
Global DNA Methylation Profiling Reveals Differentially Methylated CpGs between Salivary Gland Pleomorphic Adenomas with Distinct Clinical Course

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
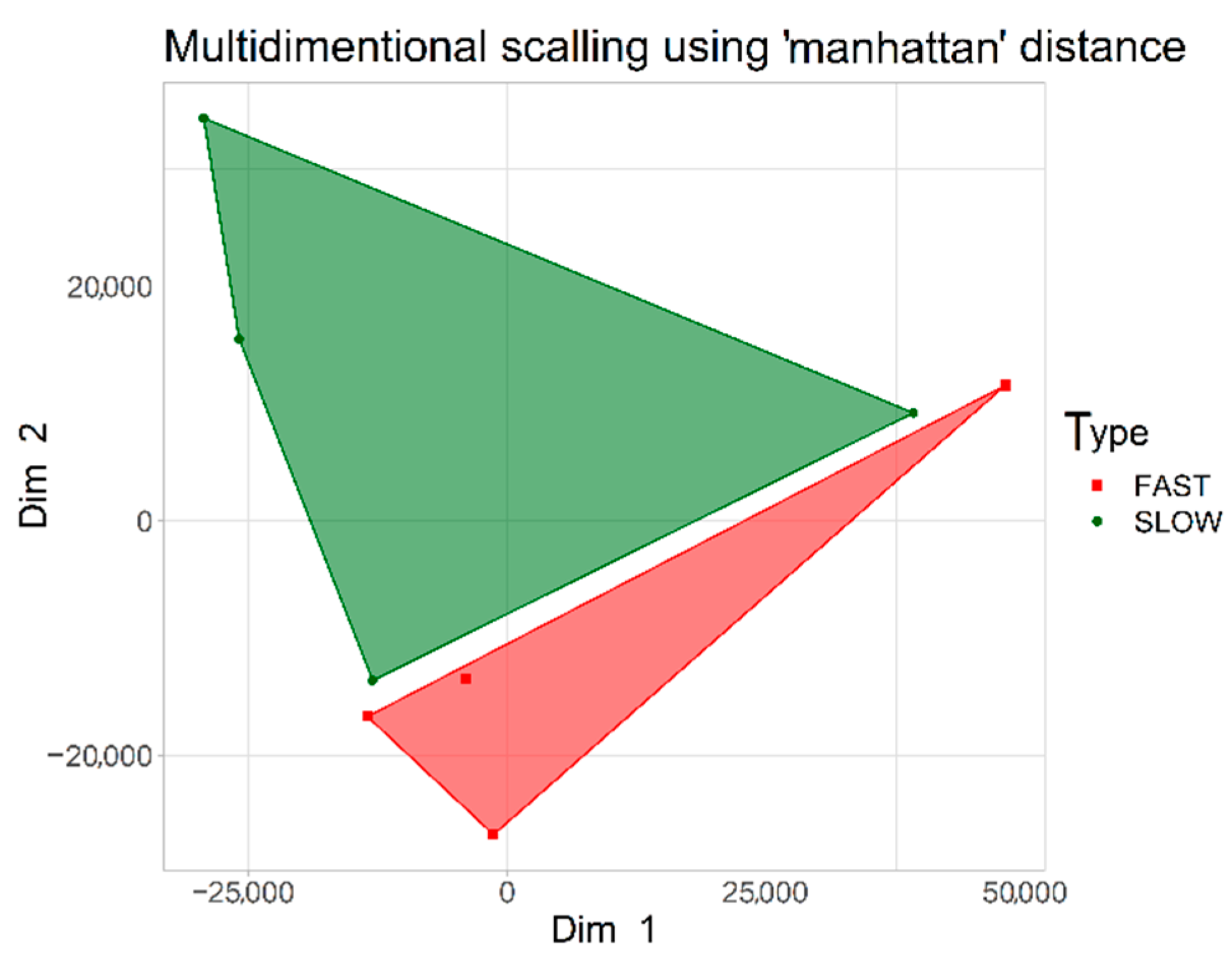
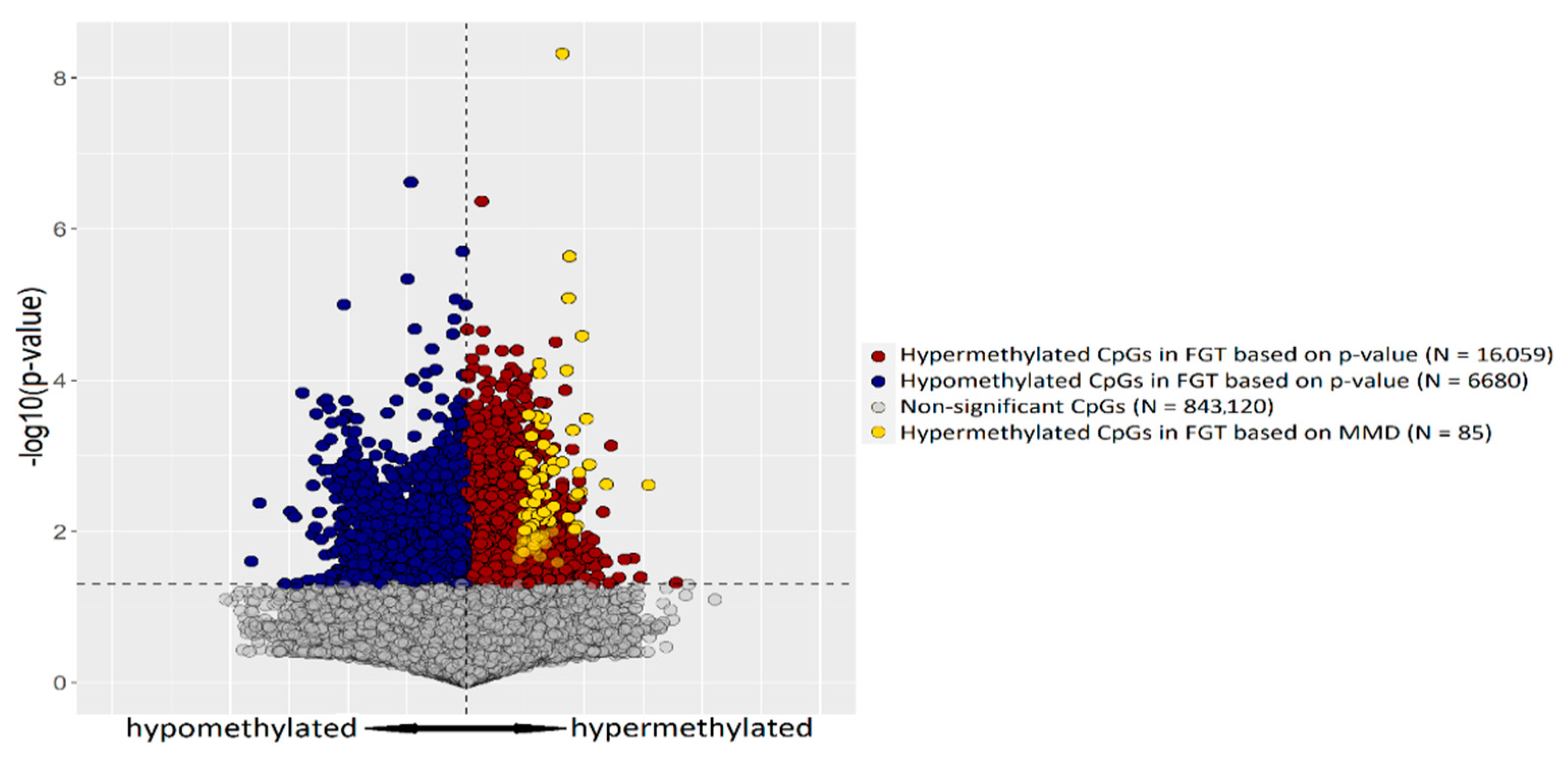
2.1. CpG Tags Selection Based on DNA Methylation Profiling
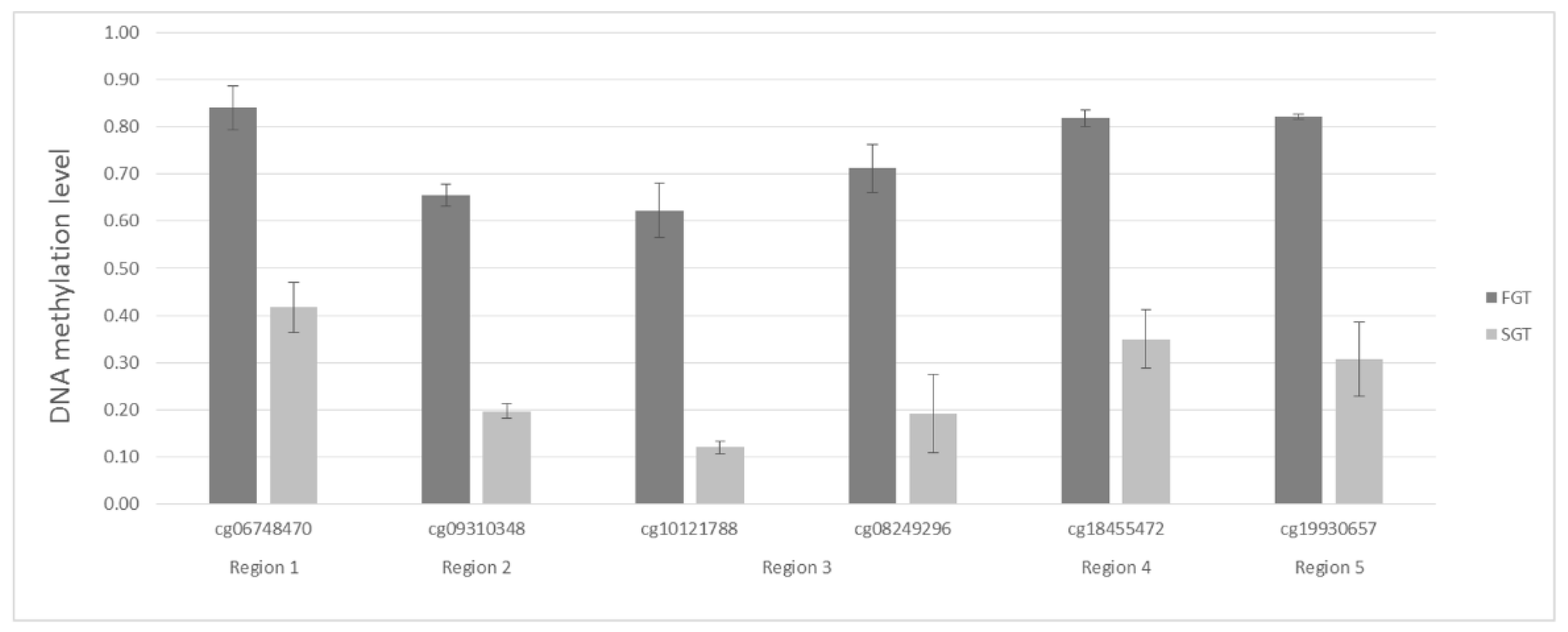
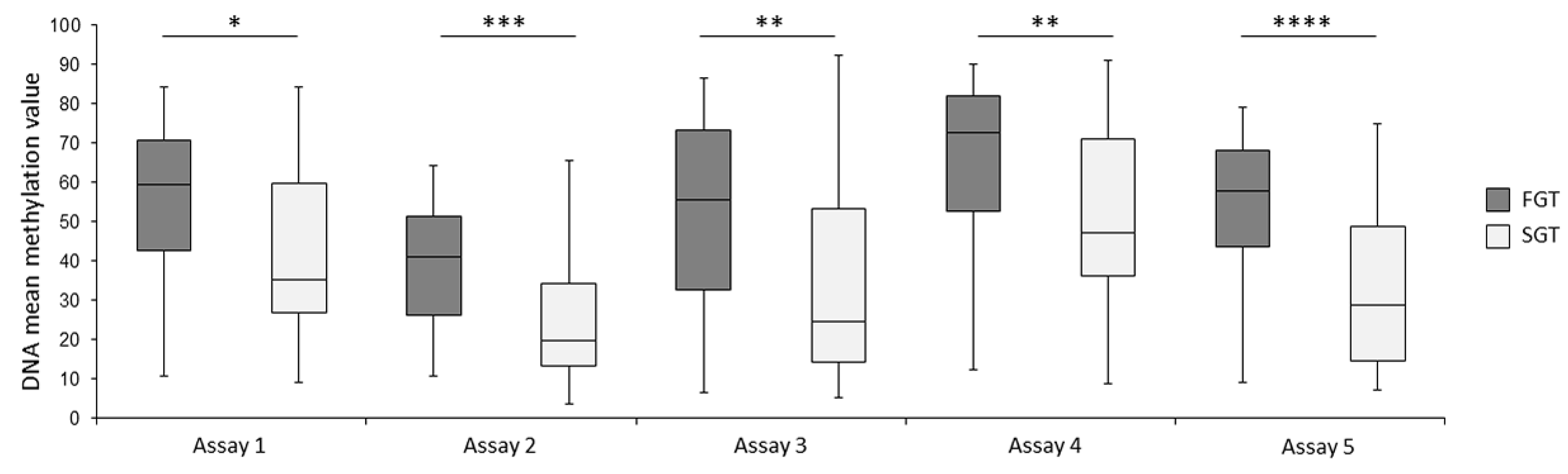
2.2. Bisulfite Pyrosequencing Confirms Differential DNA Methylation between FGT and SGT Samples
2.3. Individual CpG Dinucleotides as Potential Biomarkers
3. Discussion
4. Materials and Methods
4.1. Study Group
4.2. Global DNA Methylation Profiling
4.3. Selection of CpG Tags Differentiating Fast-Growing from Slow-Growing PAs
4.4. Validation of DNA Methylation by Bisulfite Pyrosequencing
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El-Naggar, A.K.; Chan, J.K.C.; Grandis, J.R.; Takata, T.; Slootweg, P.J. WHO Classification of Head and Neck Tumours. In WHO Classification of Tumours, 4th ed.; IARC Press: Lyon, France, 2017; Volume 9. [Google Scholar]
- de Oliveira, F.A.; Duarte, E.C.; Taveira, C.T.; Máximo, A.A.; de Aquino, É.C.; Alencar, R.d.C.; Vencio, E.F. Salivary gland tumor: A review of 599 cases in a Brazilian population. Head Neck Pathol. 2009, 3, 271–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Israel, Y.; Rachmiel, A.; Ziv, G.; Nagler, R. Benign and Malignant Salivary Gland Tumors-Clinical and Demographic Characteristics. Anticancer Res. 2016, 36, 4151–4154. [Google Scholar] [PubMed]
