
Lupeol Treatment Attenuates Activation of Glial Cells and Oxidative-Stress-Mediated Neuropathology in Mouse Model of Traumatic Brain Injury

 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Lupeol Treatment Inhibits Glial Cell Activation in the Cortex and Hippocampus of Mouse Brain with TBI
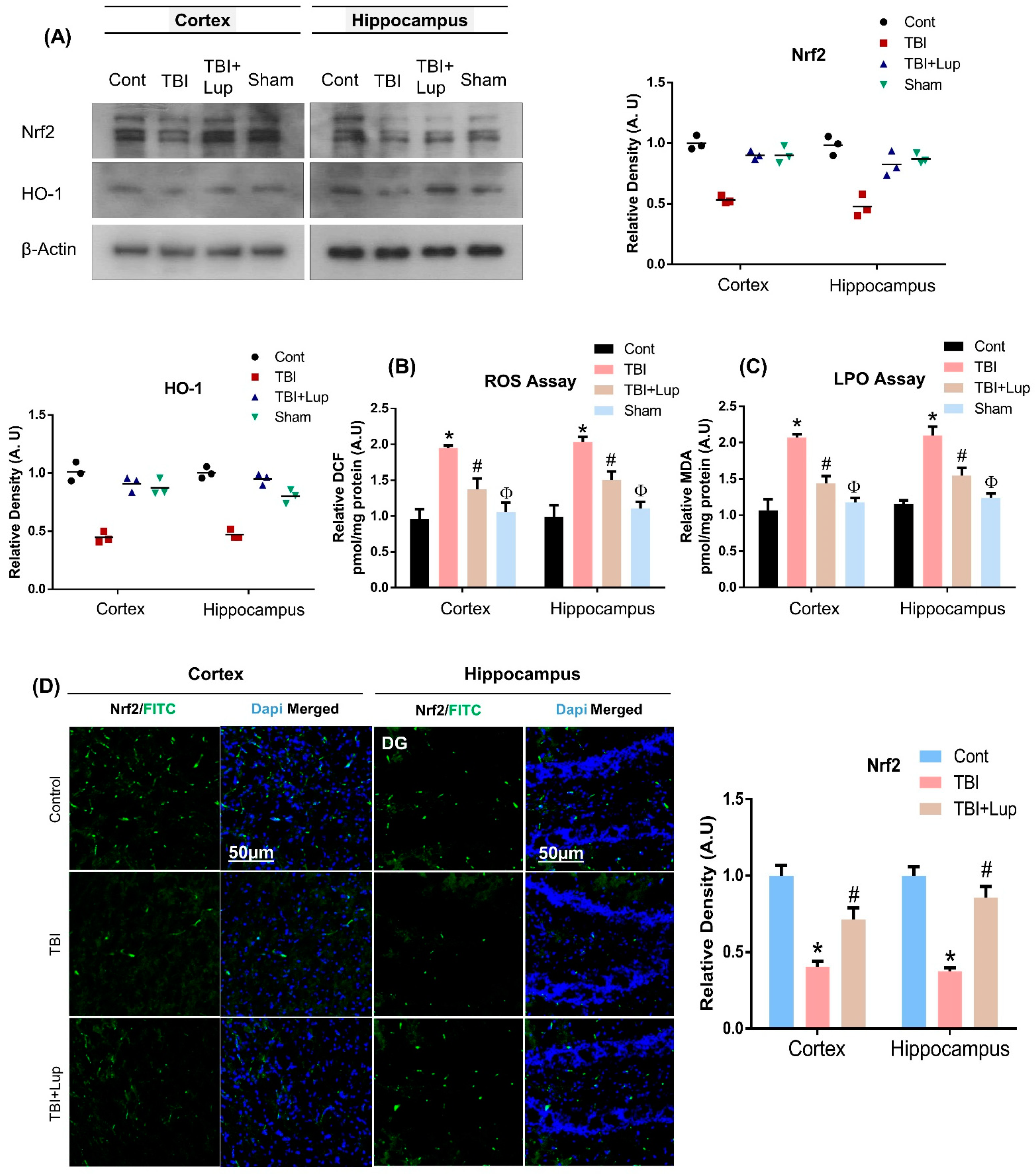
2.2. Lupeol Treatment Induces Nrf2/HO-1 Expression and Attenuates ROS and LPO in Mouse Brain with TBI
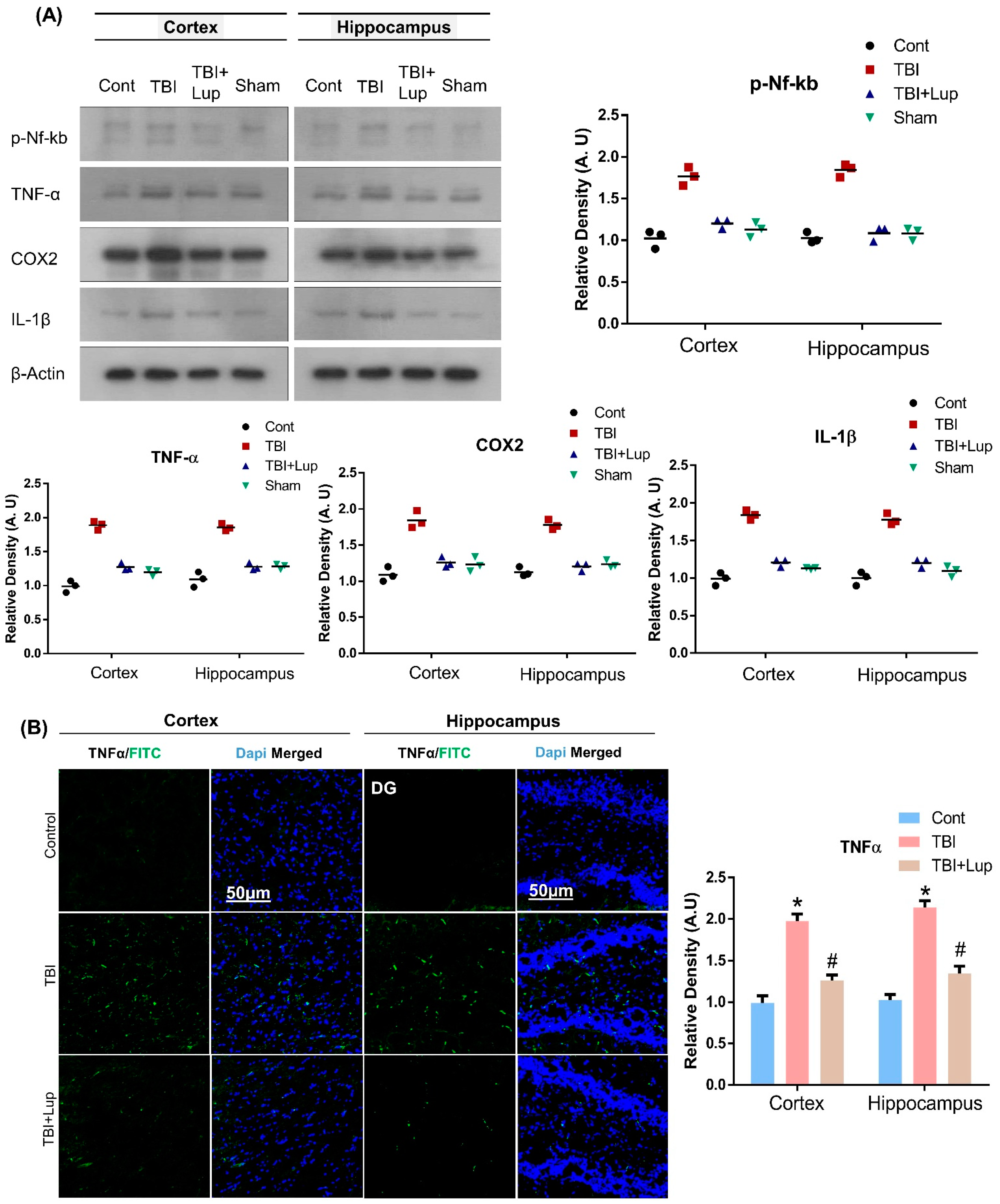
2.3. Administration of Lupeol Reduced the Expression of Inflammatory Cytokines in Mouse Brain with TBI
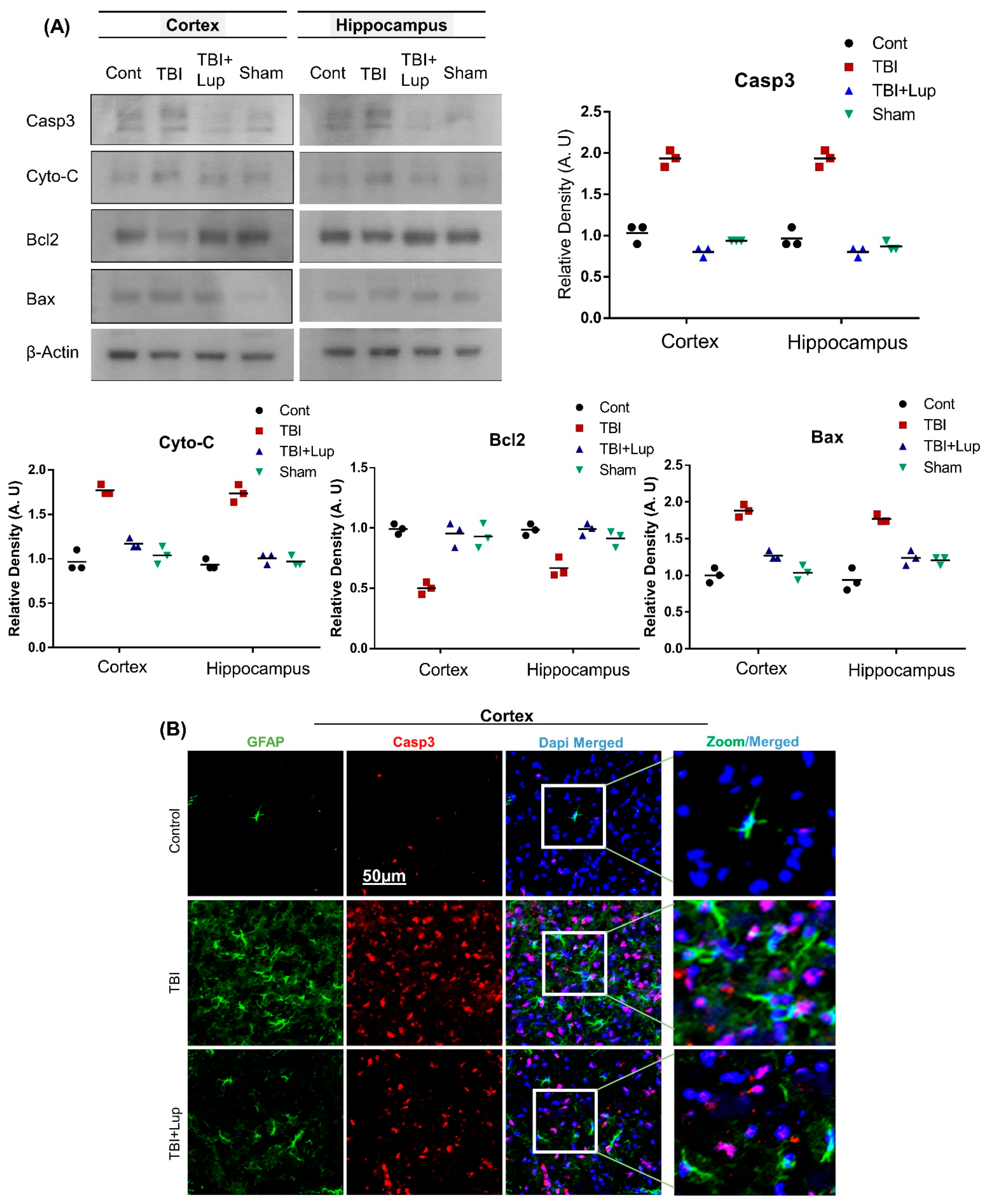
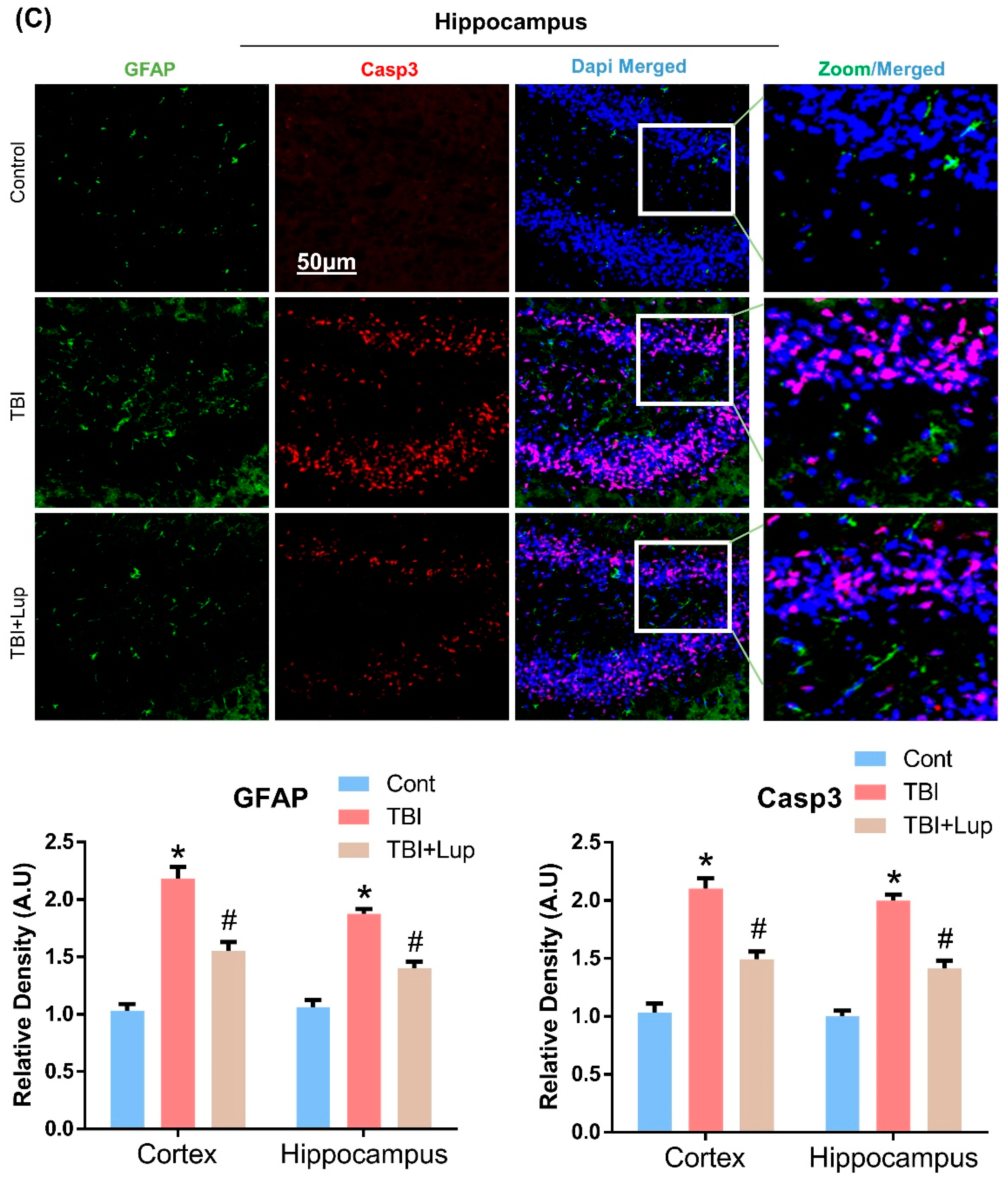
2.4. Lupeol Treatment Attenuated TBI-Induced Apoptotic Cell Death in Mouse Brain
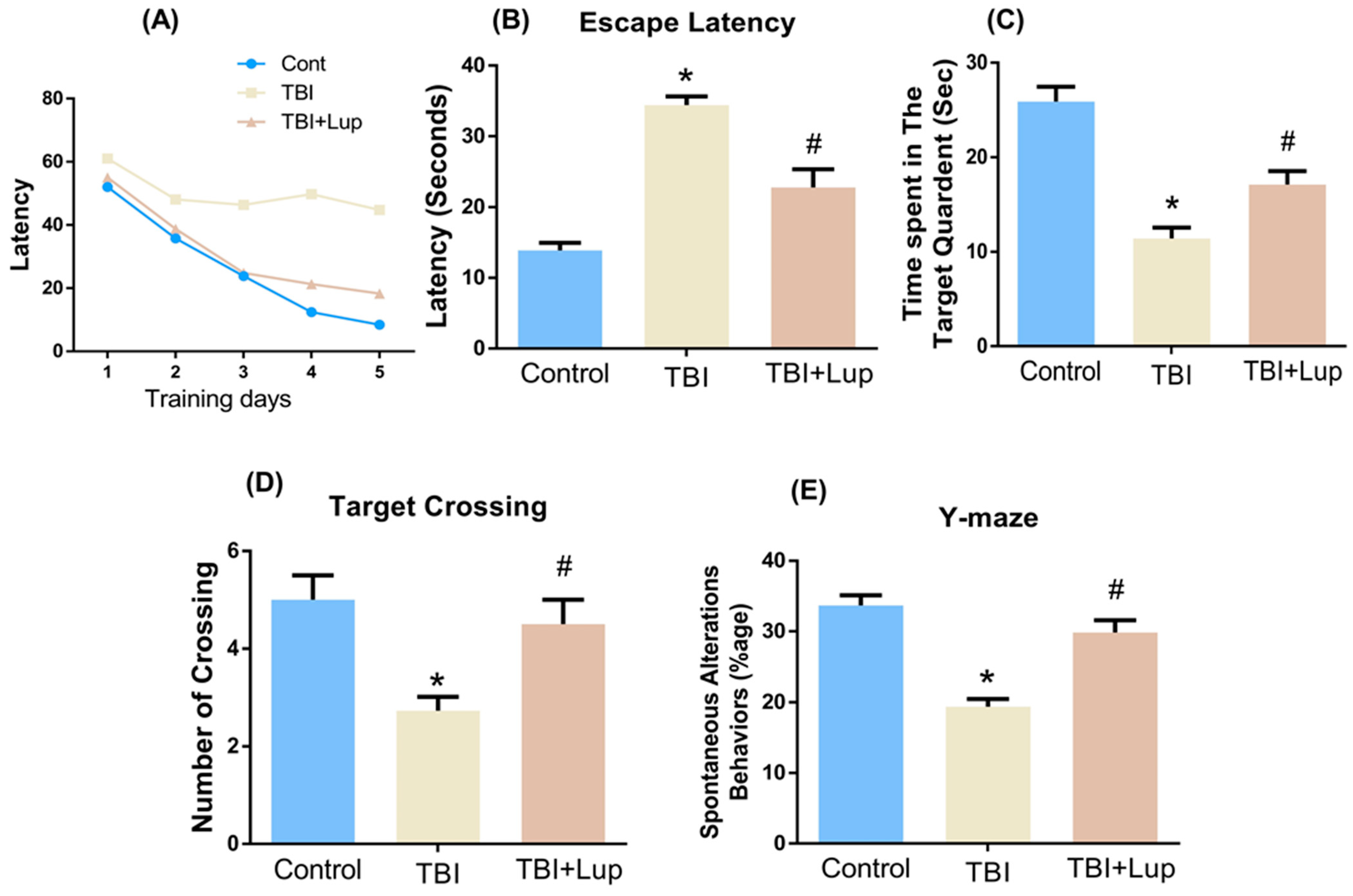
2.5. Lupeol Treatment Improved Learning Memory and Spontaneous Alteration Behavior in TBI-Induced Memory Impairment
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Traumatic Brain Injury of Mice and Drug Treatment
4.3. Morris Water Maze (MWM) Test
4.4. Y-Maze Test
4.5. Homogenization of Mouse Brain
4.6. ROS Assay
4.7. LPO Assay
4.8. Antibodies and Reagents
4.9. Immunoblotting
4.10. Immunofluorescence Analysis
4.11. Statistical Analysis
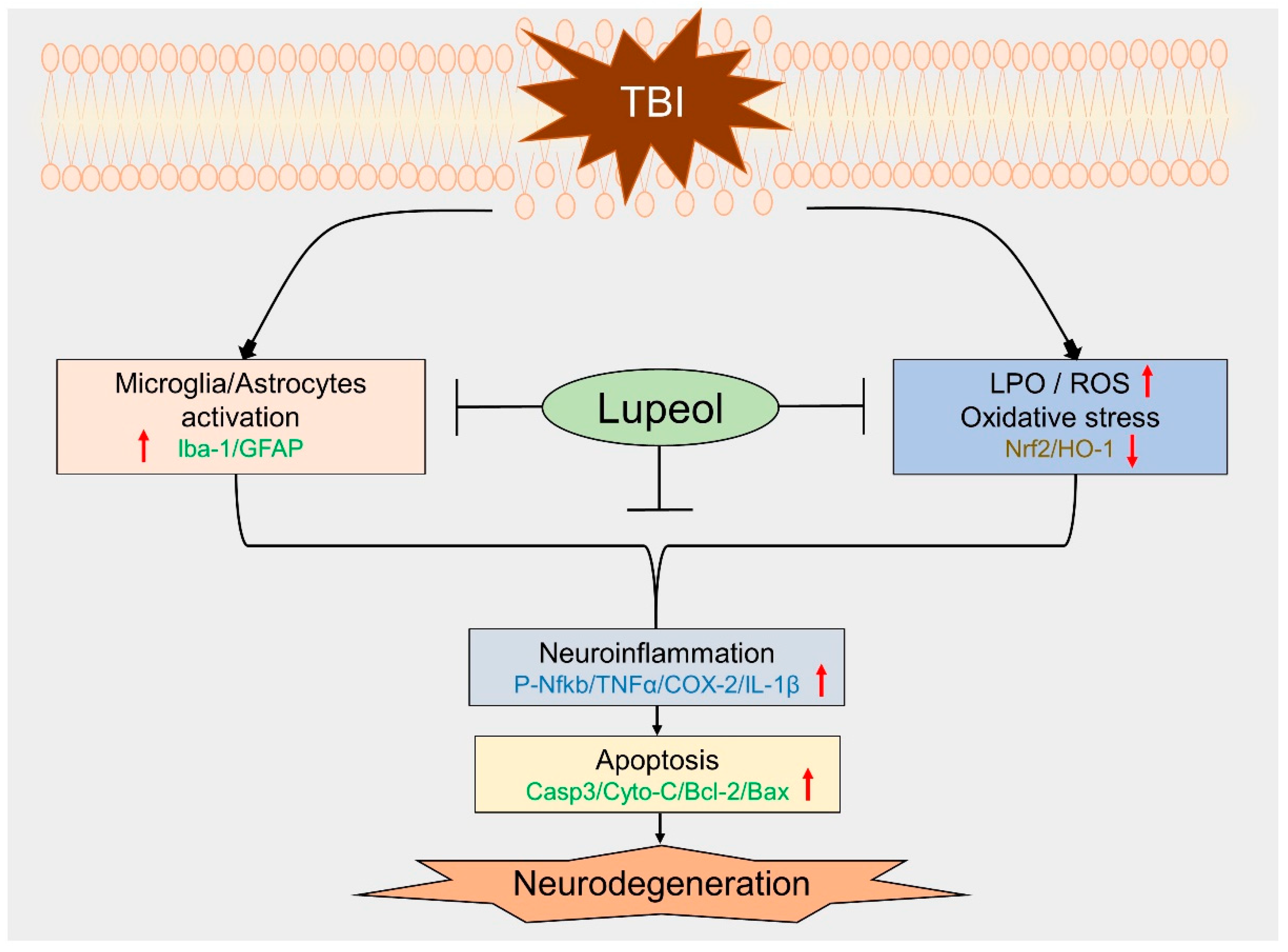
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andelic, N. The epidemiology of traumatic brain injury. Lancet Neurol. 2013, 12, 28–29. [Google Scholar] [CrossRef]
- Rosenfeld, J.V.; Maas, A.I.; Bragge, P.; Morganti-Kossmann, M.C.; Manley, G.T.; Gruen, R.L. Early management of severe traumatic brain injury. Lancet 2012, 380, 1088–1098. [Google Scholar] [CrossRef]
- Faden, A.I.; Wu, J.; Stoica, B.A.; Loane, D.J. Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br. J. Pharmacol. 2016, 173, 681–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, R.C.; Burke, J.F.; Nettiksimmons, J.; Kaup, A.; Barnes, D.E.; Yaffe, K. Dementia risk after traumatic brain injury vs nonbrain trauma: The role of age and severity. JAMA Neurol. 2014, 71, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Mohapatra, S.; Mohapatra, S.S. New perspectives on central and peripheral immune responses to acute traumatic brain injury. J. Neuroinflamm. 2012, 9, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, B.A.; Faden, A.I. Cell death mechanisms and modulation in traumatic brain injury. Neurotherapeutics 2010, 7, 3–12. [Google Scholar] [CrossRef]
- Plesnila, N.; von Baumgarten, L.; Retiounskaia, M.; Engel, D.; Ardeshiri, A.; Zimmermann, R.; Hoffmann, F.; Landshamer, S.; Wagner, E.; Culmsee, C. Delayed neuronal death after brain trauma involves p53-dependent inhibition of NF-kappaB transcriptional activity. Cell Death Differ. 2007, 14, 1529–1541. [Google Scholar] [CrossRef] [Green Version]
- Shafi, S.; Barnes, S.A.; Millar, D.; Sobrino, J.; Kudyakov, R.; Berryman, C.; Rayan, N.; Dubiel, R.; Coimbra, R.; Magnotti, L.J.; et al. Suboptimal compliance with evidence-based guidelines in patients with traumatic brain injuries. J. Neurosurg. 2014, 120, 773–777. [Google Scholar] [CrossRef]
- Timmons, S.D.; Waltzman, D.; Duhaime, A.C.; Spinks, T.J.; Sarmiento, K. Considerations for neurosurgeons: Recommendations from the CDC Pediatric Mild Traumatic Brain Injury Guideline. J. Neurosurg. 2019, 131, 979–983. [Google Scholar] [CrossRef]
