
InDels Identification and Association Analysis with Spike and Awn Length in Chinese Wheat Mini-Core Collection

 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
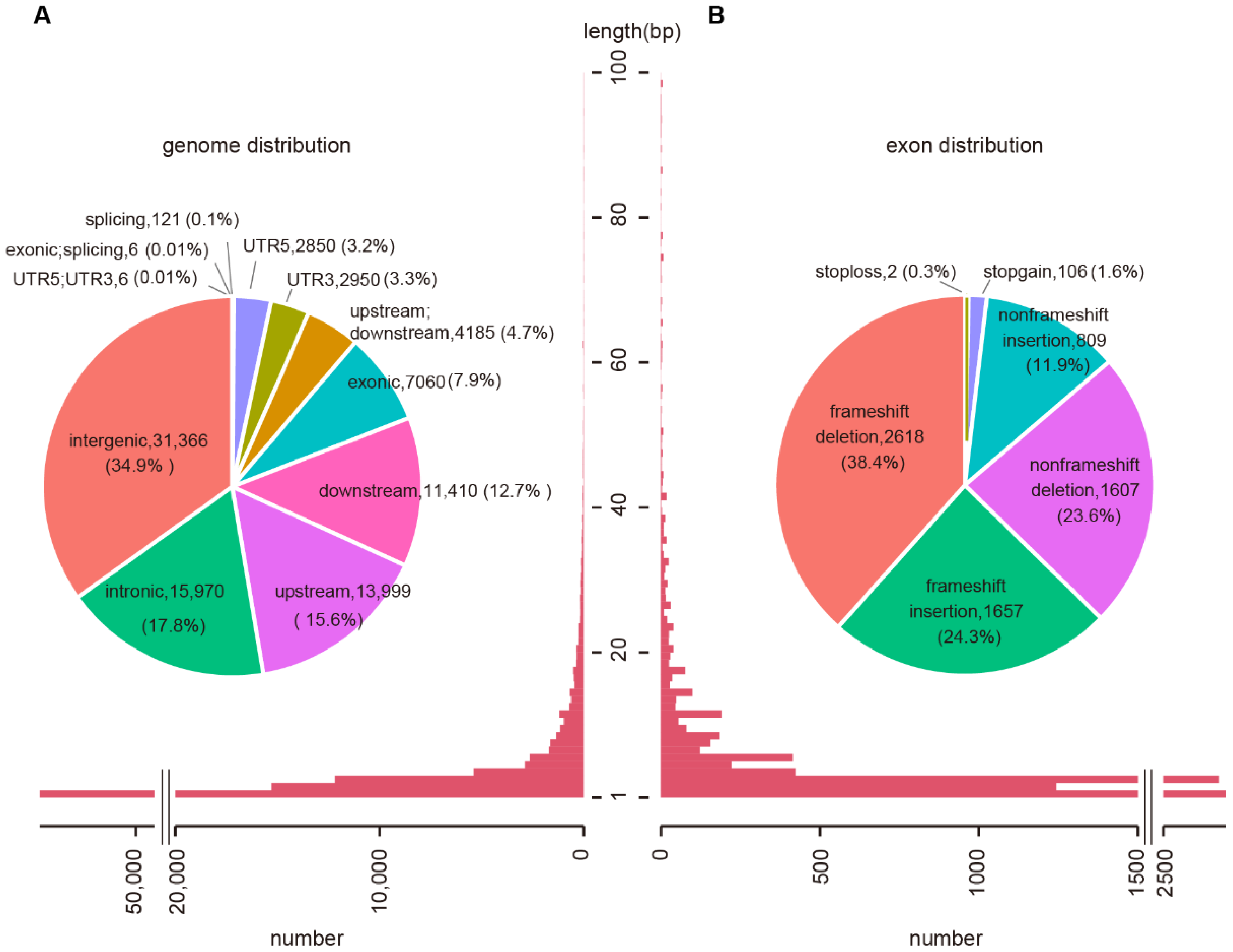
2.1. Genomic Features of Wheat InDels
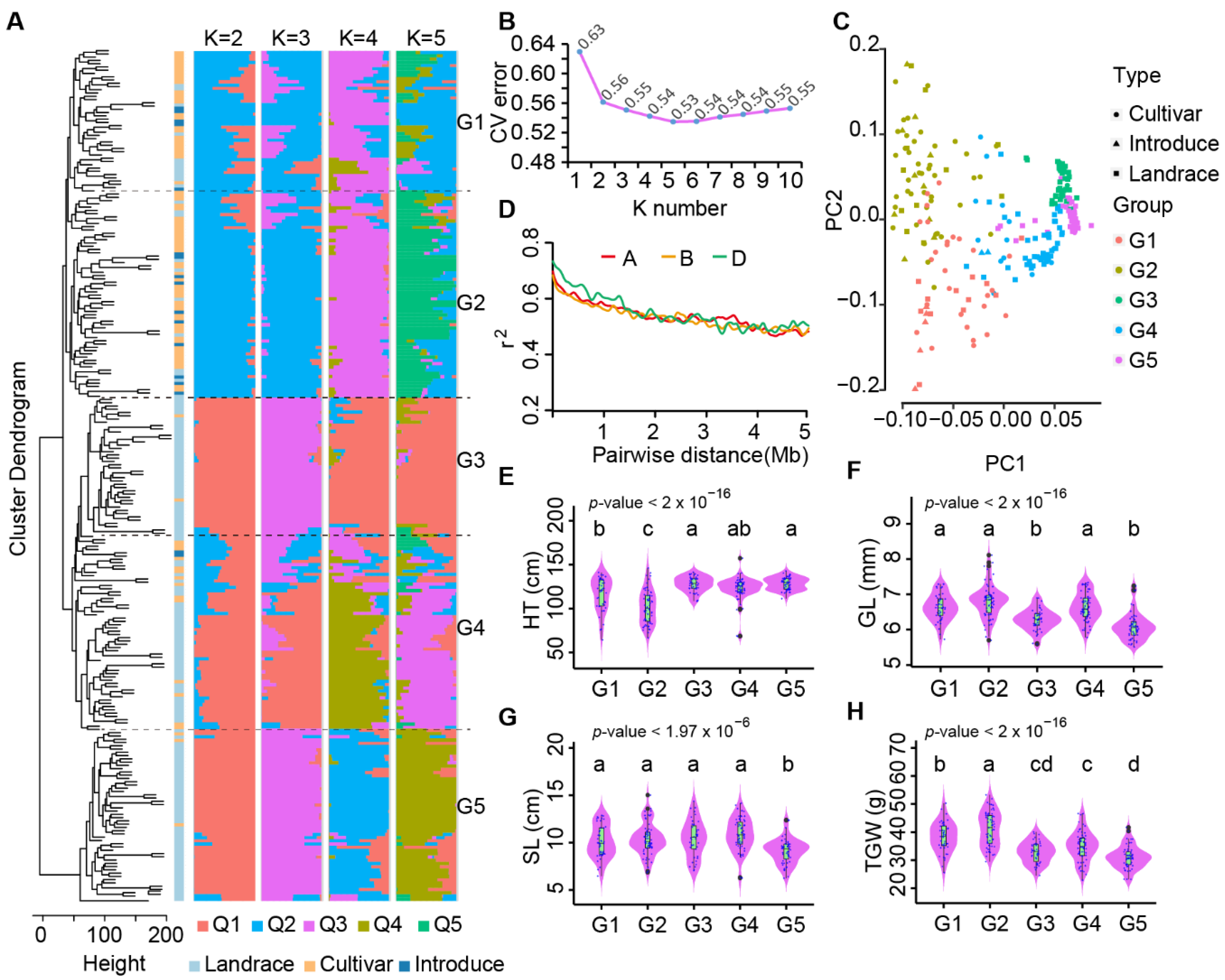
2.2. Population Structure of Chinese Mini-Core Collection Based on InDels
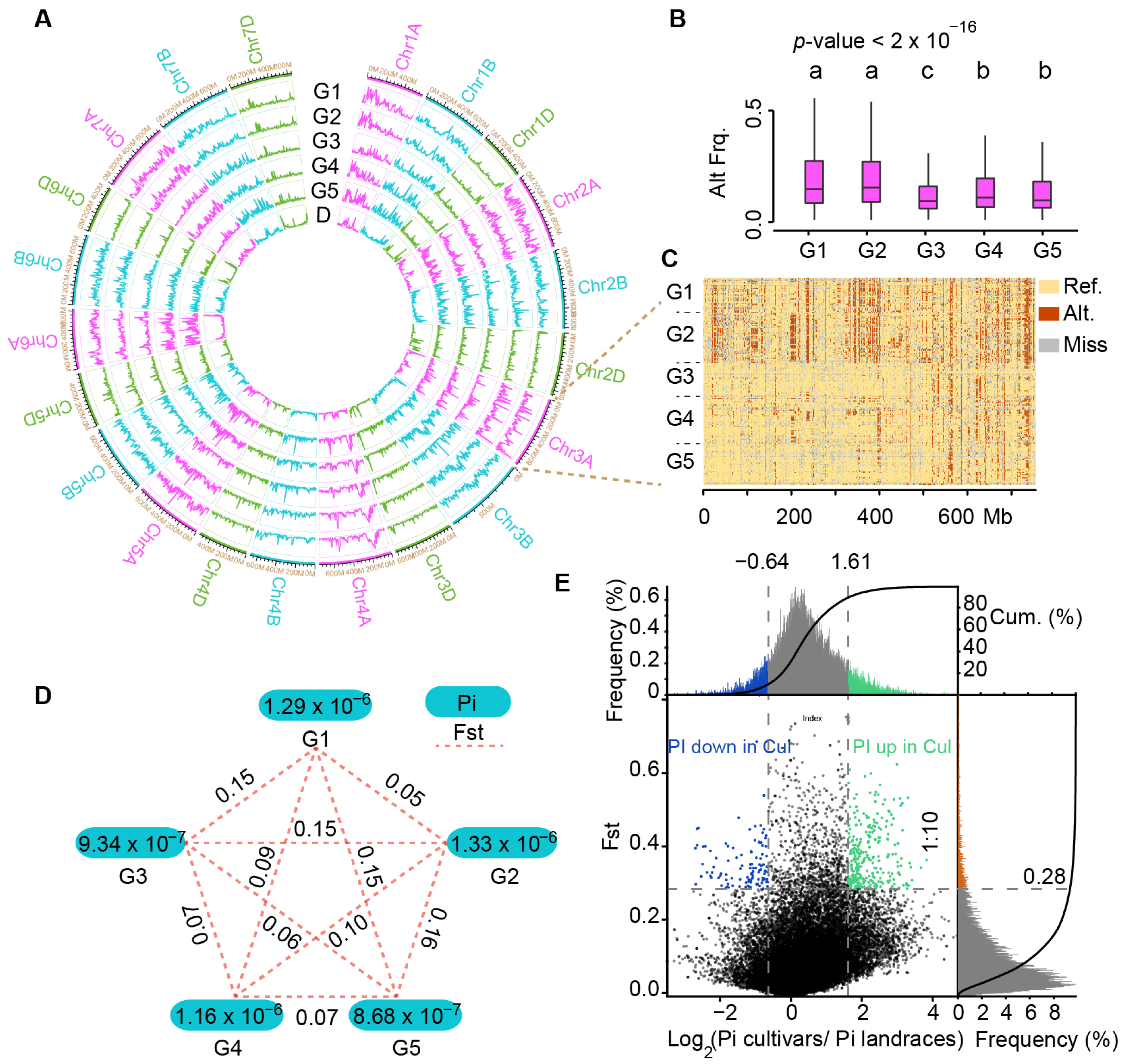
2.3. Estimation of Molecular Diversity of Chinese Mini-Core Collection Accession Using InDel Markers
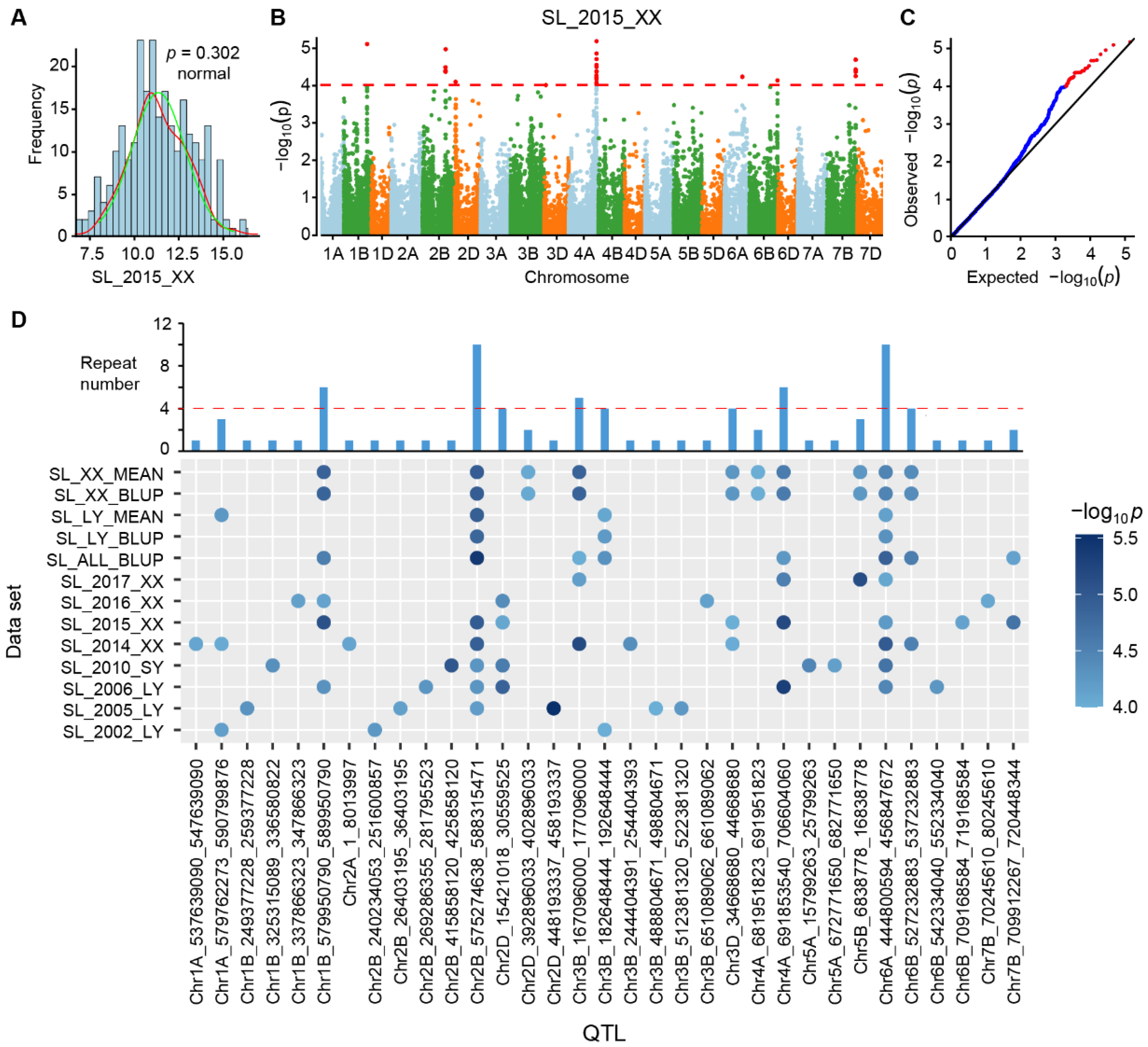
2.4. InDel-Based Genome-Wide Association Study (GWAS) on Wheat Spike Length
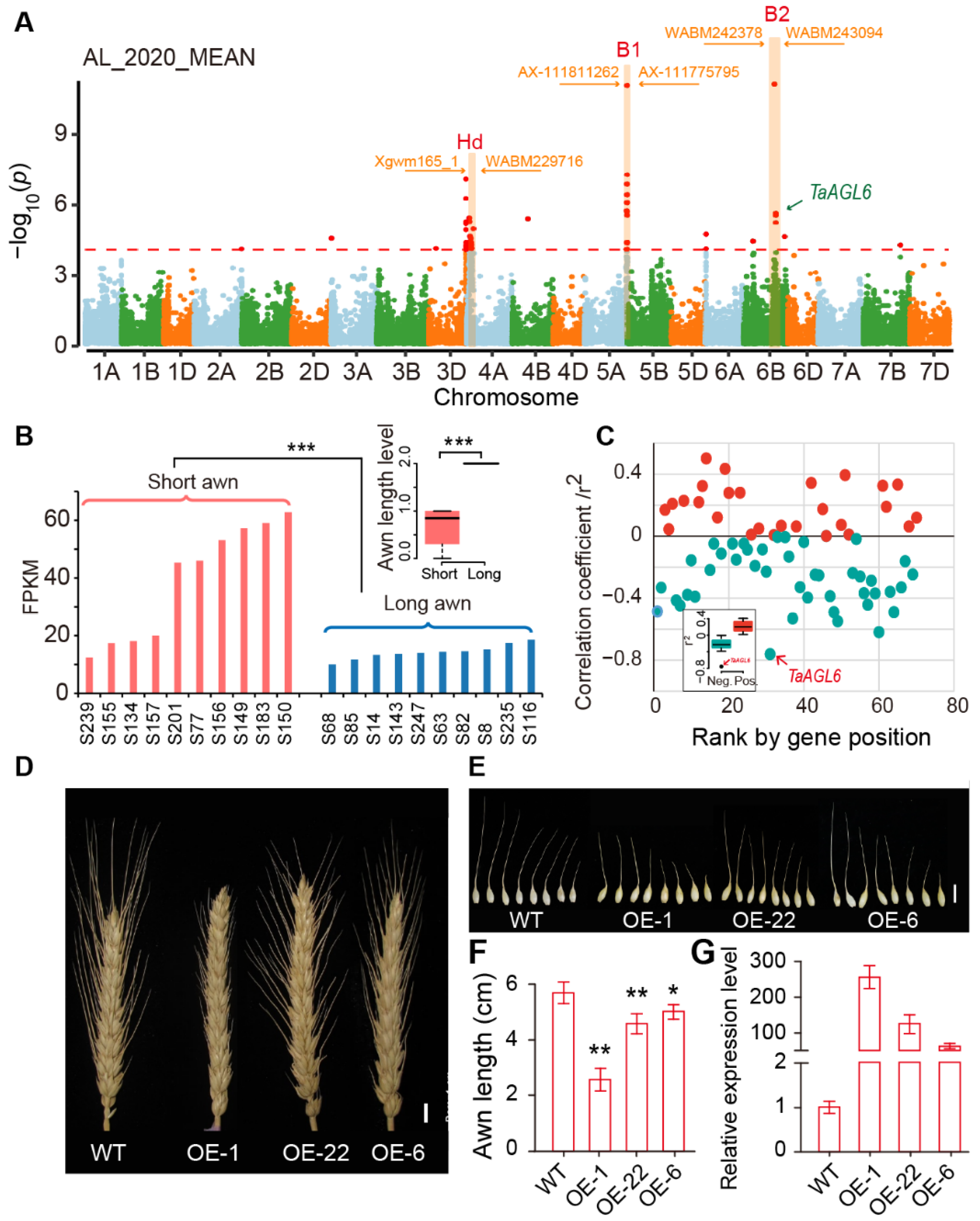
2.5. Identification of an Awn Inhibitor at the Tipped 2 (B2) Locus by GWAS
3. Discussion
3.1. InDel Diversity Was Comparable between Wheat Cultivars and Landraces
3.2. InDels Are Effective Supplements to the Analysis of Population Genetic Variation
3.3. InDels Associated with Awn Length Traits
4. Materials and Methods
4.1. Sampling and Phenotyping
4.2. Sequence Capturing and Sequencing
4.3. Sequence Quality Checking and Filtering
4.4. Sequence Alignment and InDel Detection
4.5. Population Genetics Analysis
4.6. GWAS Analysis
4.7. Construction of TaAGL6 Overexpression Transgenic Lines
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- He, Z.H.; Rajaram, S.; Xin, Z.Y.; Huang, G.Z. A History of Wheat Breeding in China; International Maize and Wheat Improvement Center: Mexico City, Mexico, 2001. [Google Scholar]
- Jin, S.B. Chinese Wheat Varieties and Their Pedigrees; Agricultural Publishing House: Beijing, China, 1983. [Google Scholar]
- Zhuang, Q. Chinese Wheat Improvement and Pedigrees Analysis; Agricultural Publishing House: Beijing, China, 2003. [Google Scholar]
- Hao, C.Y.; Dong, Y.S.; Wang, L.F.; You, G.X.; Zhang, H.N.; Ge, H.M.; Jia, J.Z.; Zhang, X.Y. Genetic diversity and construction of core collection in Chinese wheat genetic resources. Chin. Sci. Bull. 2008, 53, 1518–1526. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Hao, C.; Wang, Z.; Geng, S.; Jia, M.; Wang, F.; Han, X.; Kong, X.; Yin, L.; Tao, S.; et al. Wheat breeding history reveals synergistic selection of pleiotropic genomic sites for plant architecture and grain yield. Mol. Plant 2022, 15, 504–519. [Google Scholar] [CrossRef]