- Persson, F.; Andrén, Y.; Winnes, M.; Wedell, B.; Nordkvist, A.; Gudnadottir, G.; Dahlenfors, R.; Sjögren, H.; Mark, J.; Stenman, G. High-resolution genomic profiling of adenomas and carcinomas of the salivary glands reveals amplification, rearrangement, and fusion of HMGA2. Genes Chromosom. Cancer 2009, 48, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Andreasen, S.; Therkildsen, M.H.; Bjørndal, K.; Homøe, P. Pleomorphic adenoma of the parotid gland 1985–2010: A Danish nationwide study of incidence, recurrence rate, and malignant transformation. Head Neck 2016, 38 (Suppl. S1), E1364–E1369. [Google Scholar] [CrossRef] [PubMed]
- Piwowarczyk, K.; Bartkowiak, E.; Klimza, H.; Greczka, G.; Wierzbicka, M. Review and characteristics of 585 salivary gland neoplasms from a tertiary hospital registered in the Polish National Major Salivary Gland Benign Tumors Registry over a period of 5 years: A prospective study. Otolaryngol. Pol. 2020, 74, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Quer, M.; Guntinas-Lichius, O.; Marchal, F.; Vander Poorten, V.; Chevalier, D.; León, X.; Eisele, D.; Dulguerov, P. Classification of parotidectomies: A proposal of the European Salivary Gland Society. Eur. Arch. Oto-Rhino-Laryngol. 2016, 273, 3307–3312. [Google Scholar] [CrossRef]
- Wong, W.K.; Shetty, S. Classification of parotidectomy: A proposed modification to the European Salivary Gland Society classification system. Eur. Arch. Oto-Rhino-Laryngol. 2017, 274, 3175–3181. [Google Scholar] [CrossRef]
- Bradley, P.J. Frequency and Histopathology by Site, Major Pathologies, Symptoms and Signs of Salivary Gland Neoplasms. Adv. Otorhinolaryngol. 2016, 78, 9–16. [Google Scholar] [CrossRef]