- Hutchinson, P.J.; Kolias, A.G.; Timofeev, I.S.; Corteen, E.A.; Czosnyka, M.; Timothy, J.; Anderson, I.; Bulters, D.O.; Belli, A.; Eynon, C.A.; et al. Trial of Decompressive Craniectomy for Traumatic Intracranial Hypertension. N. Engl. J. Med. 2016, 375, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- Dusick, J.R.; Wang, C.; Cohan, P.; Swerdloff, R.; Kelly, D.F. Pathophysiology of hypopituitarism in the setting of brain injury. Pituitary 2012, 15, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichkova, A.; Rodriguez-Grande, B.; Bar, C.; Villega, F.; Konsman, J.P.; Badaut, J. Vascular impairment as a pathological mechanism underlying long-lasting cognitive dysfunction after pediatric traumatic brain injury. Neurochem. Int. 2017, 111, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.P.; Logue, M.W.; Sadeh, N.; Spielberg, J.M.; Verfaellie, M.; Hayes, S.M.; Reagan, A.; Salat, D.H.; Wolf, E.J.; McGlinchey, R.E.; et al. Mild traumatic brain injury is associated with reduced cortical thickness in those at risk for Alzheimer’s disease. Brain 2017, 140, 813–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bains, M.; Hall, E.D. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim. Biophys. Acta 2012, 1822, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Chao, H.; Anthonymuthu, T.S.; Kenny, E.M.; Amoscato, A.A.; Cole, L.K.; Hatch, G.M.; Ji, J.; Kagan, V.E.; Bayir, H. Disentangling oxidation/hydrolysis reactions of brain mitochondrial cardiolipins in pathogenesis of traumatic injury. JCI Insight 2018, 3, e97677. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; Tyurina, Y.Y.; Tang, M.; Feng, W.; Stolz, D.B.; Clark, R.S.; Meaney, D.F.; Kochanek, P.M.; Kagan, V.E.; Bayir, H. Mitochondrial injury after mechanical stretch of cortical neurons in vitro: Biomarkers of apoptosis and selective peroxidation of anionic phospholipids. J. Neurotrauma 2012, 29, 776–788. [Google Scholar] [CrossRef] [Green Version]
- Kabadi, S.V.; Faden, A.I. Neuroprotective strategies for traumatic brain injury: Improving clinical translation. Int. J. Mol. Sci. 2014, 15, 1216–1236. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Chao, H.; Li, Z.; Xu, X.; Liu, Y.; Hou, L.; Liu, N.; Ji, J. Melatonin attenuates traumatic brain injury-induced inflammation: A possible role for mitophagy. J. Pineal Res. 2016, 61, 177–186. [Google Scholar] [CrossRef]
- Menge, T.; Zhao, Y.; Zhao, J.; Wataha, K.; Gerber, M.; Zhang, J.; Letourneau, P.; Redell, J.; Shen, L.; Wang, J.; et al. Mesenchymal stem cells regulate blood-brain barrier integrity through TIMP3 release after traumatic brain injury. Sci. Transl. Med. 2012, 4, 161ra150. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.W.; McGeachy, M.J.; Bayir, H.; Clark, R.S.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J.; Mrak, R.E.; Griffin, W.S. Microglia and neuroinflammation: A pathological perspective. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, T.; Morganti-Kossmann, M.C. The role of markers of inflammation in traumatic brain injury. Front. Neurol. 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, M.L.; Hong, J.S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Loane, D.J.; Kumar, A.; Stoica, B.A.; Cabatbat, R.; Faden, A.I. Progressive neurodegeneration after experimental brain trauma: Association with chronic microglial activation. J. Neuropathol. Exp. Neurol. 2014, 73, 14–29. [Google Scholar] [CrossRef] [Green Version]
- Marklund, N.; Hillered, L. Animal modelling of traumatic brain injury in preclinical drug development: Where do we go from here? Br. J. Pharmacol. 2011, 164, 1207–1229. [Google Scholar] [CrossRef] [Green Version]
- Suliman, H.B.; Piantadosi, C.A. Mitochondrial Quality Control as a Therapeutic Target. Pharmacol. Rev. 2016, 68, 20–48. [Google Scholar] [CrossRef]
- Waldbaum, S.; Patel, M. Mitochondrial dysfunction and oxidative stress: A contributing link to acquired epilepsy? J. Bioenergy Biomembr. 2010, 42, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Clausen, F.; Lundqvist, H.; Ekmark, S.; Lewen, A.; Ebendal, T.; Hillered, L. Oxygen free radical-dependent activation of extracellular signal-regulated kinase mediates apoptosis-like cell death after traumatic brain injury. J. Neurotrauma 2004, 21, 1168–1182. [Google Scholar] [CrossRef]