- Jin, T.; Sun, Y.; Shan, Z.; He, J.; Wang, N.; Gai, J.; Li, Y. Natural variation in the promoter of GsERD15B affects salt tolerance in soybean. Plant Biotechnol. J. 2021, 19, 1155–1169. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Liu, S.; Ferjani, A.; Li, J.; Yan, J.; Yang, X.; Qin, F. Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat. Genet. 2016, 48, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Miho, F.; Yokosho, K.; Yamaji, N.; Saisho, D.; Yamane, M.; Takahashi, H.; Sato, K.; Nakazono, M. Acquisition of aluminium tolerance by modification of a single gene in barley. Nat. Commun. 2012, 3, 713. [Google Scholar] [CrossRef]
- IWGSC. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef] [Green Version]
- Pont, C.; Leroy, T.; Seidel, M.; Tondelli, A.; Duchemin, W.; Armisen, D.; Lang, D.; Bustos-Korts, D.; Goue, N.; Balfourier, F.; et al. Tracing the ancestry of modern bread wheats. Nat. Genet. 2019, 51, 905–911. [Google Scholar] [CrossRef]
- He, F.; Pasam, R.; Shi, F.; Kant, S.; Keeble-Gagnere, G.; Kay, P.; Forrest, K.; Fritz, A.; Hucl, P.; Wiebe, K.; et al. Exome sequencing highlights the role of wild-relative introgression in shaping the adaptive landscape of the wheat genome. Nat. Genet. 2019, 51, 896–904. [Google Scholar] [CrossRef]
- Juliana, P.; Montesinos-Lopez, O.A.; Crossa, J.; Mondal, S.; Perez, L.G.; Poland, J.; Huerta-Espino, J.; Crespo-Herrera, L.; Govindan, V.; Dreisigacker, S.; et al. Integrating genomic-enabled prediction and high-throughput phenotyping in breeding for climate-resilient bread wheat. Theor. Appl. Genet. 2019, 132, 177–194. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.; Liu, C.; Wang, D.; St Amand, P.; Bernardo, A.; Li, W.; He, F.; Li, L.; Wang, L.; Yuan, X.; et al. High-Resolution Genome-wide Association Study Identifies Genomic Regions and Candidate Genes for Important Agronomic Traits in Wheat. Mol. Plant 2020, 13, 1311–1327. [Google Scholar] [CrossRef]