- Antony, J.; Gopalan, V.; Smith, R.A.; Lam, A.K. Carcinoma ex pleomorphic adenoma: A comprehensive review of clinical, pathological and molecular data. Head Neck Pathol. 2012, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Ahn, D.; Sohn, J.H.; Kim, Y.H.; Lee, J.H.; Kim, H. Different correlations between tumor size and cancer-related gene profiles according to histologic type of salivary gland tumor. Int. J. Clin. Exp. Pathol. 2019, 12, 2809–2816. [Google Scholar]
- Piwowarczyk, K.; Bartkowiak, E.; Kosikowski, P.; Chou, J.T.; Wierzbicka, M. Salivary Gland Pleomorphic Adenomas Presenting with Extremely Varied Clinical Courses. A Single Institution Case-Control Study. Front. Oncol. 2021, 10, 600707. [Google Scholar] [CrossRef]
- Ito, F.A.; Jorge, J.; Vargas, P.A.; Lopes, M.A. Histopathological findings of pleomorphic adenomas of the salivary glands. Med. Oral Patol. Oral Cir. Bucal. 2009, 14, E57–E61. [Google Scholar] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [Green Version]
- Esteller, M. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 2002, 21, 5427–5440. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.; Joosten, S.C.; Feng, Z.; De Ruijter, T.C.; Draht, M.X.; Melotte, V.; Smits, K.M.; Veeck, J.; Herman, J.G.; Van Neste, L.; et al. Analysis of DNA methylation in cancer: Location revisited. Nat. Rev. Clin. Oncol. 2018, 15, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Arechederra, M.; Recalde, M.; Gárate-Rascón, M.; Fernández-Barrena, M.G.; Ávila, M.A.; Berasain, C. Epigenetic Biomarkers for the Diagnosis and Treatment of Liver Disease. Cancers 2021, 13, 1265. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Zhang, S.; Jiao, J.; Zhang, T.; Qu, W.; Muloye, G.M.; Kong, B.; Zhang, Q.; Cui, B. Promoter methylation of SEPT9 as a potential biomarker for early detection of cervical cancer and its overexpression predicts radioresistance. Clin. Epigenet. 