- Darwish, R.S.; Amiridze, N.; Aarabi, B. Nitrotyrosine as an oxidative stress marker: Evidence for involvement in neurologic outcome in human traumatic brain injury. J. Trauma 2007, 63, 439–442. [Google Scholar] [CrossRef]
- Naseem, M.; Parvez, S. Role of melatonin in traumatic brain injury and spinal cord injury. Sci. World J. 2014, 2014, 586270. [Google Scholar] [CrossRef] [PubMed]
- Fleminger, S.; Oliver, D.L.; Lovestone, S.; Rabe-Hesketh, S.; Giora, A. Head injury as a risk factor for Alzheimer’s disease: The evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 2003, 74, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.C.; Byers, A.L.; Barnes, D.E.; Li, Y.; Boscardin, J.; Yaffe, K. Mild TBI and risk of Parkinson disease: A Chronic Effects of Neurotrauma Consortium Study. Neurology 2018, 90, e1771–e1779. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Hou, S.W.; Lee, C.C.; Hsu, C.Y.; Huang, Y.S.; Su, Y.C. Increased risk of dementia in patients with mild traumatic brain injury: A nationwide cohort study. PLoS ONE 2013, 8, e62422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, S.M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 2006, 147 (Suppl. 1), S232–S240. [Google Scholar] [CrossRef] [Green Version]
- Rehman, S.U.; Shah, S.A.; Ali, T.; Chung, J.I.; Kim, M.O. Anthocyanins Reversed D-Galactose-Induced Oxidative Stress and Neuroinflammation Mediated Cognitive Impairment in Adult Rats. Mol. Neurobiol. 2017, 54, 255–271. [Google Scholar] [CrossRef]
- Gao, H.M.; Hong, J.S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef] [Green Version]
- Griffin, W.S.; Sheng, J.G.; Royston, M.C.; Gentleman, S.M.; McKenzie, J.E.; Graham, D.I.; Roberts, G.W.; Mrak, R.E. Glial-neuronal interactions in Alzheimer’s disease: The potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 1998, 8, 65–72. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Higgins, G.C.; Beart, P.M.; Shin, Y.S.; Chen, M.J.; Cheung, N.S.; Nagley, P. Oxidative stress: Emerging mitochondrial and cellular themes and variations in neuronal injury. J. Alzheimer’s Dis. 2010, 20 (Suppl. 2), S453–S473. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: An insight into cullin-ring ubiquitin ligases. Antioxid. Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, H.E.; Witte, M.; Hondius, D.; Rozemuller, A.J.; Drukarch, B.; Hoozemans, J.; van Horssen, J. Nrf2-induced antioxidant protection: A promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic. Biol. Med. 2008, 45, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Khan, A.; Ahmad, S.; Ahmad, R.; Rehman, I.U.R.; Ikram, M.; Kim, M.O. Inhibition of JNK Alleviates Chronic Hypoperfusion-Related Ischemia Induces Oxidative Stress and Brain Degeneration via Nrf2/HO-1 and NF-kappaB Signaling. Oxid. Med. Cell. Longev. 2020, 2020, 5291852. [Google Scholar] [CrossRef]
- Baierle, M.; Nascimento, S.N.; Moro, A.M.; Brucker, N.; Freitas, F.; Gauer, B.; Durgante, J.; Bordignon, S.; Zibetti, M.; Trentini, C.M.; et al. Relationship between inflammation and oxidative stress and cognitive decline in the institutionalized elderly. Oxid. Med. Cell. Longev. 2015, 2015, 804198. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Rehman, S.U.; Amin, F.U.; Kim, M.O. Enhanced neuroprotection of anthocyanin-loaded PEG-gold nanoparticles against Abeta1-42-induced neuroinflammation and neurodegeneration via the NF-KB /JNK/GSK3beta signaling pathway. Nanomedicine 2017, 13, 2533–2544. [Google Scholar] [CrossRef]
- Schmidt, O.I.; Heyde, C.E.; Ertel, W.; Stahel, P.F. Closed head injury--an inflammatory disease? Brain Res. Brain Res. Rev. 2005, 48, 388–399. [Google Scholar] [CrossRef]
- Holmin, S.; Mathiesen, T. Long-term intracerebral inflammatory response after experimental focal brain injury in rat. Neuroreport 1999, 10, 1889–1891. [Google Scholar] [CrossRef]
- Dalton, T.P.; Shertzer, H.G.; Puga, A. Regulation of gene expression by reactive oxygen. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 67–101. [Google Scholar] [CrossRef]
