
Inactivation of Osteoblast PKC Signaling Reduces Cortical Bone Mass and Density and Aggravates Renal Osteodystrophy in Mice with Chronic Kidney Disease on High Phosphate Diet

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Experimental Results
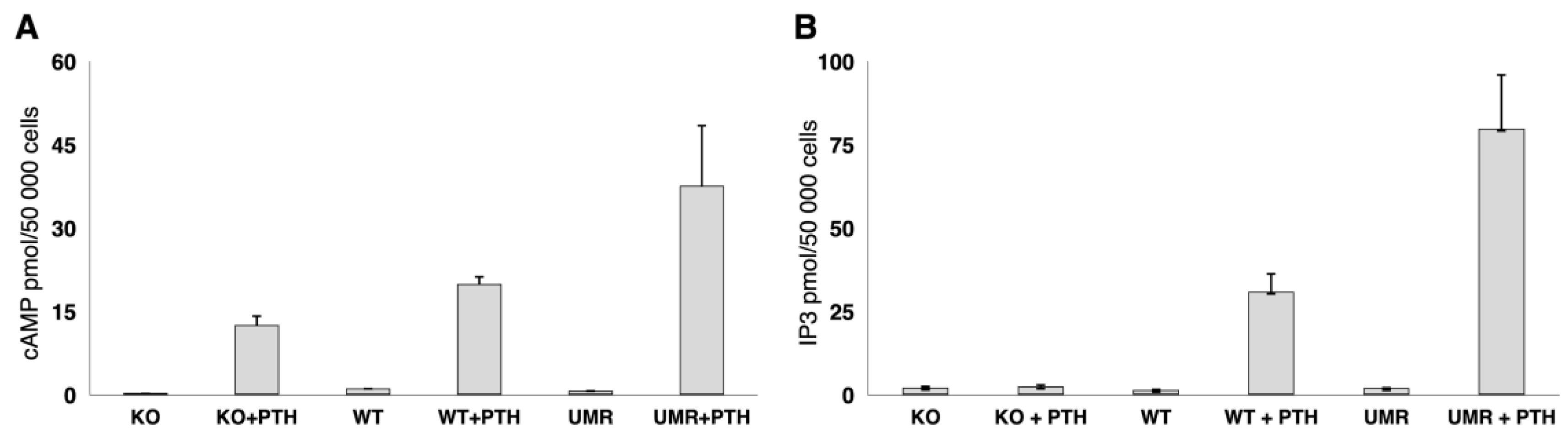
2.1.1. Osteoblast Gαq/11 Expression and PTH Response
2.1.2. Body Weight, Mortality, and Serum Biochemistry
- Body Weight:
- Mortality:
- Biochemistry:
2.1.3. Bone Morphology (µCT)
2.1.4. Bone Planar Scintigraphy Analysis
2.1.5. Bone Histomorphometry
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cells Cultures
4.3. Immunostaining
4.4. Cyclic AMP and IP3 Assay
4.5. Blood Chemistry Analysis
4.6. Micro-CT Analysis
4.7. Bone Planar Scintigraphy Analysis
4.8. Histomorphometric Analysis
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Drueke, T.B. Hyperparathyroidism in Chronic Kidney Disease. In Endotext; De Groot, L.J., Chrousos, G., Dungan, K., Feingold, K.R., Grossman, A., Hershman, J.M., Koch, C., Korbonits, M., McLachlan, R., New, M., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Hruska, K.A.; Choi, E.T.; Memon, I.; Davis, T.K.; Mathew, S. Cardiovascular risk in chronic kidney disease (CKD): The CKD-mineral bone disorder (CKD-MBD). Pediatr. Nephrol. 2010, 25, 769–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hruska, K.A.; Sugatani, T.; Agapova, O.; Fang, Y. The chronic kidney disease—Mineral bone disorder (CKD-MBD): Advances in pathophysiology. Bone 2017, 100, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.; Drueke, T.; Cunningham, J.; Goodman, W.; Martin, K.; Olgaard, K.; Ott, S.; Sprague, S.; Lameire, N.; Eknoyan, G.; et al. Definition, evaluation, and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2006, 69, 1945–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozzolino, M.; Urena-Torres, P.; Vervloet, M.G.; Brandenburg, V.; Bover, J.; Goldsmith, D.; Larsson, T.E.; Massy, Z.A.; Mazzaferro, S.; CKD-MBD Working Group of ERA-EDTA. Is chronic kidney disease-mineral bone disorder (CKD-MBD) really a syndrome? Nephrol. Dial. Transpl. 2014, 29, 1815–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, R.N. Clinical epidemiology of cardiovascular disease in chronic kidney disease. J. Ren. Care 2010, 36 (Suppl. 1), 4–8. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.A.; Djurdjev, O.; Cardew, S.; Cameron, E.C.; Levin, A. Calcium, phosphate, and parathyroid hormone levels in combination and as a function of dialysis duration predict mortality: Evidence for the complexity of the association between mineral metabolism and outcomes. J. Am. Soc. Nephrol. 2004, 15, 770–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacchetta, J.; Harambat, J.; Cochat, P.; Salusky, I.B.; Wesseling-Perry, K. The consequences of chronic kidney disease on bone metabolism and growth in children. Nephrol. Dial. Transpl. 2012, 27, 3063–3071. [Google Scholar] [CrossRef] [Green Version]