2019, 11, 120. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.-H.; Ye, Q.; Zhang, G.-B.; Jiang, J.-Y.; Zhao, H.-Y.; Shao, Y.-F.; Ye, Z.-Q.; Xuan, Z.-X.; Huang, P. Identification of differentially methylated genes as diagnostic and prognostic biomarkers of breast cancer. World J. Surg. Oncol. 2021, 19, 29. [Google Scholar] [CrossRef]
- Del Signore, A.G.; Megwalu, U.C. The rising incidence of major salivary gland cancer in the United States. Ear Nose Throat J. 2017, 96, E13–E16. [Google Scholar] [CrossRef]
- Bartkowiak, E.; Piwowarczyk, K.; Bodnar, M.; Kosikowski, P.; Chou, J.; Woźniak, A.; Wierzbicka, M. Expression of p16Ink4a protein in pleomorphic adenoma and carcinoma ex pleomorphic adenoma proves diversity of tumour biology and predicts clinical course. J. Clin. Pathol. 2021, 1–7. [Google Scholar] [CrossRef]
- Schache, A.G.; Hall, G.; Woolgar, J.A.; Nikolaidis, G.; Triantafyllou, A.; Lowe, D.; Risk, J.M.; Shaw, R.J.; Liloglou, T. Quantitative promoter methylation differentiates carcinoma ex pleomorphic adenoma from pleomorphic salivary adenoma. Br. J. Cancer 2010, 103, 1846–1851. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.H.; Zhang, C.Y.; Tian, Z.; Wang, L.Z.; Li, J. Aberrant protein expression and promoter methylation of p16 gene are correlated with malignant transformation of salivary pleomorphic adenoma. Arch. Pathol. Lab. Med. 2011, 135, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Hu, Y.; Gu, T.; Wang, L.; Tian, Z. Promoter hypermethylation may contribute to E-cadherin repression in the human salivary carcinoma ex pleomorphic adenoma. Int. J. Oncol. 