- Glushakov, A.O.; Glushakova, O.Y.; Korol, T.Y.; Acosta, S.A.; Borlongan, C.V.; Valadka, A.B.; Hayes, R.L.; Glushakov, A.V. Chronic Upregulation of Cleaved-Caspase-3 Associated with Chronic Myelin Pathology and Microvascular Reorganization in the Thalamus after Traumatic Brain Injury in Rats. Int. J. Mol. Sci. 2018, 19, 3151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilke, S.; Prehn, K.; Taud, B.; List, J.; Floel, A. Multimodal Assessment of Recurrent MTBI across the Lifespan. J. Clin. Med. 2018, 7, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, S.U.; Ali, T.; Alam, S.I.; Ullah, R.; Zeb, A.; Lee, K.W.; Rutten, B.P.F.; Kim, M.O. Ferulic Acid Rescues LPS-Induced Neurotoxicity via Modulation of the TLR4 Receptor in the Mouse Hippocampus. Mol. Neurobiol. 2019, 56, 2774–2790. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Zheng, P.; Tan, X.; Wang, Y.; Situ, B. Huperzine A alleviates neuroinflammation, oxidative stress and improves cognitive function after repetitive traumatic brain injury. Metab. Brain Dis. 2017, 32, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Choi, D.Y.; Choi, I.S.; Kim, K.H.; Kim, Y.H.; Kim, H.M.; Lee, K.; Cho, W.G.; Jung, J.K.; Han, S.B.; et al. Inhibitory effect of 4-O-methylhonokiol on lipopolysaccharide-induced neuroinflammation, amyloidogenesis and memory impairment via inhibition of nuclear factor-kappaB in vitro and in vivo models. J. Neuroinflamm. 2012, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Raghupathi, R.; Graham, D.I.; McIntosh, T.K. Apoptosis after traumatic brain injury. J. Neurotrauma 2000, 17, 927–938. [Google Scholar] [CrossRef]
- Alam, S.I.; Rehman, S.U.; Kim, M.O. Nicotinamide Improves Functional Recovery via Regulation of the RAGE/JNK/NF-kappaB Signaling Pathway after Brain Injury. J. Clin. Med. 2019, 8, 271. [Google Scholar] [CrossRef] [Green Version]
- Loane, D.J.; Stoica, B.A.; Faden, A.I. Neuroprotection for traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 343–366. [Google Scholar] [CrossRef] [Green Version]
- Sharifi-Rad, M.; Lankatillake, C.; Dias, D.A.; Docea, A.O.; Mahomoodally, M.F.; Lobine, D.; Chazot, P.L.; Kurt, B.; Tumer, T.B.; Moreira, A.C.; et al. Impact of Natural Compounds on Neurodegenerative Disorders: From Preclinical to Pharmacotherapeutics. J. Clin. Med. 2020, 9, 1061. [Google Scholar] [CrossRef] [Green Version]
- Scheff, S.W.; Ansari, M.A. Natural Compounds as a Therapeutic Intervention following Traumatic Brain Injury: The Role of Phytochemicals. J. Neurotrauma 2017, 34, 1491–1510. [Google Scholar] [CrossRef] [Green Version]
- Beserra, F.P.; Vieira, A.J.; Gushiken, L.F.S.; de Souza, E.O.; Hussni, M.F.; Hussni, C.A.; Nobrega, R.H.; Martinez, E.R.M.; Jackson, C.J.; de Azevedo Maia, G.L.; et al. Lupeol, a Dietary Triterpene, Enhances Wound Healing in Streptozotocin-Induced Hyperglycemic Rats with Modulatory Effects on Inflammation, Oxidative Stress, and Angiogenesis. Oxid. Med. Cell. Longev. 2019, 2019, 3182627. [Google Scholar] [CrossRef] [PubMed]
- Hajialyani, M.; Hosein Farzaei, M.; Echeverria, J.; Nabavi, S.M.; Uriarte, E.; Sobarzo-Sanchez, E. Hesperidin as a Neuroprotective Agent: A Review of Animal and Clinical Evidence. Molecules 2019, 24, 648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurya, S.K.; Shadab, G.; Siddique, H.R. Chemosensitization of Therapy Resistant Tumors: Targeting Multiple Cell Signaling Pathways by Lupeol, a Pentacyclic Triterpene. Curr. Pharm. Des. 2020, 26, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Lee, J.W.; Seo, J.Y.; Hwang, S.W.; Im, J.P.; Kim, J.S. Lupeol inhibits LPS-induced NF-kappa B signaling in intestinal epithelial cells and macrophages, and attenuates acute and chronic murine colitis. Life Sci. 2016, 146, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Saleem, M. Lupeol, a novel anti-inflammatory and anti-cancer dietary triterpene. Cancer Lett. 2009, 285, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, M.A.; de las Heras, B.; Garcia, M.D.; Saenz, M.T.; Villar, A. New insights into the mechanism of action of the anti-inflammatory triterpene lupeol. J. Pharm. Pharmacol. 