- Ziolkowska, H.; Paniczyk-Tomaszewska, M.; Debinski, A.; Polowiec, Z.; Sawicki, A.; Sieniawska, M. Bone biopsy results and serum bone turnover parameters in uremic children. Acta Paediatr. 2000, 89, 666–671. [Google Scholar] [CrossRef]
- Denburg, M.R.; Kumar, J.; Jemielita, T.; Brooks, E.R.; Skversky, A.; Portale, A.A.; Salusky, I.B.; Warady, B.A.; Furth, S.L.; Leonard, M.B. Fracture Burden and Risk Factors in Childhood CKD: Results from the CKiD Cohort Study. J. Am. Soc. Nephrol. 2016, 27, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Leonard, M.B. A structural approach to skeletal fragility in chronic kidney disease. Semin. Nephrol. 2009, 29, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Drueke, T.B.; Massy, Z.A. Changing bone patterns with progression of chronic kidney disease. Kidney Int. 2016, 89, 289–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, S.C.; Mavridis, D.; Johnson, D.W.; Tonelli, M.; Ruospo, M.; Strippoli, G.F.M. Comparative Effectiveness of Calcimimetic Agents for Secondary Hyperparathyroidism in Adults: A Systematic Review and Network Meta-analysis. Am. J. Kidney Dis. 2020, 76, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Bastepe, M.; Turan, S.; He, Q. Heterotrimeric G proteins in the control of parathyroid hormone actions. J. Mol. Endocrinol. 2017, 58, R203–R224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juppner, H.; Abou-Samra, A.B.; Freeman, M.; Kong, X.F.; Schipani, E.; Richards, J.; Kolakowski, L.F., Jr.; Hock, J.; Potts, J.T., Jr.; Kronenberg, H.M.; et al. A G protein-linked receptor for parathyroid hormone and parathyroid hormone-related peptide. Science 1991, 254, 1024–1026. [Google Scholar] [CrossRef]
- Cheloha, R.W.; Gellman, S.H.; Vilardaga, J.P.; Gardella, T.J. PTH receptor-1 signalling-mechanistic insights and therapeutic prospects. Nat. Rev. Endocrinol. 2015, 11, 712–724. [Google Scholar] [CrossRef] [Green Version]
- Swarthout, J.T.; D’Alonzo, R.C.; Selvamurugan, N.; Partridge, N.C. Parathyroid hormone-dependent signaling pathways regulating genes in bone cells. Gene 2002, 282, 1–17. [Google Scholar] [CrossRef]
- Hsiao, E.C.; Boudignon, B.M.; Chang, W.C.; Bencsik, M.; Peng, J.; Nguyen, T.D.; Manalac, C.; Halloran, B.P.; Conklin, B.R.; Nissenson, R.A. Osteoblast expression of an engineered Gs-coupled receptor dramatically increases bone mass. Proc. Natl. Acad. Sci. USA 2008, 105, 1209–1214. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, E.C.; Boudignon, B.M.; Halloran, B.P.; Nissenson, R.A.; Conklin, B.R. Gs G protein-coupled receptor signaling in osteoblasts elicits age-dependent effects on bone formation. J. Bone Min. Res. 2010, 25, 584–593. [Google Scholar] [CrossRef] [Green Version]