2018, 52, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Li, C.; Yuan, H.; Wang, X. Identification of prostate cancer specific methylation biomarkers from a multi-cancer analysis. BMC Bioinform. 2021, 22, 492. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, J.N. Receiver operating characteristic curve in diagnostic test assessment. J. Thorac. Oncol. 2010, 5, 1315–1316. [Google Scholar] [CrossRef] [Green Version]
- Piotrowski, I.; Zhu, X.; Saccon, T.; Ashiqueali, S.; Schneider, A.; Nunes, A.d.C.; Noureddine, S.; Sobecka, A.; Barczak, W.; Szewczyk, M.; et al. miRNAs as Biomarkers for Diagnosing and Predicting Survival of Head and Neck Squamous Cell Carcinoma Patients. Cancers 2021, 13, 3980. [Google Scholar] [CrossRef]
- Triche, T.J., Jr.; Weisenberger, D.J.; Van Den Berg, D.; Laird, P.W.; Siegmund, K.D. Low-level processing of Illumina Infinium DNA Methylation BeadArrays. Nucleic. Acids Res. 2013, 41, e90. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Fortin, J.-P.; Labbe, A.; Lemire, M.; Zanke, B.W.; Hudson, T.J.; Fertig, E.J.; Greenwood, C.M.; Hansen, K.D. Functional normalization of 450k methylation array data improves replication in large cancer studies. Genome Biol. 2014, 15, 503. [Google Scholar] [CrossRef] [Green Version]
- Bibikova, M.; Lin, Z.; Zhou, L.; Chudin, E.; Garcia, E.W.; Wu, B.; Doucet, D.; Thomas, N.J.; Wang, Y.; Vollmer, E.; et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006, 16, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 9783319242774. [Google Scholar]
- Mann Whitney Test Calculator (for Unpaired Data), Wilcoxon Signed Rank Test Calculator (for Paired Data). 2016. Available online: https://astatsa.com/WilcoxonTest/ (accessed on 28 August 2021).