2001, 53, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Khan, A.; Lee, H.J.; Ur Rehman, I.; Khan, I.; Alam, S.I.; Kim, M.O. Lupeol, a Plant-Derived Triterpenoid, Protects Mouse brains against Abeta-Induced Oxidative Stress and Neurodegeneration. Biomedicines 2020, 8, 380. [Google Scholar] [CrossRef]
- Badshah, H.; Ali, T.; Shafiq-ur, R.; Faiz-ul, A.; Ullah, F.; Kim, T.H.; Kim, M.O. Protective Effect of Lupeol Against Lipopolysaccharide-Induced Neuroinflammation via the p38/c-Jun N-Terminal Kinase Pathway in the Adult Mouse Brain. J. Neuroimmune Pharmacol. 2016, 11, 48–60. [Google Scholar] [CrossRef]
- Kumari, A.; Kakkar, P. Lupeol prevents acetaminophen-induced in vivo hepatotoxicity by altering the Bax/Bcl-2 and oxidative stress-mediated mitochondrial signaling cascade. Life Sci. 2012, 90, 561–570. [Google Scholar] [CrossRef]
- Kumari, A.; Kakkar, P. Lupeol protects against acetaminophen-induced oxidative stress and cell death in rat primary hepatocytes. Food Chem. Toxicol. 2012, 50, 1781–1789. [Google Scholar] [CrossRef]
- Eyolfson, E.; Khan, A.; Mychasiuk, R.; Lohman, A.W. Microglia dynamics in adolescent traumatic brain injury. J. Neuroinflamm. 2020, 17, 326. [Google Scholar] [CrossRef] [PubMed]
- Bylicky, M.A.; Mueller, G.P.; Day, R.M. Mechanisms of Endogenous Neuroprotective Effects of Astrocytes in Brain Injury. Oxid. Med. Cell. Longev. 2018, 2018, 6501031. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shao, A.; Yao, Y.; Tu, S.; Deng, Y.; Zhang, J. Dual roles of astrocytes in plasticity and reconstruction after traumatic brain injury. Cell Commun. Signal. 2020, 18, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef] [Green Version]
- d’Avila, J.C.; Lam, T.I.; Bingham, D.; Shi, J.; Won, S.J.; Kauppinen, T.M.; Massa, S.; Liu, J.; Swanson, R.A. Microglial activation induced by brain trauma is suppressed by post-injury treatment with a PARP inhibitor. J. Neuroinflamm. 2012, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxid. Med. Cell. Longev. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [Green Version]
- Wofford, K.L.; Loane, D.J.; Cullen, D.K. Acute drivers of neuroinflammation in traumatic brain injury. Neural Regen. Res. 2019, 14, 1481–1489. [Google Scholar] [CrossRef]
- Lozano, D.; Gonzales-Portillo, G.S.; Acosta, S.; de la Pena, I.; Tajiri, N.; Kaneko, Y.; Borlongan, C.V. Neuroinflammatory responses to traumatic brain injury: Etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr. Dis. Treat. 2015, 11, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Kempuraj, D.; Ahmed, M.E.; Selvakumar, G.P.; Thangavel, R.; Raikwar, S.P.; Zaheer, S.A.; Iyer, S.S.; Govindarajan, R.; Nattanmai Chandrasekaran, P.; Burton, C.; et al. Acute Traumatic Brain Injury-Induced Neuroinflammatory Response and Neurovascular Disorders in the Brain. Neurotox. Res. 2021, 39, 359–368. [Google Scholar] [CrossRef]
- Memet, S. NF-kappaB functions in the nervous system: From development to disease. Biochem. Pharmacol. 2006, 72, 1180–1195. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghupathi, R.; Conti, A.C.; Graham, D.I.; Krajewski, S.; Reed, J.C.; Grady, M.S.; Trojanowski, J.Q.; McIntosh, T.K. Mild traumatic brain injury induces apoptotic cell death in the cortex that is preceded by decreases in cellular Bcl-2 immunoreactivity. Neuroscience 2002, 110, 605–616. [Google Scholar] [CrossRef]