- Kao, R.S.; Abbott, M.J.; Louie, A.; O’Carroll, D.; Lu, W.; Nissenson, R. Constitutive protein kinase A activity in osteocytes and late osteoblasts produces an anabolic effect on bone. Bone 2013, 55, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Saggio, I.; Remoli, C.; Spica, E.; Cersosimo, S.; Sacchetti, B.; Robey, P.G.; Holmbeck, K.; Cumano, A.; Boyde, A.; Bianco, P.; et al. Constitutive expression of Gsalpha(R201C) in mice produces a heritable, direct replica of human fibrous dysplasia bone pathology and demonstrates its natural history. J. Bone Min. Res. 2014, 29, 2357–2368. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, L.S.; Gejman, P.V.; Friedman, E.; Kadowaki, T.; Collins, R.M.; Gershon, E.S.; Spiegel, A.M. Mutations of the Gs alpha-subunit gene in Albright hereditary osteodystrophy detected by denaturing gradient gel electrophoresis. Proc. Natl. Acad. Sci. USA 1990, 87, 8287–8290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinstein, L.S.; Shenker, A.; Gejman, P.V.; Merino, M.J.; Friedman, E.; Spiegel, A.M. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N. Engl. J. Med. 1991, 325, 1688–1695. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, M.; Tong, G.; Meng, Y.; Hao, S.; Hu, S.; Yan, W.; Yang, D. hPTH(3-34)(29-34) selectively activated PKC and mimicked osteoanabolic effects of hPTH(1-34). Bone 2020, 135, 115326. [Google Scholar] [CrossRef] [PubMed]
- Dela Cruz, A.; Mattocks, M.; Sugamori, K.S.; Grynpas, M.D.; Mitchell, J. Reduced trabecular bone mass and strength in mice overexpressing Galpha11 protein in cells of the osteoblast lineage. Bone 2014, 59, 211–222. [Google Scholar] [CrossRef]
- Ogata, N.; Kawaguchi, H.; Chung, U.I.; Roth, S.I.; Segre, G.V. Continuous activation of G alpha q in osteoblasts results in osteopenia through impaired osteoblast differentiation. J. Biol. Chem. 2007, 282, 35757–35764. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Chung, U.I.; Kondo, H.; Bringhurst, F.R.; Kronenberg, H.M. The PTH/PTHrP receptor can delay chondrocyte hypertrophy in vivo without activating phospholipase C. Dev. Cell 2002, 3, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Liu, M.; Yang, D.; Bouxsein, M.L.; Thomas, C.C.; Schipani, E.; Bringhurst, F.R.; Kronenberg, H.M. Phospholipase C signaling via the parathyroid hormone (PTH)/PTH-related peptide receptor is essential for normal bone responses to PTH. Endocrinology 2010, 151, 3502–3513. [Google Scholar] [CrossRef] [Green Version]
- Ogata, N.; Shinoda, Y.; Wettschureck, N.; Offermanns, S.; Takeda, S.; Nakamura, K.; Segre, G.V.; Chung, U.I.; Kawaguchi, H. G alpha(q) signal in osteoblasts is inhibitory to the osteoanabolic action of parathyroid hormone. J. Biol. Chem. 2011, 286, 13733–13740. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Solal, M.; Funck-Brentano, T.; Ureña Torres, P. Bone fragility in patients with chronic kidney disease. Endocr. Connect. 2020, 9, R93–R101. [Google Scholar] [CrossRef] [Green Version]