- Sing, T.; Sander, O.; Beerenwinkel, N.; Lengauer, T. ROCR: Visualizing classifier performance in R. Bioinformatics 2005, 21, 3940–3941. [Google Scholar] [CrossRef] [PubMed]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: http://www.r-project.org/index.html (accessed on 28 August 2021).
- Kuhn, M.; Wing, J.; Weston, S. Caret: Classification and Regression Training. 2016. Available online: https://CRAN.R-project.org/package=caret (accessed on 28 August 2021).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pyrosequencing Assay No. | CpG No. | CpG Name a | MMV c (FGT) | MMV c (SGT) | MMD d (MMF—MMS) | p Value |
|---|---|---|---|---|---|---|
| 1 | CpG1 | cg06748470 | 50.8 | 38.9 | 12.0 | 0.012451 |
| CpG2 | N/A b | 67.0 | 54.2 | 12.9 | 0.017414 | |
| CpG3 | cg09774749 | 48.9 | 38.0 | 10.9 | 0.015255 | |
| CpG4 | cg15208832 | 59.9 | 47.9 | 12.0 | 0.025001 | |
| CpG5 | cg00600454 | 42.1 | 32.1 | 10.0 | 0.022008 | |
| Mean CpG1-CpG5 | 53.8 | 42.2 | 11.6 | 0.017372 | ||
| 2 | CpG1 | cg09310348 | 41.6 | 26.2 | 15.4 | 0.000422 |
| CpG2 | N/A b | 35.8 | 24.2 | 11.6 | 0.000216 | |
| Mean CpG1-CpG2 | 38.7 | 25.2 | 13.5 | 0.000267 | ||
| 3 | CpG1 | N/A b | 48.6 | 31.8 | 16.8 | 0.002461 |
| CpG2 | N/A b | 56.2 | 38.8 | 17.3 | 0.003937 | |
| CpG3 | cg10121788 | 49.8 | 30.5 | 19.3 | 0.002914 | |
| Mean CpG1-CpG3 | 51.5 | 33.7 | 17.8 | 0.003122 | ||
| 4 | CpG1 | cg14066163 | 73.5 | 59.3 | 14.2 | 0.002378 |
| CpG2 | cg18455472 | 59.5 | 44.3 | 15.2 | 0.002297 | |
| Mean CpG1-CpG2 | 66.5 | 51.8 | 14.7 | 0.002068 | ||
| 5 e | CpG1 | cg19930657 | 52.4 | 32.1 | 20.3 | 0.000048 |