- Hall, E.D.; Vaishnav, R.A.; Mustafa, A.G. Antioxidant therapies for traumatic brain injury. Neurotherapeutics 2010, 7, 51–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Kong, R.H.; Zhang, L.M.; Zhang, J.N. Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br. J. Pharmacol. 2012, 167, 699–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, Z.Z.; Li, F.; Maiese, K. Oxidative stress in the brain: Novel cellular targets that govern survival during neurodegenerative disease. Prog. Neurobiol. 2005, 75, 207–246. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.N.; Sullivan, P.G.; Hall, E.D. Peroxynitrite-mediated oxidative damage to brain mitochondria: Protective effects of peroxynitrite scavengers. J. Neurosci. Res. 2007, 85, 2216–2223. [Google Scholar] [CrossRef]
- Mustafa, A.G.; Wang, J.A.; Carrico, K.M.; Hall, E.D. Pharmacological inhibition of lipid peroxidation attenuates calpain-mediated cytoskeletal degradation after traumatic brain injury. J. Neurochem. 2011, 117, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Sekhar, K.R.; Yan, X.X.; Freeman, M.L. Nrf2 degradation by the ubiquitin proteasome pathway is inhibited by KIAA0132, the human homolog to INrf2. Oncogene 2002, 21, 6829–6834. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, S.; D’Mello, V.; Caruso, D.; Abdul-Muneer, P.M. Traumatic brain injury-induced downregulation of Nrf2 activates inflammatory response and apoptotic cell death. J. Mol. Med. 2019, 97, 1627–1641. [Google Scholar] [CrossRef]
- Graham, S.H.; Chen, J.; Clark, R.S. Bcl-2 family gene products in cerebral ischemia and traumatic brain injury. J. Neurotrauma 2000, 17, 831–841. [Google Scholar] [CrossRef]
- Mancuso, C. Heme oxygenase and its products in the nervous system. Antioxid. Redox Signal. 2004, 6, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.Q.; Zhou, J.W. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 2019, 67, 1017–1035. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.N.; Yang, L.Y.; Greig, N.H.; Wang, Y.C.; Lai, C.C.; Wang, J.Y. Neuroprotective effects of pifithrin-alpha against traumatic brain injury in the striatum through suppression of neuroinflammation, oxidative stress, autophagy, and apoptosis. Sci. Rep. 2018, 8, 2368. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mao, H.; Yang, K.H.; Abel, T.; Meaney, D.F. A modified controlled cortical impact technique to model mild traumatic brain injury mechanics in mice. Front. Neurol. 2014, 5, 100. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Wang, Z.; Li, J.; Wu, H.; Peng, Y.; Fan, L.; Chen, J.; Gu, C.; Yan, F.; Wang, L.; et al. The Polarization States of Microglia in TBI: A New Paradigm for Pharmacological Intervention. Neural Plast. 2017, 2017, 5405104. [Google Scholar] [CrossRef]
- Witcher, K.G.; Bray, C.E.; Dziabis, J.E.; McKim, D.B.; Benner, B.N.; Rowe, R.K.; Kokiko-Cochran, O.N.; Popovich, P.G.; Lifshitz, J.; Eiferman, D.S.; et al. Traumatic brain injury-induced neuronal damage in the somatosensory cortex causes formation of rod-shaped microglia that promote astrogliosis and persistent neuroinflammation. Glia 2018, 66, 2719–2736. [Google Scholar] [CrossRef]
- Laird, M.D.; Shields, J.S.; Sukumari-Ramesh, S.; Kimbler, D.E.; Fessler, R.D.; Shakir, B.; Youssef, P.; Yanasak, N.; Vender, J.R.; Dhandapani, K.M. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia 2014, 62, 26–38. [Google Scholar] [CrossRef] [Green Version]
- Ullah, R.; Jo, M.H.; Riaz, M.; Alam, S.I.; Saeed, K.; Ali, W.; Rehman, I.U.; Ikram, M.; Kim, M.O. Glycine, the smallest amino acid, confers neuroprotection against D-galactose-induced neurodegeneration and memory impairment by regulating c-Jun N-terminal kinase in the mouse brain. J. Neuroinflamm. 2020, 17, 303. [Google Scholar] [CrossRef]
- Mira, R.G.; Lira, M.; Cerpa, W. Traumatic Brain Injury: Mechanisms of Glial Response. Front. Physiol. 2021, 12, 740939. [Google Scholar] [CrossRef]