- Hao, C.; Jiao, C.; Hou, J.; Li, T.; Liu, H.; Wang, Y.; Zheng, J.; Liu, H.; Bi, Z.; Xu, F.; et al. Resequencing of 145 Landmark Cultivars Reveals Asymmetric Sub-genome Selection and Strong Founder Genotype Effects on Wheat Breeding in China. Mol. Plant 2020, 13, 1733–1751. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Wang, L.; Rimbert, H.; Rodriguez, J.C.; Deal, K.R.; De Oliveira, R.; Choulet, F.; Keeble-Gagnère, G.; Tibbits, J.; Rogers, J.; et al. Optical maps refine the bread wheat Triticum aestivum cv. Chinese Spring genome assembly. Plant J. 2021, 107, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.N.; Sain, R.S.; Sharma, R.K. Genetics of spike length in durum wheat. Euphytica 2003, 130, 155–161. [Google Scholar] [CrossRef]
- Sanchez-Bragado, R.; Molero, G.; Reynolds, M.P.; Araus, J.L. Relative contribution of shoot and ear photosynthesis to grain filling in wheat under good agronomical conditions assessed by differential organ delta C-13. J. Exp. Bot. 2014, 65, 5401–5413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Bragado, R.; Molero, G.; Reynolds, M.P.; Araus, J.L. Photosynthetic contribution of the ear to grain filling in wheat: A comparison of different methodologies for evaluation. J. Exp. Bot. 2016, 67, 2787–2798. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.F.; Zhao, Y.S.; Roder, M.S.; Reif, J.C.; Ganal, M.W.; Chen, D.J.; Schnurbusch, T. Manipulation and prediction of spike morphology traits for the improvement of grain yield in wheat. Sci. Rep. 2018, 8, 14435. [Google Scholar] [CrossRef]
- Faris, J.D.; Fellers, J.P.; Brooks, S.A.; Gill, B.S. A bacterial artificial chromosome contig spanning the major domestication locus Q in wheat and identification of a candidate gene. Genetics 2003, 164, 311–321. [Google Scholar] [CrossRef]
- Sormacheva, I.; Golovnina, K.; Vavilova, V.; Kosuge, K.; Watanabe, N.; Blinov, A.; Goncharov, N.P. Q gene variability in wheat species with different spike morphology. Genet. Resour. Crop Evol. 2015, 62, 837–852. [Google Scholar] [CrossRef]
- Dixon, L.E.; Greenwood, J.R.; Bencivenga, S.; Zhang, P.; Cockram, J.; Mellers, G.; Ramm, K.; Cavanagh, C.; Swain, S.M.; Boden, S.A. TEOSINTE BRANCHED1 Regulates Inflorescence Architecture and Development in Bread Wheat (Triticum aestivum). Plant Cell 2018, 30, 563–581. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, L.; Zhao, M.; Guo, L.; Guo, X.; Zhao, D.; Batool, A.; Dong, B.; Xu, H.; Cui, S.; et al. Wheat FRIZZY PANICLE activates VERNALIZATION1-A and HOMEOBOX4-A to regulate spike development in wheat. Plant Biotechnol. J. 2021, 19, 1141–1154. [Google Scholar] [CrossRef]