- Borzych, D.; Rees, L.; Ha, I.S.; Chua, A.; Valles, P.G.; Lipka, M.; Zambrano, P.; Ahlenstiel, T.; Bakkaloglu, S.A.; Spizzirri, A.P.; et al. The bone and mineral disorder of children undergoing chronic peritoneal dialysis. Kidney Int. 2010, 78, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, A.; Bhatt, N.; Rossetti, S.; Beto, J. Management of Hyperphosphatemia in End-Stage Renal Disease: A New Paradigm. J. Ren. Nutr. 2021, 31, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, M.; Yang, D.; Bouxsein, M.L.; Saito, H.; Galvin, R.J.; Kuhstoss, S.A.; Thomas, C.C.; Schipani, E.; Baron, R.; et al. Suppression of Wnt signaling by Dkk1 attenuates PTH-mediated stromal cell response and new bone formation. Cell Metab. 2010, 11, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dela Cruz, A.; Grynpas, M.D.; Mitchell, J. Elevated Galpha11 expression in osteoblast lineage cells promotes osteoclastogenesis and leads to enhanced trabecular bone accrual in response to pamidronate. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E811–E820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolov, I.G.; Joki, N.; Nguyen-Khoa, T.; Ivanovski, O.; Phan, O.; Lacour, B.; Drueke, T.B.; Massy, Z.A.; Dos Reis, L.M.; Jorgetti, V.; et al. Chronic kidney disease bone and mineral disorder (CKD-MBD) in apolipoprotein E-deficient mice with chronic renal failure. Bone 2010, 47, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Mac Way, F.; Lessard, M.; Lafage-Proust, M.H. Pathophysiology of chronic kidney disease-mineral and bone disorder. Jt. Bone Spine 2012, 79, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Aleksova, J.; Ng, K.W.; Jung, C.; Zeimer, H.; Dwyer, K.M.; Milat, F.; MacIsaac, R.J. Bone health in chronic kidney disease-mineral and bone disorder: A clinical case seminar and update. Intern. Med. J. 2018, 48, 1435–1446. [Google Scholar] [CrossRef]
- Babayev, R.; Nickolas, T.L. Can one evaluate bone disease in chronic kidney disease without a biopsy? Curr. Opin. Nephrol. Hypertens. 2014, 23, 431–437. [Google Scholar] [CrossRef]
- Bacchetta, J.; Bernardor, J.; Garnier, C.; Naud, C.; Ranchin, B. Hyperphosphatemia and Chronic Kidney Disease: A Major Daily Concern Both in Adults and in Children. Calcif. Tissue Int. 2021, 108, 116–127. [Google Scholar] [CrossRef]
- Iseri, K.; Dai, L.; Chen, Z.; Qureshi, A.R.; Brismar, T.B.; Stenvinkel, P.; Lindholm, B. Bone mineral density and mortality in end-stage renal disease patients. Clin. Kidney J. 2020, 13, 307–321. [Google Scholar] [CrossRef]
- de Jonge, F.A.; Pauwels, E.K.; Hamdy, N.A. Scintigraphy in the clinical evaluation of disorders of mineral and skeletal metabolism in renal failure. Eur. J. Nucl. Med. 1991, 18, 839–855. [Google Scholar] [CrossRef]
- Fogelman, I.; Bessent, R.G.; Turner, J.G.; Citrin, D.L.; Boyle, I.T.; Greig, W.R. The use of whole-body retention of Tc-99m diphosphonate in the diagnosis of metabolic bone disease. J. Nucl. Med. 1978, 19, 270–275. [Google Scholar] [PubMed]