- Kumar, A.; Loane, D.J. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain Behav. Immun. 2012, 26, 1191–1201. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFkappaB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayakumar, A.R.; Tong, X.Y.; Ruiz-Cordero, R.; Bregy, A.; Bethea, J.R.; Bramlett, H.M.; Norenberg, M.D. Activation of NF-kappaB mediates astrocyte swelling and brain edema in traumatic brain injury. J. Neurotrauma 2014, 31, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflamm. 2008, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttolomondo, A.; Pecoraro, R.; Pinto, A. Studies of selective TNF inhibitors in the treatment of brain injury from stroke and trauma: A review of the evidence to date. Drug Des. Dev. Ther. 2014, 8, 2221–2238. [Google Scholar] [CrossRef] [Green Version]
- Shohami, E.; Bass, R.; Wallach, D.; Yamin, A.; Gallily, R. Inhibition of tumor necrosis factor alpha (TNFalpha) activity in rat brain is associated with cerebroprotection after closed head injury. J. Cereb. Blood Flow Metab. 1996, 16, 378–384. [Google Scholar] [CrossRef] [Green Version]
- Ullah, R.; Park, T.J.; Huang, X.; Kim, M.O. Abnormal amyloid beta metabolism in systemic abnormalities and Alzheimer’s pathology: Insights and therapeutic approaches from periphery. Ageing Res. Rev. 2021, 71, 101451. [Google Scholar] [CrossRef]
- Jantzen, P.T.; Connor, K.E.; DiCarlo, G.; Wenk, G.L.; Wallace, J.L.; Rojiani, A.M.; Coppola, D.; Morgan, D.; Gordon, M.N. Microglial activation and beta -amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J. Neurosci. 2002, 22, 2246–2254. [Google Scholar] [CrossRef]
- Jellinger, K.A. Head injury and dementia. Curr. Opin. Neurol. 2004, 17, 719–723. [Google Scholar] [CrossRef]
- Pasinetti, G.M.; Aisen, P.S. Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer’s disease brain. Neuroscience 1998, 87, 319–324. [Google Scholar] [CrossRef]
- Ali, T.; Kim, M.O. Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegeneration via PI3/Akt/GSk3beta pathway in the mouse hippocampus. J. Pineal Res. 2015, 59, 47–59. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, K.; Chatzipanteli, K.; Vitarbo, E.; Truettner, J.S.; Alonso, O.F.; Dietrich, W.D. Interleukin-1beta messenger ribonucleic acid and protein levels after fluid-percussion brain injury in rats: Importance of injury severity and brain temperature. Neurosurgery 2002, 51, 195–203; discussion 203. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Harte, M.T.; Kluck, R.M.; Wolf, B.B.; Casiano, C.A.; Newmeyer, D.D.; Wang, H.G.; Reed, J.C.; Nicholson, D.W.; Alnemri, E.S.; et al. Ordering the cytochrome c-initiated caspase cascade: Hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J. Cell Biol. 1999, 144, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Ang, B.T.; Yap, E.; Lim, J.; Tan, W.L.; Ng, P.Y.; Ng, I.; Yeo, T.T. Poly(adenosine diphosphate-ribose) polymerase expression in human traumatic brain injury. J. Neurosurg. 2003, 99, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Badshah, H.; Kim, T.H.; Kim, M.O. Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-K B/JNK signaling pathway in aging mouse model. J. Pineal Res. 2015, 58, 71–85. [Google Scholar] [CrossRef]
- Ali, T.; Rehman, S.U.; Shah, F.A.; Kim, M.O. Acute dose of melatonin via Nrf2 dependently prevents acute ethanol-induced neurotoxicity in the developing rodent brain. J. Neuroinflamm. 2018, 15, 119. [Google Scholar] [CrossRef]
- Harris, M.H.; Thompson, C.B. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000, 7, 1182–1191. [Google Scholar] [CrossRef]
- Raghupathi, R.; Strauss, K.I.; Zhang, C.; Krajewski, S.; Reed, J.C.; McIntosh, T.K. Temporal alterations in cellular Bax:Bcl-2 ratio following traumatic brain injury in the rat. J. Neurotrauma 2003, 20, 421–435. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.; Ali, T.; Kim, M.W.; Khan, A.; Jo, M.H.; Rehman, S.U.; Khan, M.S.; Abid, N.B.; Khan, M.; Ullah, R.; et al. Adiponectin homolog novel osmotin protects obesity/diabetes-induced NAFLD by upregulating AdipoRs/PPARalpha signaling in ob/ob and db/db transgenic mouse models. Metab. Clin. Exp. 2019, 90, 31–43. [Google Scholar] [CrossRef]