- Niu, J.Q.; Zheng, S.S.; Shi, X.L.; Si, Y.Q.; Tian, S.Q.; He, Y.L.; Ling, H.Q. Fine mapping and characterization of the awn inhibitor B1 locus in common wheat (Triticum aestivum L.). Crop J. 2020, 8, 613–622. [Google Scholar] [CrossRef]
- Yoshioka, M.; Iehisa, J.C.M.; Ohno, R.; Kimura, T.; Enoki, H.; Nishimura, S.; Nasuda, S.; Takumi, S. Three dominant awnless genes in common wheat: Fine mapping, interaction and contribution to diversity in awn shape and length. PLoS ONE 2017, 12, e0176148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeWitt, N.; Guedira, M.; Lauer, E.; Sarinelli, M.; Tyagi, P.; Fu, D.L.; Hao, Q.Q.; Murphy, J.P.; Marshall, D.; Akhunova, A.; et al. Sequence-based mapping identifies a candidate transcription repressor underlying awn suppression at the B1 locus in wheat. New Phytol. 2020, 225, 326–339. [Google Scholar] [CrossRef] [Green Version]
- Barton, A.B.; Pekosz, M.R.; Kurvathi, R.S.; Kaback, D.B. Meiotic recombination at the ends of chromosomes in Saccharomyces cerevisiae. Genetics 2008, 179, 1221–1235. [Google Scholar] [CrossRef] [Green Version]
- Lei, L.; Zhu, X.; Wang, S.; Zhu, M.; Carver, B.F.; Yan, L. TaMFT-A1 is associated with seed germination sensitive to temperature in winter wheat. PLoS ONE 2013, 8, e73330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.Y.; Liu, M.S.; Li, J.R.; Guan, C.M.; Zhang, X.S. The wheat TaGI1, involved in photoperiodic flowering, encodes an Arabidopsis GI ortholog. Plant Mol. Biol. 2005, 58, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Dubcovsky, J. Wheat FT protein regulates VRN1 transcription through interactions with FDL2. Plant J. 2008, 55, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Yan, X.; Wang, Y.; Liu, H.; Cui, D.; Chen, F. Haplotypes of the TaGS5-A1 Gene Are Associated with Thousand-Kernel Weight in Chinese Bread Wheat. Front. Plant Sci. 2016, 7, 783. