences 23, no. 10: 5516. https://doi.org/10.3390/ijms23105516