- Rehman, S.U.; Ikram, M.; Ullah, N.; Alam, S.I.; Park, H.Y.; Badshah, H.; Choe, K.; Kim, M.O. Neurological Enhancement Effects of Melatonin against Brain Injury-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration via AMPK/CREB Signaling. Cells 2019, 8, 760. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a Citrus Flavonoid, Attenuates LPS-Induced Neuroinflammation, Apoptosis and Memory Impairments by Modulating TLR4/NF-kappaB Signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.; Ali, T.; Rehman, S.U.; Khan, M.S.; Alam, S.I.; Ikram, M.; Muhammad, T.; Saeed, K.; Badshah, H.; Kim, M.O. Neuroprotective Effect of Quercetin Against the Detrimental Effects of LPS in the Adult Mouse Brain. Front. Pharmacol. 2018, 9, 1383. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Yoon, G.H.; Chung, S.S.; Abid, M.N.; Kim, T.H.; Lee, H.Y.; Kim, M.O. Novel osmotin inhibits SREBP2 via the AdipoR1/AMPK/SIRT1 pathway to improve Alzheimer’s disease neuropathological deficits. Mol. Psychiatry 2017, 22, 407–416. [Google Scholar] [CrossRef]
- Amin, F.U.; Shah, S.A.; Badshah, H.; Khan, M.; Kim, M.O. Anthocyanins encapsulated by PLGA@PEG nanoparticles potentially improved its free radical scavenging capabilities via p38/JNK pathway against Abeta1-42-induced oxidative stress. J. Nanobiotechnol. 2017, 15, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, T.; Kim, M.J.; Rehman, S.U.; Ahmad, A.; Kim, M.O. Anthocyanin-Loaded PEG-Gold Nanoparticles Enhanced the Neuroprotection of Anthocyanins in an Abeta1-42 Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 6490–6506. [Google Scholar] [CrossRef]
- Zhou, P.; Xie, W.; Meng, X.; Zhai, Y.; Dong, X.; Zhang, X.; Sun, G.; Sun, X. Notoginsenoside R1 Ameliorates Diabetic Retinopathy through PINK1-Dependent Activation of Mitophagy. Cells 2019, 8, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badshah, H.; Ali, T.; Kim, M.O. Osmotin attenuates LPS-induced neuroinflammation and memory impairments via the TLR4/NFkappaB signaling pathway. Sci. Rep. 2016, 6, 24493. [Google Scholar] [CrossRef]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef]
- Ali, T.; Rehman, S.U.; Khan, A.; Badshah, H.; Abid, N.B.; Kim, M.W.; Jo, M.H.; Chung, S.S.; Lee, H.G.; Rutten, B.P.F.; et al. Adiponectin-mimetic novel nonapeptide rescues aberrant neuronal metabolic-associated memory deficits in Alzheimer’s disease. Mol. Neurodegener. 2021, 16, 23. [Google Scholar] [CrossRef]
- Park, J.S.; Kim, S.T.; Kim, S.Y.; Jo, M.G.; Choi, M.J.; Kim, M.O. A novel kit for early diagnosis of Alzheimer’s disease using a fluorescent nanoparticle imaging. Sci. Rep. 2019, 9, 13184. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, R.; Khan, A.; Rehman, I.U.; Lee, H.J.; Khan, I.; Kim, M.O. Lupeol Treatment Attenuates Activation of Glial Cells and Oxidative-Stress-Mediated Neuropathology in Mouse Model of Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 23, 6086. https://doi.org/10.3390/ijms23116086
Ahmad R, Khan A, Rehman IU, Lee HJ, Khan I, Kim MO. Lupeol Treatment Attenuates Activation of Glial Cells and Oxidative-Stress-Mediated Neuropathology in Mouse Model of Traumatic Brain Injury. International Journal of Molecular Sciences. 2022; 23(11):6086. https://doi.org/10.3390/ijms23116086
Chicago/Turabian StyleAhmad, Riaz, Amjad Khan, Inayat Ur Rehman, Hyeon Jin Lee, Ibrahim Khan, and Myeong Ok Kim. 2022. "Lupeol Treatment Attenuates Activation of Glial Cells and Oxidative-Stress-Mediated Neuropathology in Mouse Model of Traumatic Brain Injury" International Journal of Molecular Sciences 23, no. 11: 6086. https://doi.org/10.3390/ijms23116086