| Pyrosequencing Assay No. | CpG No. | CpG Name a | Sensitivity | Specificity | AUC c | p Value (DeLong’s Test) | Cutoff d | NPV e | PPV f |
|---|---|---|---|---|---|---|---|---|---|
| 1 | CpG1 | cg06748470 | 0.75 | 0.69 | 0.65 | 0.02762 | 41.95 | 0.75 | 0.69 |
| CpG2 | N/A b | 0.78 | 0.67 | 0.64 | 0.03868 | 56.81 | 0.76 | 0.68 | |
| CpG3 | cg09774749 | 0.75 | 0.67 | 0.65 | 0.03353 | 38.41 | 0.74 | 0.68 | |
| CpG4 | cg15208832 | 0.72 | 0.69 | 0.63 | 0.05576 | 51.76 | 0.73 | 0.68 | |
| CpG5 | cg00600454 | 0.75 | 0.67 | 0.64 | 0.04811 | 31.24 | 0.74 | 0.68 | |
| Mean CpG1-CpG5 | 0.75 | 0.69 | 0.64 | 0.03933 | 43.48 | 0.75 | 0.69 | ||
| 2 | CpG1 | cg09310348 | 0.75 | 0.64 | 0.72 | 0.00042 | 25.39 | 0.74 | 0.66 |
| CpG2 | N/A b | 0.81 | 0.67 | 0.73 | 0.00023 | 26.43 | 0.79 | 0.69 | |
| Mean CpG1-CpG2 | 0.83 | 0.64 | 0.73 | 0.00025 | 25.67 | 0.68 | 0.81 | ||
| 3 | CpG1 | N/A b | 0.78 | 0.67 | 0.69 | 0.00502 | 28.15 | 0.76 | 0.68 |
| CpG2 | N/A b | 0.69 | 0.72 | 0.68 | 0.00787 | 50.14 | 0.72 | 0.69 | |
| CpG3 | cg10121788 | 0.75 | 0.69 | 0.69 | 0.00557 | 34.28 | 0.75 | 0.69 | |
| Mean CpG1-CpG3 | 0.78 | 0.67 | 0.68 | 0.00660 | 31.67 | 0.76 | 0.68 | ||
| 4 | CpG1 | cg14066163 | 0.81 | 0.59 | 0.69 | 0.00353 | 62.89 | 0.77 | 0.64 |
| CpG2 | cg18455472 | 0.69 | 0.64 | 0.69 | 0.00359 | 51.92 | 0.69 | 0.64 | |
| Mean CpG1-CpG2 | 0.83 | 0.56 | 0.69 | 0.00317 | 50.13 | 0.79 | 0.64 | ||
| 5 | CpG1 | cg19930657 | 0.75 | 0.72 | 0.76 | 0.00353 | 44.15 | 0.77 | 0.64 |
| Sensitivity | Specificity | Cutoff | NPV | PPV | AUC (Bias) | Bootstrap AUC | AIC | |
|---|---|---|---|---|---|---|---|---|
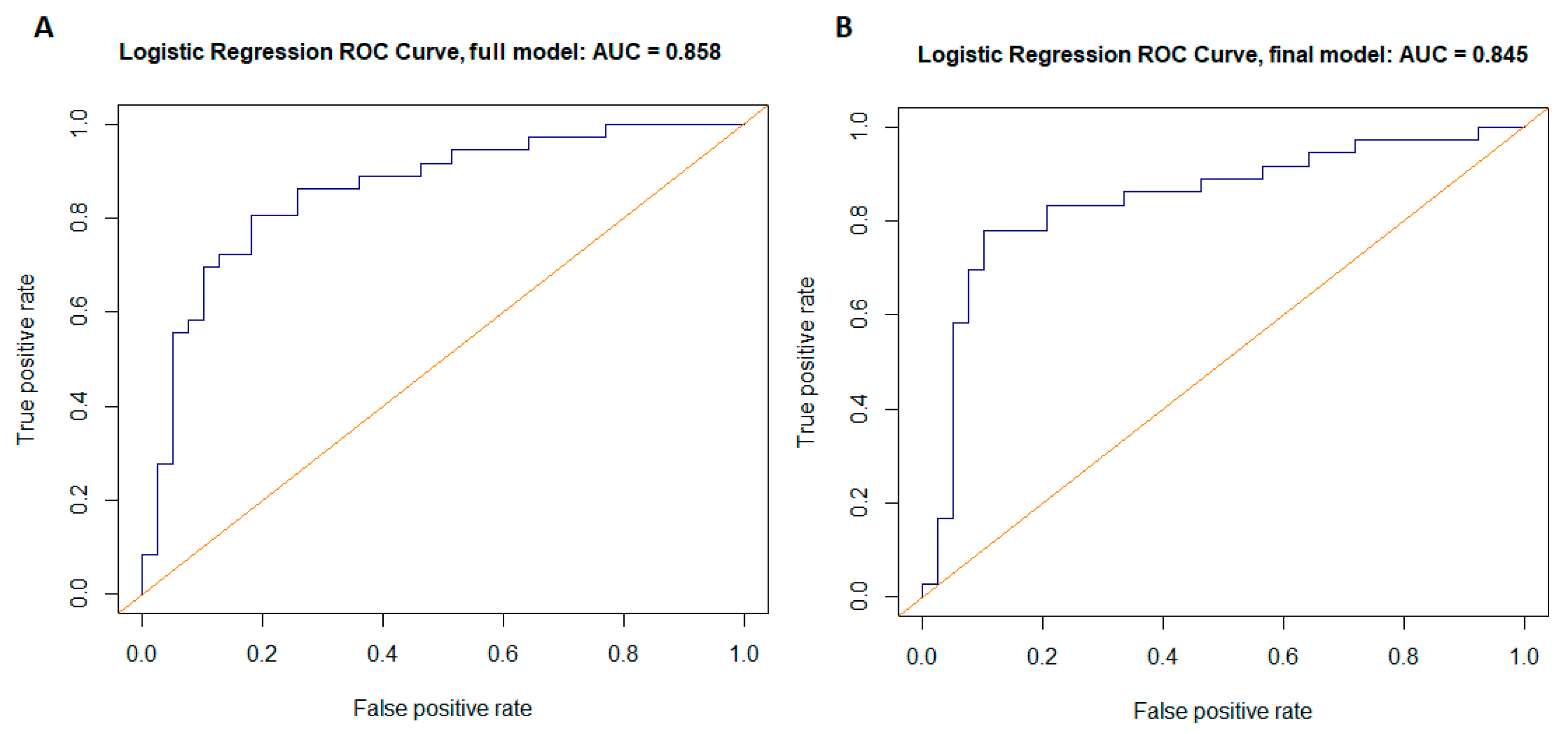