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhao, B.; Yuan, D.; Duan, M.; Qian, Q.; Tang, L.; Wang, B.; Liu, X.; Zhang, J.; Wang, J.; et al. Rice zinc finger protein DST enhances grain production through controlling Gn1a/OsCKX2 expression. Proc. Natl. Acad. Sci. USA 2013, 110, 3167–3172. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.Y.; Liu, L.T.; Li, T.; Yan, S.; Kuang, B.J.; Huang, S.J.; Yan, C.J.; Wang, T. OsKinesin-13A is an active microtubule depolymerase involved in glume length regulation via affecting cell elongation. Sci. Rep. 2015, 5, 9457. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, K.; Hirotsu, N.; Madoka, Y.; Murakami, N.; Hara, N.; Onodera, H.; Kashiwagi, T.; Ujiie, K.; Shimizu, B.; Onishi, A.; et al. Loss of function of the IAA-glucose hydrolase gene TGW6 enhances rice grain weight and increases yield. Nat. Genet. 2013, 45, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Schierenbeck, M.; Alqudah, A.M.; Lohwasser, U.; Tarawneh, R.A.; Simón, M.R.; Börner, A. Genetic dissection of grain architecture-related traits in a winter wheat population. BMC Plant Biol. 2021, 21, 417. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, S.; Xue, S.; Yang, S.; Huang, J.; Wang, L. Phylogenetic and CRISPR/Cas9 Studies in Deciphering the Evolutionary Trajectory and Phenotypic Impacts of Rice ERECTA Genes. Front. Plant Sci. 2018, 9, 473. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Bragado, R.; Kim, J.W.; Rivera-Amado, C.; Molero, G.; Araus, J.L.; Savin, R.; Slafer, G.A. Are awns truly relevant for wheat yields? A study of performance of awned/awnless isogenic lines and their response to source-sink manipulations. Field Crop Res. 2020, 254, 107827. [Google Scholar] [CrossRef]
- Huang, D.Q.; Zheng, Q.; Melchkart, T.; Bekkaoui, Y.; Konkin, D.J.F.; Kagale, S.; Martucci, M.; You, F.M.; Clarke, M.; Adamski, N.M.; et al. Dominant inhibition of awn development by a putative zinc-finger transcriptional repressor expressed at the B1 locus in wheat. New Phytol. 2020, 225, 340–355. [Google Scholar] [CrossRef] [Green Version]