- Park-Holohan, S.J.; Blake, G.M.; Fogelman, I. Quantitative studies of bone using (18)F-fluoride and (99m)Tc-methylene diphosphonate: Evaluation of renal and whole-blood kinetics. Nucl. Med. Commun. 2001, 22, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Enevoldsen, L.H.; Heaf, J.; Hojgaard, L.; Zerahn, B.; Hasbak, P. Increased technetium-99 m hydroxy diphosphonate soft tissue uptake on bone scintigraphy in chronic kidney disease patients with secondary hyperparathyroidism: Correlation with hyperphosphataemia. Clin. Physiol. Funct. Imaging 2017, 37, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Frost, M.L.; Compston, J.E.; Goldsmith, D.; Moore, A.E.; Blake, G.M.; Siddique, M.; Skingle, L.; Fogelman, I. (18)F-fluoride positron emission tomography measurements of regional bone formation in hemodialysis patients with suspected adynamic bone disease. Calcif. Tissue Int. 2013, 93, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Messa, C.; Goodman, W.G.; Hoh, C.K.; Choi, Y.; Nissenson, A.R.; Salusky, I.B.; Phelps, M.E.; Hawkins, R.A. Bone metabolic activity measured with positron emission tomography and [18F]fluoride ion in renal osteodystrophy: Correlation with bone histomorphometry. J. Clin. Endocrinol. Metab. 1993, 77, 949–955. [Google Scholar]
- Wettschureck, N.; Rutten, H.; Zywietz, A.; Gehring, D.; Wilkie, T.M.; Chen, J.; Chien, K.R.; Offermanns, S. Absence of pressure overload induced myocardial hypertrophy after conditional inactivation of Galphaq/Galpha11 in cardiomyocytes. Nat. Med. 2001, 7, 1236–1240. [Google Scholar] [CrossRef]
- Zhang, M.; Xuan, S.; Bouxsein, M.L.; von Stechow, D.; Akeno, N.; Faugere, M.C.; Malluche, H.; Zhao, G.; Rosen, C.J.; Efstratiadis, A.; et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J. Biol. Chem. 2002, 277, 44005–44012. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.R.; Lund, R.J.; Mathew, S.; Hruska, K.A. Low turnover osteodystrophy and vascular calcification are amenable to skeletal anabolism in an animal model of chronic kidney disease and the metabolic syndrome. J. Am. Soc. Nephrol. 2005, 16, 917–928. [Google Scholar] [CrossRef] [Green Version]
- Nikolov, I.G.; Joki, N.; Nguyen-Khoa, T.; Guerrera, I.C.; Maizel, J.; Benchitrit, J.; Machado dos Reis, L.; Edelman, A.; Lacour, B.; Jorgetti, V.; et al. Lanthanum carbonate, like sevelamer-HCl, retards the progression of vascular calcification and atherosclerosis in uremic apolipoprotein E-deficient mice. Nephrol. Dial. Transpl. 2012, 27, 505–513. [Google Scholar] [CrossRef]
- Gagnon, R.F.; Gallimore, B. Characterization of a mouse model of chronic uremia. Urol. Res. 1988, 16, 119–126. [Google Scholar] [CrossRef]
- Zaloszyc, A.; Schmitt, C.P.; Sayeh, A.; Higel, L.; Gros, C.I.; Bornert, F.; Aubertin-Kirch, G.; Dillenseger, J.P.; Goetz, C.; Constantinesco, A.; et al. Frequent, quantitative bone planar scintigraphy for determination of bone anabolism in growing mice. PeerJ 2021, 9, e12355. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, A.M.; Drezner, M.K.; Glorieux, F.H.; Kanis, J.A.; Malluche, H.; Meunier, P.J.; Ott, S.M.; Recker, R.R. Bone histomorphometry: Standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Min. Res. 1987, 2, 595–610. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| WT a | KO b | WTCKD c | KOCKD d | WTHP e | KOHP f | WTCKD-HP g | KOCKD-HP | |