| Full model | 0.81 | 0.82 | 0.53 | 0.82 | 0.8 | 0.858 (0.11) | 0.744 | 99.006 |
| Final model | 0.78 | 0.90 | 0.57 | 0.81 | 0.87 | 0.845 (0.04) | 0.806 | 87.421 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiwerska, K.; Kowal-Wisniewska, E.; Ustaszewski, A.; Bartkowiak, E.; Jarmuz-Szymczak, M.; Wierzbicka, M.; Giefing, M. Global DNA Methylation Profiling Reveals Differentially Methylated CpGs between Salivary Gland Pleomorphic Adenomas with Distinct Clinical Course. Int. J. Mol. Sci. 2022, 23, 5962. https://doi.org/10.3390/ijms23115962
Kiwerska K, Kowal-Wisniewska E, Ustaszewski A, Bartkowiak E, Jarmuz-Szymczak M, Wierzbicka M, Giefing M. Global DNA Methylation Profiling Reveals Differentially Methylated CpGs between Salivary Gland Pleomorphic Adenomas with Distinct Clinical Course. International Journal of Molecular Sciences. 2022; 23(11):5962. https://doi.org/10.3390/ijms23115962
Chicago/Turabian StyleKiwerska, Katarzyna, Ewelina Kowal-Wisniewska, Adam Ustaszewski, Ewelina Bartkowiak, Malgorzata Jarmuz-Szymczak, Malgorzata Wierzbicka, and Maciej Giefing. 2022. "Global DNA Methylation Profiling Reveals Differentially Methylated CpGs between Salivary Gland Pleomorphic Adenomas with Distinct Clinical Course" International Journal of Molecular Sciences 23, no. 11: 5962. https://doi.org/10.3390/ijms23115962
APA StyleKiwerska, K., Kowal-Wisniewska, E., Ustaszewski, A., Bartkowiak, E., Jarmuz-Szymczak, M., Wierzbicka, M., & Giefing, M. (2022). Global DNA Methylation Profiling Reveals Differentially Methylated CpGs between Salivary Gland Pleomorphic Adenomas with Distinct Clinical Course. International Journal of Molecular Sciences, 23(11), 5962. https://doi.org/10.3390/ijms23115962