- Wurschum, T.; Jahne, F.; Phillips, A.L.; Langer, S.M.; Longin, C.F.H.; Tucker, M.R.; Leiser, W.L. Misexpression of a transcriptional repressor candidate provides a molecular mechanism for the suppression of awns by Tipped 1 in wheat. J. Exp. Bot. 2020, 71, 3428–3436. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Z.; Yu, K.; Jin, D.; Sun, L.H.; Chu, J.F.; Wu, W.Y.; Xin, P.Y.; Gregova, E.; Li, X.; Sun, J.Z.; et al. Natural variations in the promoter of Awn Length Inhibitor 1 (ALI-1) are associated with awn elongation and grain length in common wheat. Plant J. 2020, 101, 1075–1090. [Google Scholar] [CrossRef]
- Balfourier, F.; Bouchet, S.; Robert, S.; De Oliveira, R.; Rimbert, H.; Kitt, J.; Choulet, F.; Appels, R.; Feuillet, C.; Keller, B.; et al. Worldwide phylogeography and history of wheat genetic diversity. Sci. Adv. 2019, 5, eaav0536. [Google Scholar] [CrossRef] [Green Version]
- Juliana, P.; Poland, J.; Huerta-Espino, J.; Shrestha, S.; Crossa, J.; Crespo-Herrera, L.; Toledo, F.H.; Govindan, V.; Mondal, S.; Kumar, U.; et al. Improving grain yield, stress resilience and quality of bread wheat using large-scale genomics. Nat. Genet. 2019, 51, 1530–1539. [Google Scholar] [CrossRef]
- Lu, K.; Wei, L.; Li, X.; Wang, Y.; Wu, J.; Liu, M.; Zhang, C.; Chen, Z.; Xiao, Z.; Jian, H.; et al. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 2019, 10, 1154. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Jamann, T.M.; Balint-Kurti, P.J.; Holland, J.B. QTL mapping using high-throughput sequencing. Methods Mol. Biol. 2015, 1284, 257–285. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Lu, H.; Du, H.; Wang, H.; Chen, W.; Chen, Z.; He, Q.; Ou, S.; Zhang, H.; Li, X.; et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations. Cell 2021, 184, 3542–3558.e3516. [Google Scholar] [CrossRef]
- Kou, Y.; Liao, Y.; Toivainen, T.; Lv, Y.; Tian, X.; Emerson, J.J.; Gaut, B.S.; Zhou, Y. Evolutionary Genomics of Structural Variation in Asian Rice (Oryza sativa) Domestication. Mol. Biol. Evol. 2020, 37, 3507–3524. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lin, T.; Meng, X.; Du, H.; Zhang, J.; Liu, G.; Chen, M.; Jing, Y.; Kou, L.; Li, X.; et al. A route to de novo domestication of wild allotetraploid rice. Cell 2021, 184, 1156–1170.e1114. [Google Scholar] [CrossRef]