|---|---|---|---|---|---|---|---|---|
| Body weight (g) | 30.6 ± 2.6 | 29.3 ± 2.1 | 27.4 ± 1.6 | 28.7 ± 2.2 | 33.2 ± 2.8 a | 30.7 ± 3.4 e | 28.6 ± 2.1 | 25.0 ± 2.7 b,g |
| Creatinine (g/L) | 0.70 ± 0.3 | 0.73 ± 0.2 | 2.01 ± 0.7 a | 2.67 ± 0.9 b | 1.08 ± 0.3 | 0.95 ± 0.3 | 3.06 ± 0.7 a | 2.69 ± 0.7 b |
| Urea (mmol/L) | 7.0 ± 0.6 | 6.9 ± 1.1 | 21.9 ± 3.1 a | 24.9 ± 6.4 b | 7.0 ± 1.0 | 7.7 ± 2.2 | 16.3 ± 2.3 a | 17.7 ± 3.0 b |
| Albumine (g/L) | 22.9 ± 2.1 | 20.6 ± 2.1 | 21.2 ± 2.6 | 21.5 ± 2.9 | 20.5 ± 4.4 | 20.1 ± 3.0 | 22.8 ± 2.2 | 21.9 ± 2.5 |
| Calcium (mmol/L) | 2.36 ± 0.1 | 2.37 ± 0.1 | 2.53 ± 0.1 | 2.64 ± 0.04 b | 2.10 ± 0.3 a | 2.42 ± 0.2 e | 2.57 ± 0.2 a | 2.64 ± 0.2 b |
| Phosphate (mmol/L) | 0.98 ± 0.2 | 1.03 ± 0.1 | 1.35 ± 0.3 a | 1.41 ± 0.4 b | 1.78 ± 0.3 a | 1.67 ± 0.2 b | 1.62 ± 0.08 a | 1.99 ± 0.5 b |
| PTH (ng/L) | 54 ± 37 | 70 ± 25 | 106 ± 95 | 173 ± 108 b | 229 ± 155 a | 293 ± 170 b | 1907 ± 1099 a | 1687 ± 789.1 b |
| WT a | KO b | WTCKD c | KOCKD d | WTHP e | KOHP f | WTCKD-HP g | KOCKD-HP | |
|---|---|---|---|---|---|---|---|---|
| n = 6 | n = 10 | n = 9 | n = 6 | n = 8 | n = 10 | n = 7 | n = 9 | |
| Femoral length (mm) | 16.0 ± 0.2 | 15.9± 0.3 | 15.9 ± 0.2 | 16.1 ± 0.3 | 16.1 ± 0.2 | 15.8 ± 0.3 | 16.2 ± 0.1 | 15.7 ± 0.3 |
| Cortical parameters (Diaphysis) | ||||||||
| Ct.TMD (mg/cc) | 1269 ± 62 | 1144 ± 79 a | 1131 ± 47 a | 1159 ± 37 | 1202 ± 61 | 1202 ± 49 b | 1214 ± 94 | 1174 ± 70 |
| Ct.Th (μm) | 197 ± 141 | 179 ± 15 a | 191 ± 12 | 183 ± 13 | 195 ± 10 | 194 ± 17 b | 209 ± 105 | 179 ± 16 g |
| Ct.Ar (mm2) | 0.83 ± 0.06 | 0.77 ± 0.06 | 0.79 ± 0.04 | 0.80 ± 0.08 | 0.85 ± 0.06 | 0.82 ± 0.06 | 0.90 ± 0.07 a | 0.73 ± 0.09 g |
| Cortical parameters (Metaphysis) | ||||||||
| Ct.TMD (mg/cc) | 1142 ± 42 | 1059 ± 59 a | 993 ± 70 a | 1031 ± 48 | 1089 ± 60 | 1091 ± 27 | 1029 ± 122 a | 1067 ± 75 |
| Ct.Th (μm) | 158 ± 12 | 141 ± 17 | 159 ± 14 | 146 ± 5 | 171 ± 16 | 156 ± 17 | 169 ± 17 | 148 ± 29 g |
| Ct.Ar (mm2) | 0.76 ± 0.05 | 0.72 ± 0.07 | 0.80 ± 0.04 | 0.73 ± 0.03 | 0.89 ± 0.06 a | 0.80 ± 0.06 e | 0.86 ± 0.12 a | 0.72 ± 0.16 g |
| Trabecular parameters | ||||||||
| Tb TMD (mg/cc) | 765 ± 76 | 733 ± 90 | 690 ± 49 a | 712 ± 37 | 679 ± 54 a | 7116 ± 41 | 729 ± 102 | 754 ± 61 |
| BV/TV (%) | 9.1 ± 1.6 | 10.3 ± 2.7 | 9.6 ± 1.9 | 11.6 ± 2.6 | 7.6 ± 1.9 | 7.9 ± 1.7 | 9.5 ± 3.0 | 7.8 ± 4.4 |
| Tb.Th (μm) | 28.1 ± 3.1 | 27.4 ± 3.1 | 25.4 ± 2.2 | 28.4 ± 3.0 | 23.3 ± 3.6 a | 25.0 ± 2.3 | 26.2 ± 4.6 | 27.9 ± 4.9 |
| Tb.N (/mm) | 3.2 ± 0.3 | 3.7 ± 0.8 | 3.8 ± 0.8 | 4.1 ± 0.6 | 3.3 ± 0.7 | 3.2 ± 0.5 | 3.7 ± 11 | 2.6 ± 1.0 b,g |