- Alonge, M.; Wang, X.; Benoit, M.; Soyk, S.; Pereira, L.; Zhang, L.; Suresh, H.; Ramakrishnan, S.; Maumus, F.; Ciren, D.; et al. Major Impacts of Widespread Structural Variation on Gene Expression and Crop Improvement in Tomato. Cell 2020, 182, 145–161.e123. [Google Scholar] [CrossRef]
- Zhang, Z.; Mao, L.; Chen, H.; Bu, F.; Li, G.; Sun, J.; Li, S.; Sun, H.; Jiao, C.; Blakely, R.; et al. Genome-Wide Mapping of Structural Variations Reveals a Copy Number Variant That Determines Reproductive Morphology in Cucumber. Plant Cell 2015, 27, 1595–1604. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.X.; Rajaby, R.; Wei, L.L.; Zhang, L.; Yang, Z.Q.; Yang, Q.Y.; Sung, W.K. Calling large indels in 1047 Arabidopsis with IndelEnsembler. Nucleic Acids Res. 2021, 49, 10879–10894. [Google Scholar] [CrossRef]
- Vervelde, G. The agricultural value of awns in cereals. Neth. J. Agric. Sci. 1953, 1, 2–10. [Google Scholar] [CrossRef]
- Luo, J.; Liu, H.; Zhou, T.; Gu, B.; Huang, X.; Shangguan, Y.; Zhu, J.; Li, Y.; Zhao, Y.; Wang, Y.; et al. An-1 encodes a basic helix-loop-helix protein that regulates awn development, grain size, and grain number in rice. Plant Cell 2013, 25, 3360–3376. [Google Scholar] [CrossRef] [Green Version]
- Gu, B.; Zhou, T.; Luo, J.; Liu, H.; Wang, Y.; Shangguan, Y.; Zhu, J.; Li, Y.; Sang, T.; Wang, Z.; et al. An-2 Encodes a Cytokinin Synthesis Enzyme that Regulates Awn Length and Grain Production in Rice. Mol. Plant 2015, 8, 1635–1650. [Google Scholar] [CrossRef] [PubMed]
- Bessho-Uehara, K.; Wang, D.R.; Furuta, T.; Minami, A.; Nagai, K.; Gamuyao, R.; Asano, K.; Angeles-Shim, R.B.; Shimizu, Y.; Ayano, M.; et al. Loss of function at RAE2, a previously unidentified EPFL, is required for awnlessness in cultivated Asian rice. Proc. Natl. Acad. Sci. USA 2016, 113, 8969–8974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toriba, T.; Hirano, H.Y. The DROOPING LEAF and OsETTIN2 genes promote awn development in rice. Plant J. 2014, 77, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Watkins, A.; Ellerton, S. Variation and genetics of the awn inTriticum. J. Genet. 1940, 40, 243–270. [Google Scholar] [CrossRef]