| Tb.Sp (μm) | 283 ± 29 | 256 ± 62 | 248 ± 54 | 221 ± 38 | 295 ± 66 | 298 ± 51 | 272 ± 103 | 394 ± 143 b,g |
| DA | 1.20 ± 0.06 | 1.30 ± 0.15 | 1.15 ± 0.07 | 1.26 ± 0.10 | 1.18 ± 0.06 | 1.20 ± 0.06 | 1.22 ± 0.11 | 1.21 ± 0.06 |
| SMI | 2.32 ± 0.18 | 2.00 ± 0.31 | 2.46 ± 0.21 | 2.12 ± 0.63 | 2.77 ± 0.48 a | 2.43 ± 0.26 b | 2.49 ± 0.58 | 2.36 ± 0.31 |
| Scintigraphic Index at Week 10 (Counts·s−1·Pixel−1·MBq−1·g−1 × 105) | Scintigraphic Index at Week 22 (Counts·s−1·Pixel−1·MBq−1·g−1 × 105) | |
|---|---|---|
| WT a | 11.75 ± 0.61 (n = 33) | 6.47 ± 1.55 (n = 9) |
| KO b | 14.80 ± 0.70 (n = 35) a | 16.19 ± 1.63 (n = 7) |
| WTCKD c | 6.28 ± 0.83 (n = 13) a | |
| KOCKD d | 12.10 ± 1.19 (n = 10) b,c | |
| WTHP e | 5.31 ± 1.46 (n = 6) | |
| KOHP f | 12.00 ± 2.19 (n = 8) | |
| WTCKD-HP g | 5.60 ± 0.74 (n = 7) a | |
| KOCKD-HP h | 10.11 ± 1.52 (n = 9) b |
| WT | KO | WTCKD | KOCKD | |
|---|---|---|---|---|
| BV/TV(%) | 26.9 ± 5.5 | 24.7 ± 4.2 | 25.9 ± 3.2 | 32.2 ± 14.6 |
| BS/BV(%) | 4.0 ± 0.4 | 4.8 ± 0.5 | 3.8 ± 0.6 | 3.6 ± 1.2 |
| Trabecular Thickness (µm) | 50.2 ± 5.1 | 42.0 ± 4.4 | 54.1 ± 7.4 | 63.3 ± 30.3 |
| Trabecular Number | 5.3 ± 0.7 | 5.9 ± 0.5 | 4.9 ± 0.9 | 5.1 ± 0.7 |
| Trabecular Separation (µm) | 139.0 ± 28.0 | 139.4 ± 28.0 | 157.9 ± 38.9 | 134.4 ± 38.8 |
| OV/BV (%) | 2.8 ± 1.7 | 4.6 ± 1.4 | 3.1 ± 2.4 | 4.2 ± 1.4 |
| OS/BS(%) | 11.1 ± 4.2 | 22.1 ± 9.5 | 14.8 ± 7.4 | 18.7 ± 3.8 |
| Ob.S/BS (%) | 23.0 ± 3.0 | 31.9 ± 8.6 | 20.8 ± 11.5 | 29.5 ± 5.5 |
| MAR (µm/d) | 1.7 ± 0.3 | 2.1 ± 0.4 | 1.5 ± 0.3 | 1.6 ± 0.4 |
| Mineralizing Surface (%) | 7.0 ± 3.0 | 12.9 ± 6.6 | 4.1 ± 3.6 | 8.5 ± 3.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaloszyc, A.; Choquet, P.; Sayeh, A.; Bartosova, M.; Schaefer, B.; Huegel, U.; Aubertin-Kirch, G.; Healy, C.; Severac, F.; Rizzo, S.; et al. Inactivation of Osteoblast PKC Signaling Reduces Cortical Bone Mass and Density and Aggravates Renal Osteodystrophy in Mice with Chronic Kidney Disease on High Phosphate Diet. Int. J. Mol. Sci. 2022, 23, 6404. https://doi.org/10.3390/ijms23126404
Zaloszyc A, Choquet P, Sayeh A, Bartosova M, Schaefer B, Huegel U, Aubertin-Kirch G, Healy C, Severac F, Rizzo S, et al. Inactivation of Osteoblast PKC Signaling Reduces Cortical Bone Mass and Density and Aggravates Renal Osteodystrophy in Mice with Chronic Kidney Disease on High Phosphate Diet. International Journal of Molecular Sciences. 2022; 23(12):6404. https://doi.org/10.3390/ijms23126404
Chicago/Turabian StyleZaloszyc, Ariane, Philippe Choquet, Amira Sayeh, Maria Bartosova, Betti Schaefer, Ulrike Huegel, Gaëlle Aubertin-Kirch, Christopher Healy, François Severac, Sébastien Rizzo, and et al. 2022. "Inactivation of Osteoblast PKC Signaling Reduces Cortical Bone Mass and Density and Aggravates Renal Osteodystrophy in Mice with Chronic Kidney Disease on High Phosphate Diet" International Journal of Molecular Sciences 23, no. 12: 6404. https://doi.org/10.3390/ijms23126404