- Jordan, K.W.; Wang, S.; Lun, Y.; Gardiner, L.-J.; MacLachlan, R.; Hucl, P.; Wiebe, K.; Wong, D.; Forrest, K.L.; Sharpe, A.G.; et al. A haplotype map of allohexaploid wheat reveals distinct patterns of selection on homoeologous genomes. Genome Biol. 2015, 16, 48. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.Y.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Wang, F.; Geng, S.; Guan, J.; Tao, S.; Jia, M.; Sun, G.; Wang, Z.; Wang, K.; Ye, X.; et al. The wheat AGL6-like MADS-box gene is a master regulator for floral organ identity and a target for spikelet meristem development manipulation. Plant Biotechnol. J. 2022, 20, 75–88. [Google Scholar] [CrossRef]
- Li, F.; Wen, W.; He, Z.; Liu, J.; Jin, H.; Cao, S.; Geng, H.; Yan, J.; Zhang, P.; Wan, Y.; et al. Genome-wide linkage mapping of yield-related traits in three Chinese bread wheat populations using high-density SNP markers. Theor. Appl. Genet. 2018, 131, 1903–1924. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Deng, Z.; Kong, X.; Wang, F.; Guan, J.; Cui, D.; Sun, G.; Liao, R.; Fu, M.; Che, Y.; et al. InDels Identification and Association Analysis with Spike and Awn Length in Chinese Wheat Mini-Core Collection. Int. J. Mol. Sci. 2022, 23, 5587. https://doi.org/10.3390/ijms23105587
Wang Z, Deng Z, Kong X, Wang F, Guan J, Cui D, Sun G, Liao R, Fu M, Che Y, et al. InDels Identification and Association Analysis with Spike and Awn Length in Chinese Wheat Mini-Core Collection. International Journal of Molecular Sciences. 2022; 23(10):5587. https://doi.org/10.3390/ijms23105587
Chicago/Turabian StyleWang, Zhenyu, Zhongyin Deng, Xingchen Kong, Fang Wang, Jiantao Guan, Dada Cui, Guoliang Sun, Ruyi Liao, Mingxue Fu, Yuqing Che, and et al. 2022. "InDels Identification and Association Analysis with Spike and Awn Length in Chinese Wheat Mini-Core Collection" International Journal of Molecular Sciences 23, no. 10: 5587. https://doi.org/10.3390/ijms23105587
APA StyleWang, Z., Deng, Z., Kong, X., Wang, F., Guan, J., Cui, D., Sun, G., Liao, R., Fu, M., Che, Y., Hao, C., Geng, S., Zhang, X., Zhou, P., Mao, L., Liu, S., & Li, A. (2022). InDels Identification and Association Analysis with Spike and Awn Length in Chinese Wheat Mini-Core Collection. International Journal of Molecular Sciences, 23(10), 5587. https://doi.org/10.3390/ijms23105587